
Abstract
In many Gram-negative bacteria, including Salmonella enterica serovar Typhimurium (S. Typhimurium), the sigma factor RpoS/σS accumulates during stationary phase of growth and associates with the core RNA polymerase enzyme (E) to promote transcription initiation of genes involved in general stress resistance and starvation survival. Whereas σ factors are usually inactivated upon interaction with anti-σ proteins, σS binding to the Crl protein increases σS activity by favouring its association to E. Taking advantage of evolution of the σS sequence in bacterial species that do not contain a crl gene, like Pseudomonas aeruginosa, we identified and assigned a critical arginine residue in σS to the S. Typhimurium σS-Crl binding interface. We solved the solution structure of S. Typhimurium Crl by NMR and used it for NMR binding assays with σS and to generate in silico models of the σS-Crl complex constrained by mutational analysis. The σS-Crl models suggest that the identified arginine in σS interacts with an aspartate of Crl that is required for σS binding and is located inside a cavity enclosed by flexible loops, which also contribute to the interface. This study provides the basis for further structural investigation of the σS-Crl complex.
Similar content being viewed by others

Introduction
In bacteria, a primary housekeeping sigma factor and one or more alternative σ factors associate with the catalytically active RNA polymerase (RNAP) core enzyme (α2ββ’ω, E), to form the holoenzyme Eσ and direct transcription initiation of specific subsets of genes1,2. In many Gram-negative bacteria, σS/RpoS is produced during late exponential phase, or in response to stress, to modify global gene transcription and to allow stationary phase survival and general stress resistance3,4,5. In the wide host-range pathogen S. Typhimurium, σS is not only required for general stress resistance, but also for virulence, biofilm formation and development of the red dry and rough (rdar) morphotype, a colony morphology caused by the production of amyloid fibers (curli) and cellulose6,7,8.
The efficiency of formation of the housekeeping and alternative Eσ can be modulated by regulatory factors that bind E and/or σ5,9. So far, Crl is the only known σS-dedicated regulatory factor that enhances σS activity through a direct interaction, favouring EσS formation7,10,11,12,13,14,15. Analyses of sequenced bacterial genomes revealed that crl is less widespread and less conserved at the sequence level than rpoS16. Nevertheless, Crl family members perform the same biological function and share a common mechanism of σS binding17,18. Moreover, the X-ray crystal structure of Crl from Proteus mirabilis (CrlPM) and mutational analyses strongly suggest that σS binds to a Crl cavity enclosed by flexible loops16,17,18. In contrast to housekeeping σ factors, a three dimensional structure is not available for σS. However, sequence conservation between σS and housekeeping σ suggests that, like housekeeping sigma factors, σS contains four structural domains connected by flexible linkers19,20. Domain 2 (σ2), the most highly conserved domain of σ factors19,20, is the only σS domain involved in Crl binding and two regions of σS2 are required for interaction16,21. In the structural model of S. Typhimurium σS (σSSTM)16, these two regions are accessible and close together, consisting of an α-helix (α2) and the DPE motif located within a long loop just on top of helix α2 (Fig. 1a,b).

The Crl binding region of S. Typhimurium σS and sequence comparison of σS from species harbouring crl or lacking crl.
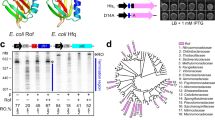
(a) Cartoon representation of the structural model16 of full-length σSSTM, in which domains σS2, σS3 and σS4 are shown. The helix α216,21 and the DPE motif21 within σS2 are highlighted. (b) Zoomed view of σS2 in which the side chains of critical residues are depicted in cyan and yellow. (c) WebLogo (http://weblogo.threeplusone.com/create.cgi) of σS residues 71–91 (numbered as in σSSTM) generated with the σS sequences listed in Supplementary Figure S2, from bacterial genomes containing crl or lacking crl. (d) Sequence alignment of σS helix α2 from S. Typhimurium and P. aeruginosa, both with their own numbering. Residues that differ between σSPA and σSSTM are highlighted in the alignment and represented with the same colour code as in the surface representation of S. Typhimurium σS2 shown above.
Many bacterial species containing rpoS do not harbour crl in their genome, such as Pseudomonas aeruginosa22, in which σS is involved in stress resistance and in the production of virulence factors8. We show here that Pseudomonas aeruginosa σS (σSPA) does not interact with Crl, despites the conservation of the DPE motif. Most interestingly, substitution of one single residue in the helix α2 of σSPA was sufficient to confer to σSPA the ability to bind Crl and our data assigned this residue to the σS-Crl binding interface. By NMR, we solved the solution structure of S. Typhimurium Crl (CrlSTM) and used it for NMR binding assays with σSSTM. Furthermore, in silico models of the σSSTM-CrlSTM complex were generated based on mutational analyses. The output models show that two specific salt bridges can be formed between Crl and σS, in agreement with our previous biophysical data suggesting that σS-Crl complex formation is driven by electrostatic interactions18.
Results
Crl does not activate σS from Pseudomonas aeruginosa
σS2 is the only domain involved in the interaction with Crl16,18,21 and two regions, close together on the structural model of σSSTM16 (Fig. 1a,b), were identified as the Crl binding regions: the helix α2, corresponding to residues 74 to 85 in σSSTM16,21 and the DPE motif, corresponding to residues 135 to 137 and initially identified in σS from E. coli21. Consistently, a fragment of σSSTM domain 2 lacking this motif, σSSTM (1–136) and σSSTM variants at position D135 and E137, were not able to interact with Crl in bacterial two hybrid (BACTH) assays (Supplementary Fig. S1), confirming that the DPE motif in σSSTM is involved in Crl binding.
However, the DPE motif is conserved in σS of P. aeruginosa (σSPA) that does not have crl22 and the sequence of the helix α2 differs from σSSTM by only four residues (Fig. 1c,d and Supplementary Fig. S2), prompting us to examine whether σSPA can be activated by Crl. σSPA activity and its response to Crl were evaluated in a S. Typhimurium strain in which the native rpoS gene was replaced by the rpoS allele from P. aeruginosa (rpoSPA). Development of the rdar morphotype of S. Typhimurium is highly dependent on σS and Crl7 (compare spots 1, 2 and 11, Fig. 2). The S. Typhimurium strain harbouring the rpoSPA allele was able to develop the rdar morphotype, in contrast to the ΔrpoS mutant of S. Typhimurium (compare spots 3 and 11, Fig. 2), indicating that σSPA was expressed and functional in this strain. However, the rdar morphotype of the rpoSPA strain was similar to that of the S. Typhimurium Δcrl mutant and was not affected by a Δcrl mutation (compare spots 2, 3 and 4, Fig. 2), suggesting that σSPA did not respond to Crl. We also followed the expression of the rpoS-dependent katN-lacZ transcriptional fusion13 and of the σS protein, in wild type and Δcrl S. Typhimurium strains harbouring the rpoSSTM and rpoSPA alleles (Fig. 3 and Supplementary Fig. S3). The growth kinetics of strains harbouring rpoSSTM and rpoSPA were similar and expression of katN-lacZ and σS was induced in stationary phase, as expected13. The lower expression level of katN-lacZ in the rpoSPA strain, compared to that in the rpoSSTM strain, could be due to differences in the expression level or intrinsic activities of σSPA with respect to σSSTM. Activation of katN-lacZ expression by Crl in Salmonella harbouring the rpoSSTM allele was maximal at the entry to stationary phase when σS begins to accumulate (Fig. 3b), as previously reported13. Indeed, at the entry to stationary phase, katN-lacZ expression was decreased and delayed by the Δcrl mutation. In contrast, no significant effect of the Δcrl mutation on katN-lacZ expression was detected in Salmonella containing the rpoSPA allele (Fig. 3a), even though Crl amounts were similar in the rpoSPA and rpoSSTM strains (Supplementary Fig. S3c).

In vivo activity of σS variants and their sensitivity to Crl activation.
Development of the red dry and rough (rdar) morphotype by S. Typhimurium strains harbouring wild-type and mutant rpoS alleles and the effect of a Δcrl mutation: spot 1, wild-type strain ATCC14028; spot 2, ATCC14028 Δcrl; spot 3, ATCC14028 rpoSPA; spot 4, ATCC14028 rpoSPA Δcrl; spot 5, ATCC14028 rpoSSTM-R82L; spot 6, ATCC14028 rpoSSTM-R82L Δcrl; spot 7, ATCC14028 rpoSPA-L87R; spot 8, ATCC14028 rpoSPA-L87R Δcrl; spot 9, ATCC14028 rpoSSTM-R82E; spot 10, ATCC14028 rpoSSTM-R82E Δcrl; spot 11, ATCC14028 ΔrpoS; spot 12, ATCC14028 ΔrpoS Δcrl.

Expression kinetics of the katN-lacZ transcriptional fusion in S. Typhimurium strains harbouring different rpoS alleles.
Growth (dashed lines) and β-galactosidase activities (solid lines) of the S. Typhimurium strains indicated, harbouring wild-type and mutant rpoS alleles from P. aeruginosa (a) and S. Typhimurium (b). Aliquots were taken at different time intervals during growth and β-galactosidase activity was measured in Miller units. Aliquots were also used for σS immunodetection (Supplementary Fig. S3b). The growth phase was determined by the measurement of culture turbidity at OD 600 nm. The experiments were repeated twice with similar results.
To determine whether the failure of σSPA to respond to Crl activation resulted from a lack of interaction between the two proteins, we used the BACTH in vivo assay (Fig. 4) and isothermal titration calorimetry (ITC) in vitro assay (Supplementary Fig. S4a). Unlike σSSTM16,18, σSPA did not interact with Crl, suggesting that unidentified σSSTM residues, not conserved in σSPA, are crucial for Crl binding.

BACTH interaction analyses between Crl from S. Typhimurium and σS wild-type and variant proteins from S. Typhimurium and P. aeruginosa.
(a) Interaction between CrlSTM-T18 and the T25-σS hybrid proteins indicated was quantified by measuring β-galactosidase activity in Miller units. Results are the mean of at least three independent experiments and standard deviations are indicated with black bars. (b) Immunodetection of T25-σS fusion proteins by antibodies directed against the T25 polypeptide. Lane 1, σSSTM; lane 2, σSPA; lane 3, σSPA H83Y; lane 4, σSPA L87R; lane 5, σSPA Q89L; lane 6, σSPA K90R; lane 7, σSSTM; lane 8, σSSTM R82L; lane 9, σSSTM R82E.
A single amino acid substitution renders σSPA sensitive to Crl activation
To further refine our understanding of σS2 residues involved in Crl binding, the amino acid sequence of σS2 was compared in bacterial species harbouring crl in their genome and in those lacking crl (Fig. 1c and Supplementary Fig. S2). Whereas the DPE motif was well conserved, the sequence of the helix α2 was more variable, especially in σS from strains lacking crl. In this region, the sequence between σSSTM and σSPA differs by four surface-exposed residues (Y78, R82, L84 and R85 in σSSTM that correspond to H83, L87, Q89 and K90 in σSPA, respectively) (Fig. 1d). Two of these (R82 and L84 in σSSTM) are conserved in all σS proteins from species harbouring crl, but less conserved in σS from species lacking crl. To determine whether the non-conserved residues at position 83, 87, 89 and 90 in σSPA were responsible for the defect in Crl binding, we constructed σSPA variants in which the σSSTM sequence was restored at these positions and assessed their ability to interact with Crl in the BACTH assay (Fig. 4). Expression levels of σSPA wild type and variants were similar (Fig. 4b). Interestingly, one variant, σSPA L87R, was able to interact with Crl (Fig. 4a), suggesting that an arginine at position 87 in σSPA (corresponding to position 82 in σSSTM) is of paramount importance for Crl binding. This finding was further confirmed in vitro by ITC (Supplementary Fig. S4c,d). Interestingly, σSPA L87R and wild-type σSSTM showed similar affinity for Crl (Supplementary Table S1). The major difference observed between σSPA L87R and σSSTM was in the value of ΔbH, which was less negative for σSPA L87R than for σSSTM. This suggests that the number and type of intermolecular interactions in Crl-σSPA L87R and Crl-σSSTM complexes might be slightly different, due to non-conserved σS residues affecting directly or indirectly the σS-Crl binding interface. However, the ΔbS and ΔbG values were similar for σSPA L87R and σSSTM, endorsing the key role of an arginine at position 87 in the σSPA variant.
To assess whether the interaction between σSPA L87R and Crl was functional (i.e. whether Crl activates σSPA L87R), we monitored the rdar morphotype of S. Typhimurium harbouring the chromosomal rpoSPA-L87R allele expressing σSPA L87R. Morphotypes of the rpoSPA-L87R and wild type Salmonella strains were similar and dependent on crl (compare spots 1, 7 and 2, 8, Fig. 2), suggesting that σSPA L87R was able to respond to Crl activation. Consistently, katN-lacZ expression level in the rpoSPA-L87R strain was decreased by the Δcrl mutation (Fig. 3a). Altogether these results demonstrated that substitution L87R renders σSPA sensitive to Crl activation.
Residue R82 in σSSTM is required for Crl binding and activation
To evaluate the impact in Crl binding of σSSTM residue R82, corresponding to L87 in σSPA, (Fig. 1d), the ability of the σSSTM R82L variant to interact with Crl was assessed in BACTH and ITC assays (Fig. 4 and Supplementary Fig. S4b). Both assays showed that σSSTM R82L does not interact with Crl. Consistently, development of the rdar morphotype and katN-lacZ expression levels were similar in the rpoSSTM-R82L strain (whatever its crl status) and the Δcrl strain harbouring the wild-type rpoSSTM allele (spots 5, 6 and 2, Fig. 2 and Fig. 3b), indicating that σSSTM R82L was not activated by Crl. Far-UV CD spectra showed a similar secondary and tertiary structure for σSSTM, σSSTM R82L, σSPA and σSPA L87R (Supplementary Fig. S4e,f), indicating that the σS conformation was similar in the four proteins. In the absence of Crl, katN-lacZ expression level was similar in the rpoSSTM-R82L and rpoSSTM strains (Fig. 3b), suggesting that σS stability and its interaction with the core RNAP were not affected by the R82L substitution. To assess the effects of a more drastic amino acid substitution at position 82, the σSSTM R82E variant was characterized. Expression level and activity of this variant were similar to those of σSSTM R82L (spots 9, 10 and 5, 6, Fig. 2 and Supplementary Fig. S5). Altogether, these findings suggested that σSSTM R82 plays a key role in Crl binding and activation.
Solution structure of Salmonella Crl
We previously reported the X-ray crystal structure of Crl from Proteus Mirabilis (CrlPM) (PDB 4Q1118), which suggested a high degree of flexibility of the protein. To get more insights into the dynamics of Crl, we solved the solution structure of CrlSTM by NMR23 (Fig. 5, Supplementary Table S2). Structural alignment with CrlPM indicated that the fold of CrlSTM is conserved with a core consisting of a five-stranded β-sheet flanked by two helices, α1 and α3, with a cavity on top, closed by loops 1 and 2 (Supplementary Fig. S6). The electrostatic surface potential of CrlSTM delimits two faces of the protein, corresponding to lateral entries of the cavity (Fig. 5b). One face is overall neutral with several basic patches, whereas the opposite face is predominantly negatively charged, like loop 3 and the inside of the cavity, which are also rather acidic.

NMR structure of S. Typhimurium Crl.
(a) Backbone ensemble structure of CrlSTM showing the 10 final low-energy conformers with the lowest CYANA target function in cartoon representation. Secondary structure elements are annotated using the numbering of CrlPM18. The two views are rotated by 180°. (b) The electrostatic surface potential was calculated with Delphi44 using the conformer 1. Colours are red to blue for acidic to basic potential. Critical residues, including D36, which delineate the entrance of the cavity, are indicated.
The NMR ensemble structure (Fig. 5a) showed that several regions at the periphery of the core display structural disorder. NMR signals in loop 1 (L19-F33) were broad, possibly due to conformational exchange at the millisecond timescale. Still a number of NOE contacts were found with α1 and β2, showing that it is not completely disordered. Due to the absence of the small helix α2 found in CrlPM, loop 1 of CrlSTM explores a wider space and contributes to forming a deeper cavity than in CrlPM (Supplementary Fig. S6). Residues in loop 2 displayed sharp signals but only few NOE contacts, indicating that this region is disordered and flexible. The difference of dynamics in loop 2 as compared to the structured regions was also corroborated by 15N relaxation experiments, where it displays lower R2 rates that deviate from simulated R2 values (Supplementary Fig. S7). Finally the region corresponding to helix α4 in CrlPM is not structured in CrlSTM (Supplementary Fig. S6b). Indeed, only few inter-residue NOE contacts were found in the P120-P128 region, but they provided evidence of the proximity between the C-terminus and helix α3. Strikingly, signals of several residues in the core β-sheet were broad, denoting conformational fluctuations that might be coupled to those in loop 1. The corresponding side chains could not be constrained during structure calculation, prominently that of W82 in strand β4, which points towards the cavity in the crystal structure, but appears to flip out in the NMR structures (Fig. 6c).

Structural analysis by NMR of the D36A CrlSTM mutant.
(a) Selected region of assigned 1H-15N HSQC spectra of 13C15N-labeled D36A (red) and wild-type (black) CrlSTM displaying chemical shift perturbations due to the D36A substitution. Red lines link D36A to the corresponding wild-type CrlSTM signals. (b) Plot of combined 1H15N amide and 13Cα and 13C’ backbone CSPs as a function of residue numbers in CrlSTM. Secondary structures are indicated by background coloring (red for α-helices and blue for β-strands) and annotated on top. Loops are denoted by the letter L. (c) Mapping of amide CSPs on a cartoon representation of an NMR ensemble structure (10 conformers) of CrlSTM. Nitrogen atoms are shown by red and orange spheres for residues with ΔδHN > 0.1 ppm and >0.05 ppm, respectively. The side chains of R24, D36 and W82 are shown as lines in green, cyan and magenta, respectively. The corresponding side chains of a model built from the X-ray structure of CrlPM (PDB 4Q11) are indicated in sticks with the same colours.
Structural analysis of the CrlSTM D36A mutant
As shown previously, the CrlSTM D36A variant neither activates nor binds to σSSTM18, but it was not clear if this was due to structural alterations, since previous biophysical data suggested that the substitution could lead to partial loss of secondary and tertiary structure. Therefore we investigated the structural integrity of CrlSTM D36A by analysing its backbone chemical shifts. Signal overlap between wild-type and D36A CrlSTM allowed to partly transpose chemical shift assignments from wild type to D36A CrlSTM (Fig. 6a). But chemical shift perturbations (CSPs) were not restricted to the region of the mutation (Fig. 6b) and de novo backbone assignment had to be carried out. The data showed that there is no major difference for 13C’ or 13Cα chemical shifts, excepted for D36 and C37 (Fig. 6b), indicating that the secondary structure and overall fold are conserved in the mutant. In contrast, amide chemical shifts were significantly perturbed all over the sequence, even if the largest CSPs were also observed around the mutation. They seem to be relayed from D36 in strand β1 to β4, via β2 and β3 and to loops 1 and 3 (Fig. 6c). CSPs in loops 1 can be traced back to the salt bridge formed between the R24 guanidinium and the D36 carboxylate in the X-ray structure of CrlPM as well as in most NMR conformers of CrlSTM (Fig. 6c). When Asp is replaced by Ala, this interaction is disrupted, allowing loop 1 more conformational freedom. Loop 3 could be affected by breaking the hydrogen bond between D36 and the W82 indole observed in the crystal structure of CrlPM (Fig. 6c). This hydrogen bond is not present in the wild-type CrlSTM NMR structure, but it cannot be ruled out that it is transiently formed in solution. Amide CSPs inside the β-sheet, but far from position D36, could be explained by a slight reorganization of the hydrogen bond network. Altogether, these results endorse the role of residue D36 in σS binding.
NMR analysis of the CrlSTM binding interface for σSSTM
We next characterized the influence of σSSTM on CrlSTM NMR spectra. 1H-13C HSQC spectra displayed line broadening, i.e. a decrease of intensities, in particular in the methyl region on addition of σS (Supplementary Fig. S8). Differential broadening was observed in loop 2 and helix α3. However, since methyl groups are mainly pointing to the inside of the structure and are not homogeneously distributed throughout the sequence, they may not be very sensitive probes for the Crl-σS interaction, which was suggested to rely on electrostatic interactions18. σSSTM also induced overall line broadening in CrlSTM 1H-15N HSQC spectra, as a consequence of faster transverse relaxation in the Crl-σS complex than in free Crl and additional line broadening for several residues (Fig. 7a,b, e.g. residue N43), due to exchange between free and complexed Crl. These are mainly clustered in loop 2 (Fig. 7b,c) which contains R51, one of the key residues for σS binding17,18. Since this region appears to be flexible in free Crl, the dynamics of loop 2 certainly plays a role in the formation of the CrlSTM-σSSTM complex. Helix α1 and loop 1 also seem to be affected by σSSTM (Fig. 7b,c).

σSSTM-induced perturbations in NMR spectra of CrlSTM.
(a) Selected region of the 1H-15N TROSY spectrum of 15N2H-labeled CrlSTM in the absence (black) and presence of 0.25 equivalents of unlabelled σSSTM (green) showing the intensity decrease of amide signals for some residues like N43. (b) Plot of intensity ratios as a function of residue number. Background colours indicate the boundaries of CrlSTM secondary structures (red for α-helices, blue for β-strands). The mean value and mean minus one standard deviation (SD) are shown in continuous and dashed lines. (b) The nitrogen atoms of residues with the lowest intensity ratios (I/I0 < mean—SD) in the presence of σS are shown in green spheres on the NMR structure of CrlSTM, represented by three conformers to illustrate the structural variability in loop regions (in red). Two critical residues for σSSTM binding, D36 and R51, are represented in blue sticks.
Modeling of the σS-Crl complex based on mutational analysis
Charged residues in Crl, D36 and R51, were previously found to be essential for σS binding18. The results above corroborate the hypothesis that the Crl-σS complex formation is likely driven by electrostatic interactions by demonstrating that one positively charged residue, R82 in σSSTM, is of paramount importance for Crl binding and activation. In addition, two acidic residues in σS, D135 and E137, were spotted as likely candidates for interaction with Crl (Supplementary Fig. S1 and 21).
To integrate these data, we modelled the Crl-σS complex from a structural model of S. Typhimurium σS216 and the CrlSTM NMR structure. In a first step we performed normal mode analysis (NMA) on both Crl and σS to detect collective low-frequency motions that could provide conformations more favourable for complex formation than the starting structures of isolated proteins. In the case of σS, although shearing movements take place between helix α2 and the DPE loop, residues R82 and E137 do not move wide apart and belong to a common interaction surface (Supplementary Fig. S9). In the case of Crl, collective motions of the three loops remodel the cavity either by closing it or widening it, which would help accommodating σS2 (Supplementary Fig. S10).
In a second step, Crl-σS complexes were obtained in silico, using the information of critical binding residues and two different docking strategies. In the first strategy, we used the ZDock server24 in combination with refinement on the RosettaDock server25, that do not take into account conformational changes and flexibility of proteins (Models A to E, Supplementary Fig. S11). The second strategy used the Haddock Webserver26,27 to integrate the high degree of flexibility of the NMR structure of Crl (Supplementary Fig. S12).
In models A and B (Supplementary Fig. S11), σS R82 interacts with the Crl residues E25 or E102, respectively. These residues are not conserved in Crl family members22 and the Crl E25A and E102A variants interacted with σSSTM in the same manner as wild type Crl (Supplementary Fig. S1) indicating that E25 and E102 are not required for σS binding. It is noteworthy that in these two models the DPE motif does not have any interacting partner. Altogether these data suggest that models A and B do not represent the Crl-σS interface. Models C and D are also unlikely since E137 in σS interacts with R24 in Crl, a residue dispensible for σS binding18. Furthermore, in model C, R82 in σS interacts with E25 in Crl, which is not involved in σS binding (Supplementary Fig. S1) and in both models R51 in Crl does not have any possible charged interacting partner in σS.
From the five models generated by the first docking strategies, model E appears the more likely. In this model two salt bridges are formed involving the critical binding residues R82 and E137 in σS and D36 and R51 in Crl (Fig. 8). Moreover, several van der Waals and hydrogen bond interactions between the σS helix α2 and both loop 1 and β1 of Crl and the σS loop containing the DPE motif and loop 2 of Crl, can further contribute to the σS-Crl complex (Supplementary Fig. S13), in agreement with NMR data which suggest that also loop 1 in Crl is affected upon σSSTM binding.

Cartoon representation of the proposed σS-Crl interface.
The most likely model (Model E, Supplementary Fig. S11) is shown with Crl depicted in cyan and σS in wheat. The region containing the two salt bridges between σS and Crl is zoomed. In the zoom view, the side chain of the charged residues, involved in salt bridges, are colored as follows: in Crl, R51 in orange, D36 in magenta and in σS, R82 in green and the DPE motif in yellow.
In the second series of docking experiments using Haddock Webserver26,27, pairs of active residues with complementary charges straightforwardly formed salt bridges (Supplementary Fig. S12), most often Crl-D36/σS-R82. It was not possible to restrain the Crl-R51/σS-E137 pair to form a salt bridge, but in a number of clusters the two loops that contain these two residues were in close contact, corroborating the relevance of model E for the σS-Crl interface and in agreement with the NMR interaction experiments that pointed to the role of loop 2 for complex formation.
Residue D87 was previously pointed as important for Crl binding in E. coli21. The amino acid sequence of σ2 is identical in σSSTM and σS from E. coli and, in the σSSTM structural model, D87 is located at the edge of helix α2, with its side chain directed on the opposite face with respect to residue R82, as imposed by the geometry of an α-helix (Supplementary Fig. S14). Therefore D87 is unlikely to interact directly with Crl. Consistent with this hypothesis, in the study by Banta et al.21, some amino acid substitutions of D87, such as D87C, did not drastically affect the σS interaction with Crl.
Discussion
In many Gram-negative bacteria, σS/RpoS is the master regulator of gene expression in stress conditions and during stationary phase. σS is exquisitely and tightly regulated by many mechanisms that keep its production level and activity under strict control3,4,5. Crl is a unique regulatory factor, specifically dedicated to σS, which enhances its activity, helping the association of σS with E15. Nevertheless, there are some rpoS-containing species, including P. aeruginosa, that do not harbour a crl gene16 and in which σS activity may be controlled by alternative mechanisms or functional homologs of Crl.
The strong sequence conservation of σS2, the only σS domain that binds Crl16,18,21, prompted us to assess possible activation of σSPA by Crl. We show here that σSPA is not activated by Crl due to its inability to interact with Crl. Taking advantage of the evolution of the σS sequence in P. aeruginosa and other species lacking crl, we identified residues conserved in σS sequences from crl proficient species and potentially implicated in Crl recognition. Among these, a surface-exposed arginine in σSSTM, R82, was assigned to the σS-Crl interface. This residue is not conserved in σSPA, which instead contains a leucine. Importantly, substitution of this leucine by an arginine rendered σSPA sensitive to Crl activation. It is noteworthy that, in some σS proteins from species that do not harbour crl, the arginine residue is conserved (Supplementary Fig. S2). It would be interesting to determine whether these σS proteins interact with Crl and if not, whether they could be used to identify additional σS residues involved in Crl binding by the strategy described in this study for σSPA.
The in silico models of the σS-Crl complex show that salt bridges can indeed be formed for the two pairs of residues Crl-D36/σS-R82 and Crl-R51/σS-E137. In some models they can be formed simultaneously. This leads to a picture of an ideal binding interface in which helix α2 of σS, containing R82, would dock into the cavity of Crl containing D36, disrupting the intermolecular R24-D36 contact and the DPE motif and loop 2 of Crl would make contact on the outside, driven by electrostatic interactions between Crl-R51 and σS-D135/E137 (Fig. 8).
What renders the σS-Crl system very intriguing is its transitory and dynamic binding mechanism, which is unclear so far. Our NMR data together with the in silico modelling shed some light on how σS and Crl may interact and form a transient complex. The chemical shift perturbations in the NMR spectrum of Crl in the presence of σS indicate that loop 2 senses the presence of σS, but extend beyond the region directly involved in σS binding, including helix α1, loop 1 and helix α3. These findings suggest that local structural rearrangements might take place in the flexible loops that allow breathing of the cavity as indicated by normal mode analysis of the Crl structure. Such rearrangements might contribute not only to the formation of the σS-Crl complex, but also to its dissociation, once Crl has accomplished its work. Moreover, in free Crl, residue D36 is involved in an intramolecular interaction with R24. To form a new salt bridge with σS-R82, the first one has to be broken. The perturbations observed in the NMR spectra of the Crl D36A variant show how the disruption of this network is sensed by the whole Crl structure, in particular by loop 1. It is tempting to speculate that this variant mimics the molecular processes that Crl undergoes upon σS binding, as we previously hypothesized18.
How does Crl binding to σS increase the σS association rate with E? Why is the σS-Crl interaction so transient? These questions are still open. One possibility is that Crl triggers a conformational change in σS favouring its association with E. There is no high resolution 3D structure for free σ factors, but several biochemical and structural studies using the housekeeping σ70 have shown that σ factors undergo pronounced conformational changes upon E binding, allowing domains σ2 and σ4 to be spaced correctly for promoter binding2,20. These findings have led to the proposal that σ factors must be in a more compact conformation when free in the cell than in the Eσ complex. Consistent with this hypothesis, free σ are not able to bind promoters efficiently. This concept was further supported by the results obtained with engineered cysteine mutants of σ28, which showed that this σ factor has a compact conformation when free in solution28.
Modulation of the free σS conformation might be a common way to regulate both the stability and activity of σS. σS is degraded by the ATP-dependent complex ClpXP protease3,4,5. However, σS binding by the RssB protein is required for delivery to ClpXP3,4,5. It has been postulated that RssB binding triggers a conformational opening of σS that exposes a ClpXP binding site, that is otherwise occluded in a closed conformation of free σS5. Therefore, if the conformation of σS in the cell is rather compact, Crl binding to σS2 may alleviate intramolecular interactions between σS2 and other σS domains, favouring an open conformation for just the time required for σS to bind E, but transiently enough to avoid σS degradation by ClpXP. Further investigation of the structure of the σS-Crl complex, for which a starting base is provided in the present study and of the free σS conformation will assess the relevance of this scenario.
Methods
Bacterial strains, bacteriophage, plasmids and growth conditions
Strains and plasmids used for this work are listed in Supplementary Table S5. Bacteriophage P22HT105/1int was used to transfer mutations and the katN-lacZ fusion between Salmonella strains by transduction29. Green plates, for P22-infected cells or lysogens screening, were prepared as described previously30. Strains were grown in Luria-Bertani (LB) medium31 at 37°C under aeration. Development of the rdar morphotype was monitored on CR plates (LB agar without NaCl supplemented with Congo red 40 μg/ml and Coomassie brilliant blue R250 20 μg/ml), at 28°C as described7. Antibiotics were used at the following concentrations: ampicillin (Ap) 100 μg/mL; carbenicillin (Cb) 100 μg/mL; chloramphenicol (Cm) 15 μg/mL for the chromosomal resistance gene and 30 μg/mL for the plasmid resistance gene; kanamycin (Km) 50 μg/mL; and tetracycline (Tet) 20 μg/mL.
rpoS allelic exchange in Salmonella
Allelic exchange of rpoS in S. Typhimurium ATCC14028 was achieved with a two-step Red-recombinase-based recombineering procedure32,33,34,35. The procedure involves replacement of the tetRA module of strain VFC326 by PCR-amplified DNA fragments of the rpoS allele from pVFC629, pVFD410, pVFD412 and pVFD399 (Supplementary Table S5 and S6) through positive selection of tetracycline-sensitive recombinants. All strains were confirmed to contain the expected mutation by DNA sequencing.
Protein production and BACTH assays
The N-terminal (his)6-tagged σSPA wild type and variant L87R, σSSTM R82L variant and CrlSTM were produced in E. coli strain BL21 (DE3) harbouring plasmid derivatives of pETM11 (Supplementary Table S5). Production and purification of the proteins were carried out as previously described18. 15N-, 13C15N- or 15N2H-labeled wild type (his)6- CrlSTM and 15N13C-labeled CrlSTM (his)6-D36A protein samples for NMR experiments were produced in minimum M9 medium31 supplemented with 15NH4Cl and unlabelled or 13C- or 2H-labeled glucose following the same protocol as18. Samples were subsequently dialyzed into NMR buffer (50 mM sodium or potassium phosphate, 300 mM NaCl or KCl, 2 mM dithiotreitol, at pH 8 or 7.5).
For bacterial adenylate cyclase-based two hybrid assay, the E. coli cya strain DHT1 was transformed with derivatives of plasmids pKT25 and pUT18 encoding σS and Crl proteins fused to the C-terminal part of T25 and the N-terminal part of T18, respectively (Supplementary Table S5). Co-transformants were plated onto MacConkey maltose plates supplemented with Cb, Km and 0.5 mM IPTG to assess the Mal phenotype and on LB plates supplemented with X-Gal (40 μg/ml) Cb, Km and IPTG (0.5 mM) to assess the Lac phenotype. Plates were incubated at 30 °C for 2 days and then isolated colonies were grown in LB supplemented with Cb, Km and IPTG, at 30 °C for 20 hours. β-galactosidase activities were measured as described by Miller and are expressed in Miller units36.
NMR experiments
NMR measurements were carried out at 293 K on a Bruker Avance III spectrometer with a magnetic field of 18.8 T (800 MHz 1H frequency) equipped with a cryogenic TCI probe. The magnetic field was locked with 7% or 100% 2H2O. Spectra were processed with Topspin 3.1 (Bruker Biospin) or NMRPipe37 and analysed with CCPNMR 2.2 software38. Chemical shift assignments of CrlSTM are reported elsewhere23. For structure determination, 2D 1H-15N HSQC and 3D 1H-15N NOESY-HSQC spectra were recorded from a 300 μM 15N-Crl sample and 1H-13C HSQC, 2D 1H-1H NOESY and 3D 1H-13C NOESY-HSQC spectra from a 250 μM 13C15N-Crl sample in deuterium oxide buffer. NOESY mixing times were 80 ms.
Sequential backbone assignment of CrlSTM D36A (380 μM, 293 K) was carried out with a minimal set of triple resonance experiments: HNCA and HN(CO)CA were recorded at 14.1 T, CBCA(CO)NH and HNCO) at 18.8 T. Chemical shift perturbations induced by the D36A mutation were calculated as combined 1H and 15N perturbations ΔδHN for a given residue i:

The scaling factor 1/10 corresponds to the gyromagnetic ratio difference between 15N and 1H.
NMR structure calculation
NMR structures of wild-type CrlSTM were calculated using torsion angle dynamics in CYANA 2.239. Backbone torsion angle restraints were generated with TALOS-N40 using CrlSTM backbone chemical shifts. Ambiguous distance restraints were collected from three sets of NOESY spectra and purged from 3D peaks without possible assignments in the 1H dimension bound to a heteroatom. The disordered N-terminal His-tag (His(−20)-His(0)) was excluded from structure calculation. Structure statistics were obtained from the Protein Structure Validation Server, version 1.5 (http://psvs-1_5-dev.nesg.org/) (Supplementary Table S2). Normal Mode Analysis was performed on single conformers on the ElNémo webserver41.
Protein-protein docking
Rigid-body docking was carried out first on the ZDock server24, which employs a fast Fourier transform (FFT) algorithm, to generate the initial models (about 100) for the σS-Crl complex with CrlSTM as the receptor and a homology model of S. Typhimurium σS2 as the ligand16. Five models selected from ZDock were further refined using RosettaDock25, which performs a searching for the lowest-energy binding interface structures giving as ouput the 10 best-scoring models from 1000 total models. The presence of a ‘docking funnel’ was verified, considering that at least three of the first five lowest-energy binding interface models have a value of I_rmsd < 4 Å42 (Supplementary Table S3).
Flexible docking was carried out on the guru interface of the Haddock Webserver26,27 using single conformers from the NMR structure ensemble of CrlSTM and the homology model of S. Typhimurium σS2. D36 and R51 in Crl and R82, D135 and E137 in σS were defined as active residues. Passive residues were automatically defined around active residues. Loops 1 (E25-R32) and 2 (N43-E52) in Crl and the DPE loop (K133-F142) in σS were defined as fully flexible. 1000 initial structures were generated. 200 final structures were refined in water and clustered according to RMSD criterion. Statistics for clusters obtained for the conformers 1 and 2 are given in Supplementary Table S4.
Illustrations
Visualization and graphic rendering of protein structures were prepared with PyMOL43.
Other methods
Methods for DNA manipulation, immunoblot analysis of proteins and CD and ITC experiments are described in Supplementary Methods.
Additional Information
How to cite this article: Cavaliere, P. et al. Binding interface between the Salmonella σS/RpoS subunit of RNA polymerase and Crl: hints from bacterial species lacking crl. Sci. Rep. 5, 13564; doi: 10.1038/srep13564 (2015).
References
Gruber, T. M. & Gross, C. A. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu Rev Microbiol 57, 441–66 (2003).
Feklistov, A., Sharon, B. D., Darst, S. A. & Gross, C. A. Bacterial sigma factors: a historical, structural and genomic perspective. Annu Rev Microbiol 68, 357–76 (2014).
Klauck, E., Typas, A. & Hengge, R. The sigmaS subunit of RNA polymerase as a signal integrator and network master regulator in the general stress response in Escherichia coli. Sci Prog 90, 103–27 (2007).
Battesti, A., Majdalani, N. & Gottesman, S. The RpoS-mediated general stress response in Escherichia coli. Annu Rev Microbiol 65, 189–213 (2011).
Hengge, R. The general stress response in Gram-negative bacteria. Bacterial Stress Responses (Second Edition, eds. G. Storz & R. Hengge ), ASM Press, Washington D.C., 251–289 (2011).
Romling, U. Characterization of the rdar morphotype, a multicellular behaviour in Enterobacteriaceae. Cell Mol Life Sci 62, 1234–46 (2005).
Robbe-Saule, V. et al. Crl activates transcription initiation of RpoS-regulated genes involved in the multicellular behavior of Salmonella enterica serovar Typhimurium. J Bacteriol 188, 3983–94 (2006).
Dong, T. & Schellhorn, H. E. Role of RpoS in virulence of pathogens. Infect Immun 78, 887–97 (2010).
Osterberg, S., del Peso-Santos, T. & Shingler, V. Regulation of alternative sigma factor use. Annu Rev Microbiol 65, 37–55 (2011).
Pratt, L. A. & Silhavy, T. J. Crl stimulates RpoS activity during stationary phase. Mol Microbiol 29, 1225–36 (1998).
Bougdour, A., Lelong, C., Geiselmann, J. Crl, a low temperature-induced protein in Escherichia Coli that binds directly to the stationary phase σ subunit of RNA polymerase J Biol Chem 279, 19540–19550 (2004).
Gaal, T., Mandel, M. J., Silhavy, T. J. & Gourse, R. L. Crl facilitates RNA polymerase holoenzyme formation. J Bacteriol 188, 7966–70 (2006).
Robbe-Saule, V., Lopes, M. D., Kolb, A. & Norel, F. Physiological effects of Crl in Salmonella are modulated by sigmaS level and promoter specificity. J Bacteriol 189, 2976–87 (2007).
Typas, A., Barembruch, C., Possling, A. & Hengge, R. Stationary phase reorganisation of the Escherichia coli transcription machinery by Crl protein, a fine-tuner of σS activity and levels. EMBO J 26, 1569–1578 (2007).
England, P. et al. Binding of the unorthodox transcription activator, Crl, to the components of the transcription machinery. J Biol Chem 283, 33455–64 (2008).
Monteil, V. et al. Crl binds to domain 2 of sigma(S) and confers a competitive advantage on a natural rpoS mutant of Salmonella enterica serovar Typhi. J Bacteriol 192, 6401–10 (2010).
Banta, A. B. et al. Structure of the RNA polymerase assembly factor Crl and identification of its interaction surface with sigma S. J Bacteriol 196, 3279–88 (2014).
Cavaliere, P. et al. Structural and functional features of Crl proteins and identification of conserved surface residues required for interaction with the RpoS/sigmaS subunit of RNA polymerase. Biochem J 463, 215–24 (2014).
Lonetto, M., Gribskov, M. & Gross, C. A. The sigma 70 family: sequence conservation and evolutionary relationships. J Bacteriol 174, 3843–9 (1992).
Murakami, K. S. & Darst, S. A. Bacterial RNA polymerases: the wholo story. Curr Opin Struct Biol 13, 31–9 (2003).
Banta, A. B. et al. Key features of sigmaS required for specific recognition by Crl, a transcription factor promoting assembly of RNA polymerase holoenzyme. Proc Natl Acad Sci USA 110, 15955–15960 (2013).
Monteil, V., Kolb, A., D’Alayer, J., Beguin, P. & Norel, F. Identification of conserved amino acid residues of the Salmonella sigmaS chaperone Crl involved in Crl-sigmaS interactions. J Bacteriol 192, 1075–87 (2010).
Cavaliere, P., Norel, F. & Sizun, C. 1H, 13C and 15N resonance assignments of σS activating protein Crl from Salmonella enterica serovar Typhimurium. Biomol NMR Assign Epub ahead of print (2015) 10.1007/s12104-015-9617-z.
Pierce, B. G. et al. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30, 1771–3 (2014).
Lyskov, S. et al. Serverification of molecular modeling applications: the Rosetta Online Server that Includes Everyone (ROSIE). PLoS One 8, e63906 (2013).
de Vries, S. J., van Dijk, M. & Bonvin, A. M. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc 5, 883–97 (2010).
Wassenaar, T. A. et al. WeNMR: Structural Biology on the Grid. J Grid Comp 10, 743–767 (2012).
Sorenson, M. K. & Darst, S. A. Disulfide cross-linking indicates that FlgM-bound and free sigma28 adopt similar conformations. Proc Natl Acad Sci USA 103, 16722–7 (2006).
Schmieger, H. Phage P22-mutants with increased or decreased transduction abilities. Mol Gen Genet 119, 75–88 (1972).
Sternberg, N. L. & Maurer, R. Bacteriophage-mediated generalized transduction in Escherichia coli and Salmonella typhimurium. Methods Enzymol 204, 18–43 (1991).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. (1989).
Bochner, B. R., Huang, H. C., Schieven, G. L. & Ames, B. N. Positive selection for loss of tetracycline resistance. J Bacteriol 143, 926–33 (1980).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97, 6640–5 (2000).
Gerlach, R. G., Jackel, D., Holzer, S. U. & Hensel, M. Rapid oligonucleotide-based recombineering of the chromosome of Salmonella enterica. Appl Environ Microbiol 75, 1575–80 (2009).
Levi-Meyrueis, C. et al. Repressor activity of the RpoS/sigmaS-dependent RNA polymerase requires DNA binding. Nucleic Acids Res 43, 1456–68 (2015).
Miller, J. H. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. (1972).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–93 (1995).
Vranken, W. F. et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–96 (2005).
Guntert, P. Automated NMR structure calculation with CYANA. Methods Mol Biol 278, 353–78 (2004).
Shen, Y. & Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. Journal of biomolecular NMR 56, 227–41 (2013).
Suhre, K. & Sanejouand, Y. H. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic acids research 32, W610–4 (2004).
Chaudhury, S. et al. Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS One 6, e22477 (2011).
Schrodinger, L. L. C. The PyMOL Molecular Graphics System, Version 1.3 (2010). Available at: http://pymol.sourceforge.net (19/08/10).
Rocchia, W., Alexov, E. & Honig, B. Extending the applicability of the nonlinear Poisson-Boltzmann equation: Multiple dielectric constants and multivalent ions. Journal of Physical Chemistry B 105, 6507–6514 (2001).
Acknowledgements
We thank O. Francetic, A. Pugsley and all members of the laboratory for their kind support. We thank G. André-Leroux for advice regarding the docking programs. We are also grateful to the collection of the Institut Pasteur for providing P. aeruginosa and D. Ladant for the T25 antibody. Funding: This work was supported by the French National Research Agency [grant ANR- 11-BSV3-009], the IR-RMN-THC (CNRS FR3050) for access to the 950 MHz spectrometer at Gif-sur-Yvette for preliminary experiments and by grants from the Institut Pasteur and the Centre National de la Recherche Scientifique. The WeNMR project (European FP7 e-Infrastructure grant, contract no. 261572, www.wenmr.eu), supported by the European Grid Initiative (EGI) through the national GRID Initiatives of Belgium, France, Italy, Germany, the Netherlands, Poland, Portugal, Spain, UK, South Africa, Malaysia, Taiwan, the Latin America GRID infrastructure via the Gisela project and the US Open Science Grid (OSG) are acknowledged for the use of web portals, computing and storage facilities.
Author information
Authors and Affiliations
Contributions
Designed the study: P.C., C.S., J.B., C.M. and F.N. Performed the experiments: P.C., C.S., F.L.A., M.N., V.M. and F.B. Analysed the data: P.C., C.S., J.B., C.M. and F.N. Wrote the manuscript: P.C., C.S., C.M. and F.N.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Competing interests
Coordinates for the NMR structure of S. Typhimurium were deposited at the Protein Data Bank under accession code 2MZ8.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Cavaliere, P., Sizun, C., Levi-Acobas, F. et al. Binding interface between the Salmonella σS/RpoS subunit of RNA polymerase and Crl: hints from bacterial species lacking crl. Sci Rep 5, 13564 (2015). https://doi.org/10.1038/srep13564
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep13564
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
