
Abstract
Here we report a facile approach to synthesize highly optically active oxindole-type analogues with both high yield and enantioselectivity. This single-step synthesis strategy represents a substantial improvement upon existing methods that are often involved with multi-step routes and have suboptimal atomic economy. One such compound, namely Q4c, showed remarkable in vivo anti-inflammatory activity with efficiency approaching to that of a steroidal compound dexamethasone. Moreover, Q4c alleviated pain in mouse models with comparable activity to morphine. Further investigation suggested that nitric oxide signaling pathway is involved in the anti-inflammatory and analgesic activities of Q4c. Notably, this is the first time that chiral oxindole-type analogues have been identified to be both anti-inflammatory and analgesic and our study also paved the way for future development of oxindoles as drug candidates in this field.
Similar content being viewed by others
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are among the most commonly prescribed medications for reducing pain and inflammation. While NSAIDs exert their pharmacological activity by inhibiting cyclooxygenases (COXs)1, they represent a wide range of compounds with chemically distinct structures. Despite their notable anti-inflammatory activity, NSAIDs are usually associated with gastrointestinal, cardiovascular and renal complications2,3,4. Further improvements on anti-inflammatory medications require more detailed understanding of inflammatory biology and perhaps development of novel anti-inflammatory drugs with high efficacy yet low side effects.
Traditional NSAIDs usually require multi-step synthesis procedure. For example, ibuprofen, one of the most widely used NSAIDs on the market, is produced through a three-step synthesis route5. The complexity of synthesis reaction has been a major obstacle to reducing the cost of anti-inflammatory agents. Rapid, efficient synthesis methodologies play a critical role in current medicinal chemistry. Construction of chiral compounds on the basis of biologically relevant frameworks has been the focus of medicinal chemists for decades. This target-oriented organic synthesis has greatly advanced the overall speed of drug discovery6,7. To date, however, it remains a great challenge to develop a highly efficient and simply-convenient strategy, thereby contributing to the development of new therapeutic agents.
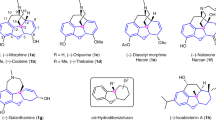
Chiral oxindole-type molecules are increasingly important lead compounds for a wide range of biologically active molecules8,9,10,11 Evaluation of optically active oxindoles as therapeutic agents requires the development of a facile, robust synthesis route. On the basis of our recent efforts to develop novel methods to synthesize biologically and pharmaceutically active chiral spirooxindole-type alkaloids by asymmetric strategies in our group (Fig. 1A,B)12,13. Interestingly, one such compound, JP-8 g, exhibited not only a wide range of anti-cancer activity13, but also potent in vivo anti-inflammatory activity14. In vitro and in vivo experiments suggested that JP-8g exerted anti-inflammatory activity through nitric oxide (NO) signaling pathway14. Most interestingly, when compared with a traditional NSAID, indomethacin, JP-8 g showed dramatically reduced acute gastric damage15. These results suggested that spirooxindoles as anti-inflammaory agents may possess some advantages over conventional NSAIDs.

Synthesis of optically and biologically active spirooxindole-type alkaloids by asymmetric strategies.
(A) SPX-F with antipyretic activity, three-step synthesis12; (B) JP-8 g with inflammatory and anti-cancer activities, three-step synthesis13,14; (C) proposed catalytic mechanism for the one-step synthesis of oxindoles in this study; (D) the structure of new oxindole-type molecules.
In order to expedite the evaluation of their therapeutic potential, we sought to establish a simple and robust approach for the synthesis of oxindole derivatives. Although our previous methods are highly efficient, SPXs12 and JPs13 series compounds still require multi-step synthesis. In this study, we first designed a single-step approach for the asymmetric synthesis of spirooxindole-type compounds and explored the feasibility of using these compounds as anti-inflammatory and analgesic drug candidates. This single-step synthesis strategy represents a substantial improvement upon existing methods that are often involved with multi-step routes and have suboptimal atomic economy.
Results
Synthesis of chiral oxindole-type compounds
Inspired by the bifunctional catalytic system-based asymmetric synthesis16,17,18, we rationalized that isatylidene malononitrile can be activated by two thiourea hydrogen atoms through weak hydrogen bonds. This can be achieved by the nucleophilic attack of enolate, which is promoted by the HOMO energy raised from the interaction between the tertiary amine moiety of catalyst and the carbonyl group of benzoylacetonitrile. As a result, this process leads to the formation of spirooxindole-type product (Fig. 1C,D), (Supplementary Fig. 1).
To explore the possibility of the proposed cyclization process, our investigation began with screening of reaction conditions to evaluate their catalytic activities (Supplementary Fig. 2). Subsequently, the results of experiments in which a variety of diversely structured spirooxindole-type compounds were ingeniously constructed under the optimized conditions are summarized in Fig. 2. In general, variation of the electronic properties of the substituent at different sites of the N-protected or unprotected isatylidenemalononitriles with different steric parameters was tolerated, giving the corresponding spiro[indoline-3,4′-pyran]s in excellent enantioselectivities (93–>99% ee, Q4a – Q4p) and good to high yields. As expected, various substituted benzoylacetonitriles at different positions on the aromatic ring could also be tolerated and gave the corresponding compounds Q4q – Q4t with excellent enantioselectivities (90–>99% ee) in yields ranging from 75 to 90%. Our synthetic method shows higher atom economy without using any metal, as well as operationally simple process with low costs will benefit for using of these potential compounds in pharmaceuticals discovery.

Synthesis of various isatylidene malononitriles under optimized conditions.
Unless noted, the reactions were conducted with 2 (0.22 mmol) and 3 (0.20 mmol) using 1.0 mol% catalyst in Et2O for 30 min at room temperature. Yield and ee values were determined by HPLC and configuration was assigned by comparison of retention time and specific rotation of obtained compounds with the data reported in previous literatures13,14. See Supplementary Fig. 3 for more details.
Structure and activity relationship (SAR) of Q series compounds
Despite structurally different from traditional NSAIDs, chiral spirooxindole framework has been discovered to exhibit antipyretic, anti-cancer and anti-inflammatory activities12,13,14. After constructing a series of compounds with optically active spiro [indoline-3,4′-pyran] frameworks, we sought to evaluate their therapeutic potential as anti-inflammatory and analgesic agents. We first studied the structure-activity relationship (SAR) of selected compounds using a mouse ear inflammation model. As shown in Fig. 3, many of these compounds showed significant suppression of ear swelling. Excitingly, one compound, Q4c, exhibited remarkable in vivo anti-inflammatory activity.

Structure-activity relationship of Q series compounds.
Synthesized spirooxindole compounds (12.5 mg/kg) were tested using mouse ear inflammation model (n = 6). A steroidal anti-inflammatory drug dexamethasone (DEX, 5.0 mg/kg) was used as a reference compound.
Anti-inflammatory and analgesic activity of Q4c
We next evaluated the time- and dose-dependency of the anti-inflammatory activity of Q4c using mouse paw inflammation model. Swelling of paws was efficiently induced by carrageenan injection during the entire 48 h period and peaked at 4–6 h (Fig. 4A, vehicle). A reference drug dexamethasone (DEX) showed significant suppression (p < 0.001, Tukey’s post-tests) of paw swelling at 4–24 h after carrageenan injection. Q4c of doses ranging from 6.25 to 25.0 mg/kg inhibited the progression of inflammation with efficacy comparable to that of DEX. Similarly, Q4c showed potent anti-inflammatory activity on mouse ear inflammation with the highest dose (50.0 mg/kg) as potent as 5.0 mg/kg DEX (Fig. 4B).

The in vivo anti-inflammatory and analgesic activity of Q4c.
(A) Carrageenan-induced paw inflammation model (n = 7). (B) Xylene-induced ear inflammation model (n = 10). (C) Tail flick pain model (n = 6). (D) Acetic acid twist body pain model (n = 6). Data are expressed as mean ± SEM. Statistical evaluation was performed by two-way ANOVA, followed by Tukey post-tests (*p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.001).
To further understand the therapeutic potential of Q4c, we sought to test whether it had analgesic activity. As seen in Fig. 4C, Q4c effectively suppressed radiant heat-stimulated tail flick. Q4c of high dose (50.0 mg/kg) exhibited efficacy approaching to that of morphine (5.0 mg/kg). In addition, we studied the time- and dose-response of Q4c activity on mouse acetic acid-induced twist model. Mice were pre-treated with vehicle and test compounds for 15 min before the administration of acetic acid. We found that Q4c showed potent in vivo analgesic activity toward the stimulation of acetic acid. It is also worth noting that this activity was largely dose dependent. Consistent with the results on tail flick pain model, high dose of Q4c (50.0 mg/kg) attenuated acetic acid-induced body twist with efficacy comparable to that of morphine (5.0 mg/kg) (Fig. 4D).
In vitro and in vivo toxicity of Q4c
To evaluate the potential of Q4c as a safe medication, we first analyzed its effect on the viability of isolated mouse primary peritoneal macrophages. Minor cytotoxicity was observed only at the highest concentration (50 μM), while Q4c at or below 40 μM had no significant effect on cell viability (Supplementary Fig. 4). We next evaluated the in vivo toxicity of Q4c on Wistar rat using acute gastric damage model. In comparison with indomethacin, Q4c exhibited significantly reduced gastric damage (Supplementary Fig. 5). Nevertheless, it is worth noting that Q4c may not bear the same in vivo target as indomethacin does. Indomethacin is a nonselective inhibitor of COX-1 and COX-2 whereas the target of Q4c is unknown. In vitro inhibition experiments suggested that the median inhibition concentrations (IC50) of Q4c on COX-1 and COX-2 are both above 50 μM, indicating that Q4c is not a potent COX inhibitor (Supplementary Fig. 6). We speculate that Q4c may either require in vivo metabolic processing for COX targeting, or bear a completely novel target for its in vivo activity.
Mechanism of actions of Q4c
To explore how Q4c exerts its anti-inflammatory and analgesic activities, we next examined the signaling pathways that may be involved in its function. Our previous study suggested that nitric oxide synthase (NOS) signaling pathway was involved in the anti-inflammatory activity of JP-8 g, another spirooxindole compound14. Therefore, we sought to determine the effect of Q4c on LPS-stimulated nitric oxide (NO) release in isolated primary macrophages. As seen in Fig. 5A, Q4c suppressed NO release in a dose-dependent manner. Q4c of 10 μM achieved more than 50% suppression. To confirm the role of NOS signaling pathway, we evaluated the effect of NOS inhibitors on the anti-inflammatory activity of Q4c using mouse paw swelling model. It was observed that 30 mg/kg of inducible NOS (iNOS) inhibitor SMT significantly (p < 0.05) reduced the anti-inflammatory activity of Q4c 5–24 h after carrageenan injection (Fig. 5B). In contrast, NF-kB inhibitor BAY 11–7082 did not appear to affect the in vivo activity of Q4c (Supplementary Fig. 7). This is consistent with our previous findings that NOS pathway, but not NF-κB pathway, is involved in the anti-inflammatory activity of spirooxindole compounds15.

Mechanism of actions of Q4c.
(A) The effect of Q4c on LPS-stimulated NO release from primary peritoneal macrophages. Nitrite levels were assessed by Griess reagent. Data are shown as mean ± SEM (n = 5). Significant difference between LPS and test groups are indicated, **p < 0.01; ***p < 0.001.(B) The effect of iNOS antagonist SMT on the in vivo anti-inflammatory activity of Q4c. Data are shown as mean ± SEM (n = 7). Significant difference from vehicle, *p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.001. Significant difference between Q4c treatments in the absence and presence of SMT, #p < 0.05. Statistical analysis was performed by two-way ANOVA, followed by Tukey’s post-tests.
Discussion
Here we described a facile method to synthesize chiral oxindole analogues in a single step and explored the feasibility of using these compounds as anti-inflammatory and analgesic drug candidates. Due to its simplicity, atomic economy, metal-free feature and high yield and enantioselectivity, this reaction could provide very valuable means for preparing biologically active spirooxindoles. Importantly, one product from this reaction, Q4c, exhibited potent in vivo anti-inflammatory and analgesic activity. Q4c is different from traditional NSAIDs in their chemical structure and in vitro inhibition experiments suggested that Q4c could not efficiently inhibit COX enzymes. Further studies indicated that Q4c exerts the anti-inflammatory activity through NOS signaling pathway, though its in vivo target and detailed mechanism of actions are yet to be elucidated. Our data also showed that Q4c had only minor in vitro cytotoxicity to primary peritoneal macrophage and caused no appreciable acute gastric damage. It is important, however, to carefully assess the safety of Q4c using other animal models before its therapeutic potential can be considered.
Methods
Synthesis of Q series compounds and evaluation of in vivo anti-inflammatory and analgesic activities of Q4c. See Supplementary Information for more details.
Ethics Statement
All animal experiments were carried out in accordance with the approved guidelines of China Council on Animal Care and Use. All animal procedures performed in this study were approved by the Institutional Animal Care and Use Committee of the Ethics Committee of Lanzhou University, China.
Animal Experiments
All animals (C57BL/6J mice and Wistar rats) were maintained and the experiments were carried out in accordance with the European Community guidelines for the use of experimental animals (86/609/EEC). All the protocols in this study were executed under the guideline of the Ethics Committee of Lanzhou University, China. Find Supplementary Information for further details.
Statistical Analysis
Statistical significance of results was analyzed by one- or two-way ANOVA using Prism software version 6.01 (GraphPad Software, San Diego, CA, USA) followed by Tukey’s post-tests, respectively. Data were presented as mean ± SEM unless noted otherwise.
Additional Information
How to cite this article: Sun, Y. et al. One-Step Synthesis of Chiral Oxindole-type Analogues with Potent Anti-inflammatory and Analgesic Activities. Sci. Rep. 5, 13699; doi: 10.1038/srep13699 (2015).
References
Smith, W. L. & Dewitt, D. L. Prostaglandin endoperoxide H synthases-1 and -2. Adv. Immunol. 62, 167–215 (1996).
Ding, K. et al. Structure-based design of potent non-peptide MDM2 inhibitors. J. Am. Chem. Soc. 127, 10130–10131 (2005).
Rottmann, M. et al. Spiroindolones, a potent compound class for the treatment of malaria. Science 329, 1175–1180 (2010).
Roth, S. H. Coming to terms with nonsteroidal anti-inflammatory drug gastropathy. Drugs 72, 873–879 (2012).
Marnett, L. J. The COXIB experience: a look in the rearview mirror. Annu. Rev. Pharmacol. Toxicol. 49, 265–290 (2009).
Kneller, R. The importance of new companies for drug discovery: origins of a decade of new drugs. Nat. Rev. Drug Discov. 9, 867–882 (2010).
Vintonyak, V. V. et al. Identification of thiazolidinones spiro-fused to indolin-2-ones as potent and selective inhibitors of the Mycobacterium tuberculosis protein tyrosine phosphatase B. Angew. Chem. Int. Ed. Engl. 49, 5902–5905 (2010).
Ding, K. et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 49, 3432–3435 (2006).
Galliford, C. V. & Scheidt, K. A. Pyrrolidinyl-spirooxindole natural products as inspirations for the development of potential therapeutic agents. Angew. Chem. Int. Ed. Engl. 46, 8748–8758 (2007).
Trost, B. M. & Brennan, M. K. Asymmetric Syntheses of Oxindole and Indole Spirocyclic Alkaloid Natural Products. Synthesis 2009, 3003–3025 (2009).
Badillo, J. J., Hanhan, N. V. & Franz, A. K. Enantioselective synthesis of substituted oxindoles and spirooxindoles with applications in drug discovery. Curr. Opin. Drug Discov. Devel. 13, 758–776 (2010).
Jiang, X. et al. A unique approach to the concise synthesis of highly optically active spirooxazolines and the discovery of a more potent oxindole-type phytoalexin analogue. J. Am. Chem. Soc. 132, 15328–15333 (2010).
Jiang, X. X. et al. Core Scaffold-Inspired Concise Synthesis of Chiral Spirooxindole-Pyranopyrimidines with Broad-Spectrum Anticancer Potency. Adv. Synth. Catal. 354, 917–925 (2012).
Sun, Y. L. et al. Anti-cancer small molecule JP-8g exhibits potent in vivo anti-inflammatory activity. Sci Rep 4, 4372 (2014).
Liu, J. et al. Spirooxindoles, a potential novel class of anti-inflammatory agents. Inflamm. Cell. Signal. 1, e372 (2014).
Taylor, M. S. & Jacobsen, E. N. Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem. Int. Ed. Engl. 45, 1520–1543 (2006).
Doyle, A. G. & Jacobsen, E. N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 107, 5713–5743 (2007).
Pan, S. C., Zhou, J. & List, B. Catalytic asymmetric acylcyanation of imines. Angew. Chem. Int. Ed. Engl. 46, 612–614 (2007).
Acknowledgements
We are grateful for the grants from the National Natural Science Foundation of China (Nos. 21202072, 21432003, 81200469 and 81401553) and Major Scientific and Technological Project of Guangdong Province (No. 2011A080300001) .
Author information
Authors and Affiliations
Contributions
X.J. and R.W. conceived the project and drafted the manuscript. X.J., Y.S., J.L., L.L., T.S., X.Z., Y.W., D.S., Z.Y. and J.L. designed the experiments, performed the syntheses and carried out the characterization and property studies. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sun, Y., Liu, J., Jiang, X. et al. One-Step Synthesis of Chiral Oxindole-type Analogues with Potent Anti-inflammatory and Analgesic Activities. Sci Rep 5, 13699 (2015). https://doi.org/10.1038/srep13699
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep13699
This article is cited by
-
Investigating anti-inflammatory and apoptotic actions of fucoidan concentrating on computational and therapeutic applications
3 Biotech (2023)
-
Green synthesis of isatin-based compounds
Research on Chemical Intermediates (2022)
-
Anti-inflammatory activity of 3-cinnamoyltribuloside and its metabolomic analysis in LPS-activated RAW 264.7 cells
BMC Complementary Medicine and Therapies (2020)
-
Discovery of hybrids of indolin-2-one and nitroimidazole as potent inhibitors against drug-resistant bacteria
The Journal of Antibiotics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
