
Abstract
The crystal structures and properties of hafnium hydride under pressure are explored using the first-principles calculations based on density function theory. The material undergoes pressure-induced structural phase transition I4/mmm→Cmma→P21/m at 180 and 250 GPa, respectively and all of these structures are metallic. The superconducting critical temperature Tc values of I4/mmm, Cmma and P21/m are 47–193 mK, 5.99–8.16 K and 10.62–12.8 K at 1 atm, 180 and 260 GPa, respectively. Furthermore, the bonding nature of HfH2 is investigated with the help of the electron localization function, the difference charge density and Bader charge analyses, which show that HfH2 is classified as a ionic crystal with the charges transferring from Hf atom to H.
Similar content being viewed by others


Introduction
Up to now, hydrogen, the simplest and most abundant element of universe is still fascinating to physics community. The studies about hydrogen are mainly reflected in two aspects. One is the metallic and superconducting of hydrogen. The other is considered as the most promising clean energy sources with the capability of replacing fossil fuels. The former is going back to the early 1930: hydrogen was predicted to be a potential high Tc superconductor at high pressures1,2,3, because of its low mass density and high elastic stiffness. However, hydrogen remains an insulator, despite considerable ongoing experimental effort up to pressure of 300 GPa4. Recently, hydrogen dense materials has been as a potential route to achieve metallization with high Tc at lower pressure. For example, high Tc with 190 K in the sulfur hydrides at 200 GPa has been found by both of theoretical predicted and experimental observation5,6,7. The latter is still limited by many practical and technological factors, which requires the development of safe and efficient hydrogen storage technology.
Hydrogen can also react with transition metal elements formed metal hydrides under certain conditions and they can keep stable under ambient conditions, such as YH2, YH3, TiH2, ZrH2, HfH2, etc8,9,10,11. In addition, for transition metal, the number of d-shell electrons per atom is import for the Tc, which is correlate with the important parameters of the Bardeen–Cooper–Schrieffer theory about superconductivity12. Transition metal hydrides not only are regarded as promising potential materials for storing hydrogen13, but also exhibit fascinating superconducting properties. For example, the superconducting transition temperature Tc of Th4H15 is 8 K at ambient conditions14. Many transition-metal dihydrides TMH2 can form CaF2 (Fm–3m space group) crystal structure. And in this structure, the metal atoms form a face-centered-cubic sublattice while the hydrogen atoms occupy the tetrahedral lattice sites. However, the group IVB dihydrides TiH2, ZrH2 and HfH2 show a basically face-centered-tetragonal (fct) cell structure (I4/mmm space group). And these dihydrides (TiH2, ZrH2 and HfH2) are applied in various fields. For instances, TiH2 can be served as the catalyst in polymerization reaction, which is interesting in the new quenchable phases15,16. ZrH2 is widely used as a neutron moderator in nuclear reactor17,18,19. Hafnium hydride, instead of boron carbide, is a perfect neutron control materials for fast reactors20.
The high-pressure development has become a vibrant area of research. Not only does pressure provide a different route to the synthesis of new compounds, but also enables many known materials to exhibit novel phenomena that cannot be found at normal conditions. Lately, theoretical study revealed that TiH2 at high pressure had a structural transformations I4/mmm→P4/nmm→P21/m and the calculated pressure of phase transition were 63 and 294 GPa, respectively21. The structures and properties of ZrH2 were also investigated in experiment under high pressure22. Despite large amounts of theoretical and experimental researches on HfH223, there is relatively little investigation on its new structures, chemical bonding nature, dynamical properties and superconductivity under high pressures. Therefore, great attentions are needed to explore the high–pressure structures of HfH2.
In this paper, we examine in detail the optimum static structures of HfH2 system at zero temperature by using the newly developed ab initio evolutionary algorithm. Moreover, we employ a first-principles method to calculate their dynamical stability and electronic band structures. Our calculated results show that HfH2 adopts the I4/mmm structure at low pressures. On compression, Cmma phase possesses the lowest enthalpy, then at higher pressures P21/m becomes energetically favorable. Band structures and density of states indicate that these structures are metallic. The estimated Tc are 47–193 mK at 1 atm, 5.99–8.16 K at 180 GPa and 10.62–12.8 K at 260 GPa, for I4/mmm, Cmma and P21/m, respectively. The electron localization function (ELF), the difference charge density and Bader charge analysis show that HfH2 is an ionic crystal with the charge transferring from Hf to H atom. Our present study attempts to provide a better understanding of the pressure-induced phase transformations and properties of HfH2 under pressure.
Results and Discussion
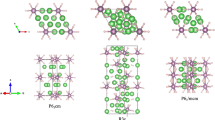
The crystal structure prediction were performed with simulation sizes ranging from one to four HfH2 formula units (f.u.) at 1 atm, 50, 80, 100, 150, 200, 250 and 300 GPa. Analysis of the predicted structures gave us a shortlist of candidate structures with space groups Fm–3m, I4/mmm, P4/nmm, Cmma, P21/m and C2/m. Figure 1 shows the structural motifs of HfH2 under high pressure. At 1 atm, we obtain two structures: a CaF2 type structure with space group Fm–3m (4 f.u./cell) and a centered tetragonal structure with space group I4/mmm (2 f.u./cell) (Fig. 1(a)). At ambient conditions, many transition-metal dihydrides exist with fcc structure (i.e., a CaF2 structure), where metal atoms form a face-centered cubic lattice and the centers of tetrahedrons are occupied by H atoms. Note that in I4/mmm structure, Hf atoms form a body-centered tetragonal (bct) sublattice with its coordination number is 8, while the H atoms are located on the planes and present a one dimensional chain. Then we observed the most stable structure which possess simple tetragonal P4/nmm (4 f.u./cell) orthogonal Cmma (4 f.u./cell) at 200 GPa. In the above two phases, the metal atoms form a bct and fcc cell for P4/nmm and Cmma, respectively. For Cmma, the coordination number of Hf becomes 9. Finally, at 300 GPa, the monoclinic P21/m phase (4 f.u. /cell) becomes the preferred one (Fig. 1(d)). And in the P21/m structure, Hf atoms can form a little distorted bcc lattice and its coordination is 12. Within P21/m structure, Hf site is not equivalent, occupying the crystallographic 2e site, while H atoms take four different 2e sites. The lattice parameters and atomic positions of selected structures at favored pressures are summarized in Table SI. In comparison, we observed that in both HfH2 and TiH221, the locations of metal and hydrogen atoms are similar for I4/mmm, P4/nmm and P21/m, respectively. In all above structures, the nearest H-H distances are 2.149 Å at 1 atm, much longer than that in the pure H2. Furthermore, with increasing pressure, H-H distances decrease and become 1.548 Å at 300 GPa, which indicate that there isn’t any bonding trend among H atoms.

Crystal Structures of HfH2.
The selected stable phases for HfH2. Green atoms depict Hf, while pink atoms present H, (a) I4/mmm at 1 atm, (b) Cmma at 200 GPa, (c) P4/nmm at 200 GPa and (d) P21/m at 300 GPa.
The phase stability of HfH2 under pressure has been investigated systematically. Enthalpy differences as a function of pressure for these competitive solid structures together relative to Fm–3m in the pressure range from 0 to 300 GPa are plotted in Fig. 2(a). It is clearly seen that I4/mmm phase has lower enthalpies than all other candidates, which indicate it is the thermodynamic ground state. Above 180 GPa, Cmma (P4/nmm) becomes the most stable structure and remains up to 250 GPa. Then under higher pressures, a monoclinic P21/m becomes more favored. In these two transitions, the coordination number of Hf increased from 8 to 9, 9 to 12, respectively. Surprisingly, we found that Cmma and P4/nmm are energetically nearly degenerated in the range of 0–300 GPa and enthalpy difference is less than 1 meV/f.u.. If I4/mmm, P4/nmm and P21/m phases satisfied the stability conditions of mechanics and dynamics, the phase transition sequence of HfH2 is similar to the TiH221. However, we found that the P4/nmm is not stable both in dynamical and mechanical properties (see the later discussions). The calculated equations of state (EOS) depicted in Fig. 2(b) shows that the I4/mmm→Cmma and Cmma→P21/m phase transitions are discontinuous changes in volume at the transition point with volume collapses of 0.9% and 7.6%, suggesting the two phase transitions are the first-order nature.

Enthalpy difference curves of HfH2 and EOS.
(a) Calculated enthalpies per HfH2 unit of various structures relative to our predicted Fm–3m phase as a function of pressure range from 0 to 300 GPa. (b) Volume plotted as a function of pressure for the I4/mmm, Cmma and P21/m structures.
It is essential to determine the dynamical stability of the studied structures. The calculated phonon dispersion curves and projected phonon density of states (PHDOS) of the I4/mmm, Cmma, P4/nmm and P21/m phases in the studied pressure range are presented in Fig. 3. The absence of any imaginary phonon frequencies in the entire Brillouin zone (Fig. 3(a–c)) confirms that I4/mmm, Cmma and P21/m structures are dynamically stable regardless of the applied pressure. By contrast, there are imaginary phonon frequencies for P4/nmm phase (Fig. 3(d)) which indicates this phase is dynamically unstable. In addition, it is shown that two separate regions of phonon bands are clearly recognized. Since hafnium is much heavier than hydrogen atom, the vibration frequency of hafnium atom is obviously lower than that of hydrogen atom. And the low-frequency bands are merely from the Hf atoms, while higher-frequency modes are solely due to the light H atoms.

The phonon band structure and projected phonon DOS charts.
(a–d) The phonon band structure and projected phonon DOS charts for I4/mmm, P4/nmm, Cmma and P21/m at different pressure.
Mechanical property of the crystalline structure is one of the basic requirement when considering the phase stability. The elastic constants of I4/mmm, Cmma, P21/m and P4/nmm structures were calculated at different pressures, as shown in Table SII. According to the mechanical stability criteria, the strain energy should be positive, which means the whole set of elastic constants matrix Cij meet the Born-Huang stability criteria24. Obviously, the elastic constants of I4/mmm, Cmma and P21/m meet the mechanical stability criteria, suggesting that the three structures are mechanically stable in our studied pressure range. However, elastic constants of P4/nmm phase cannot satisfy the stability conditions of mechanics for tetragonal crystal system owing to C66 < 0, which means it is mechanically unstable.
The electronic properties were studied by calculations of the electronic band structure and partial densities of states (PDOS) for I4/mmm, Cmma and P21/m phases at different pressures, as presented in Fig. 4. Clearly, the overlap between the conduction and the valence bands for the three structures suggests that the above phases are metallic. From the PDOS of HfH2 (Fig. 4(d–f)), we see that the occupation properties of I4/mmm, Cmma and P21/m phases are similar. And the pseudogap which is below the Fermi level, located at around −4.2 eV. The predominant feature of hybridization for H 1 s orbital and Hf 5d orbital is observed in the energy region below the pseudogap, while from the pseudogap to the Fermi level, the domination is Hf 5d states in the energy range. The metallic behavior of HfH2 indicates that this material might be a superconductor and we discussed it in the following.

Electronic band structure and partial density of states (PDOS).
(a–f) The calculated electronic band structure and PDOS for I4/mmm (0 GPa), Cmma (200 GPa) and P21/m (250 GPa).
In order to explore the bonding and analyze the ionic or covalent character of HfH2, the electron localization function (ELF) of I4/mmm, Cmma and P21/m at 100 GPa, 200 GPa and 250 GPa, are plotted in Fig. 5(a–c). The maximum ELF value between Hf and H is less than 0.3, which suggests no covalent bonding. In order to further clearly understand the bonding nature, we have calculated the difference charge density (crystal density minus superposition of isolated atomic densities) of I4/mmm (100 GPa), Cmma (200 GPa) and P21/m (250 GPa), as shown in Fig. 5(d–f). We can see charges transfer from Hf atom to H atom. The profile of the Hf is identical for I4/mmm and Cmma phases in Fig. 5(d,e), respectively. By contrast, P21/m phase in Fig. 5(f) has two Hf’s contours owing to Hf sites are not equivalent, which occupy the crystallographic 2e site (Fig. 1(d)). To gain a better understanding of the bonding characters between Hf and H atoms, we calculate the qe/atom from Hf to H by the Bader charge analysis for I4/mmm, Cmma and P21/m at different pressures, as shown in Figure SI. We can see that about 1.22 electrons and 1.12 electrons transfer from each Hf to H atom for I4/mmm and Cmma structure, at 100 and 200 GPa. For P21/m, the number of electron transfer from each Hf is 1.29 and 0.42 at 250 GPa, which originates from its two different Hf site. In addition, the pressure dependence of δ (δ donates the values of charge transfer from Hf to H atom) for the above three phases is different. The δ depended on pressure in Cmma is smaller than that in I4/mmm. While turning to P21/m, it exists two forms due to two inequitable Hf sites. The change of charge transfer may be related to the structural evolution. Overall, the ELF, the difference charge density and Bader analysis results reveal that the ionic bonds were formed between Hf and H and HfH2 was classified as a ionic cyrstal with the charge transferring from Hf to H atom.

The electron localization function and difference charge density maps.
(a–c) Electron localization function (ELF) maps of I4/mmm (100 GPa), Cmma (200 GPa) and P21/m (250 GPa), respectively. (d–f) Difference charge density (crystal density minus superposition of isolated atomic densities) of I4/mmm, Cmma and P21/m for HfH2 plotted at 100, 200 and 250 GPa, respectively. The isosurface value is set as: Blue represent positive (+0.05) while Yellow represent negative (−0.015).
We calculated the electron phonon coupling (EPC) parameter λ, the logarithmic average phonon frequency ωlog and the electronic DOS at the Fermi level N(Ef) for I4/mmm, Cmma and P21/m. The Tc was estimated by using the Allen-Dynes modified McMillan equation25  ,. For materials with λ < 1. 5, this equation has been found to be accurate. The Coulomb parameter set μ* = 0.1–0.13 are adopted for HfH2. According to our calculations, the λ are 0.33, 0.64 and 0.87 for I4/mmm (at 1 atm), Cmma (at 180 GPa) and P21/m (at 260 GPa). The estimated Tc of I4/mmm, Cmma and P21/m, are 47–193 mK at 1 atm, 5.99–8.16 K at 180 GPa and 10.62–12.8 K at 260 GPa, respectively. We note that I4/mmm has a lower Tc value relative to the other two structures. For the I4/mmm (at 1 atm), the EPC parameter λ is 0.33, which indicate the electron-phonon interaction is fairly weak. In addition, the electronic DOS at the Fermi level N(Ef) for the phase I4/mmm is relatively small (3.978 states/spin/Ry/Unit cell). So the weak electron phonon coupling λ and small N(Ef) are the main factors, which lead to the low Tc. To study the pressure dependence of the superconducting critical temperature Tc of I4/mmm, Cmma and P21/m, the λ, ωlog and N(Ef) were calculated as summarized in Table 1. With the increasing pressure, the value of ωlog is increased in I4/mmm, while it is diminished in Cmma and P21/m. For N(Ef) and λ parameters, they both decrease as pressure increased, which mainly lead to the decrease of the Tc values for the above three structures.
,. For materials with λ < 1. 5, this equation has been found to be accurate. The Coulomb parameter set μ* = 0.1–0.13 are adopted for HfH2. According to our calculations, the λ are 0.33, 0.64 and 0.87 for I4/mmm (at 1 atm), Cmma (at 180 GPa) and P21/m (at 260 GPa). The estimated Tc of I4/mmm, Cmma and P21/m, are 47–193 mK at 1 atm, 5.99–8.16 K at 180 GPa and 10.62–12.8 K at 260 GPa, respectively. We note that I4/mmm has a lower Tc value relative to the other two structures. For the I4/mmm (at 1 atm), the EPC parameter λ is 0.33, which indicate the electron-phonon interaction is fairly weak. In addition, the electronic DOS at the Fermi level N(Ef) for the phase I4/mmm is relatively small (3.978 states/spin/Ry/Unit cell). So the weak electron phonon coupling λ and small N(Ef) are the main factors, which lead to the low Tc. To study the pressure dependence of the superconducting critical temperature Tc of I4/mmm, Cmma and P21/m, the λ, ωlog and N(Ef) were calculated as summarized in Table 1. With the increasing pressure, the value of ωlog is increased in I4/mmm, while it is diminished in Cmma and P21/m. For N(Ef) and λ parameters, they both decrease as pressure increased, which mainly lead to the decrease of the Tc values for the above three structures.
Conclusion
In summary, we have extensively investigated structures and examined the structural stability of HfH2 at high pressures up to 300 GPa through ab initio evolutionary simulations. Three structures I4/mmm, Cmma and P21/m are predicted and all of them are energetically much superior to others phases. The electronic structures are characterized as conductors with band overlap for I4/mmm, Cmma and P21/m phases. The measured superconducting transition temperature Tc values for I4/mmm, Cmma and P21/m are 47–193 mK (at 1 atm), 5.99–8.16 K (at 180 GPa) and 10.62–12.8 K (260 GPa). Further analysis of the bonding nature shows that charges transfer from the hafnium to hydrogen with ionic bonds in HfH2. The current study has great implications for researching other transition metal hydrides.
Methods
We have used the evolutionary algorithm USPEX code (Universal structure predictor: Evolutionary Xtallography)26,27,28 for crystal structure prediction to extensively explore the high-pressure phases of HfH2 system at zero temperature. In the evolutionary structural predictions, the first generation of structures was always created randomly and its population size is 20–60 structures, increasing with system size. Every subsequent generation is produced from the best 60% of the previous generation. Moreover, the lowest-enthalpy structures always survived into the next generation. New structures are produced by variation operator heredity (60%), permutation (10%) and lattice mutation (30%). The energetic calculations and electronic structure calculations presented here are performed within density functional theory, carried out within the Vienna ab initio simulation package (VASP)29. The generalized gradient approximation with Perdew-Burke-Ernzerh functinal30 for the exchange correlation is employed. The projector-augmented wave (PAW)31 method is adopted with valence electrons of 5d26 s2 and 1 s1 and cutoff radii of 2.5 and 0.8 a.u. for Hf and H atoms, respectively. The electronic wave functions were expanded in a plane-wave basis set with a cutoff energy of 800 eV and appropriate Monkhorst-Pack meshes were chosen for all structures to ensure that enthalpy calculations are well converged to better than 1 meV/atom. In the geometrical optimization, all forces on atoms were converged to less than 0.005 eV/Å. We used the Bader charge analysis32,33,34 to calculate the electronic charge transfer. To determine the dynamical stability of the studied structures, the phonon calculations are carried out using a supercell approach35 with the PHONOPY code36. Electron-phonon coupling (EPC) calculations were carried out using the linear response theory through the Quantum ESPRESSO package37. The kinetic energy cutoff was set 90 Ry. And the q-point mesh of the electron-phonon interaction matrix element adopted 4 × 4 × 4, 4 × 4 × 3 and 2 × 3 × 3 for I4/mmm, Cmma and P21/m, respectively.
Additional Information
How to cite this article: Liu, Y. et al. First-principles study on the structural and electronic properties of metallic HfH2 under pressure. Sci. Rep. 5, 11381; doi: 10.1038/srep11381 (2015).
References
Ashcroft, N. Metallic Hydrogen: A High-Temperature Superconductor? Phys. Rev. Lett. 21, 1748–1749 (1968).
Barbee, T., García, A. & Cohen, M. L. First-principles prediction of high-temperature superconductivity in metallic hydrogen. Nature 340, 369–371 (1989).
Zhang, L. et al. Ab initio prediction of superconductivity in molecular metallic hydrogen under high pressure. Solid State Commun. 141, 610–614 (2007).
Loubeyre P., Occelli F. & LeToullec R. Optical studies of solid hydrogen to 320 GPa and evidence for black hydrogen. Nature 416, 613–617 (2002).
Duan, D. et al. Pressure-induced metallization of dense (H2S)2H2 with high-Tc superconductivity. Sci. rep. 4, 6968, (2014).
A. P. Drozdov, M. I. Eremets & Troyan, I. A. Conventional superconductivity at 190 K at high pressures arXiv:1412.0460 (2014).
Duan, D. et al. Pressure-induced decomposition of solid hydrogen sulfide. arXiv preprint arXiv:1501, 01784, (2015).
Zabel, H. et al. Hydrogen in thin epitaxial metal films and superlattices: structure, magnetism and transport. J. Magn. Magn. Mater. (Netherlands) 198, 264–266 (1998).
Kalita, P. E. et al. Equation of state of TiH2 up to 90 GPa: A synchrotron x-ray diffraction study and ab initio calculations. J. Appl. Phys. 108, 043511, (2010).
Bowman, R., Venturini, E., Craft, B., Attalla, A. & Sullenger, D. Electronic structure of zirconium hydride: A proton NMR study. Phys. Rev. B 27, 1474–1488 (1983).
Sidhu, S. S. & McGuire, J. C. An X-Ray Diffraction Study of the Hafnium-Hydrogen System. J. Appl. Phys. 23, 1257 (1952).
Slocombe, D. R., Kuznetsov, V. L., Grochala, W., Williams, R. J. & Edwards, P. P. Superconductivity in transition metals. Philosophical transactions. Series A, Mathematical, physical and engineering sciences 373, (2015).
Züttel, A. Materials for hydrogen storage. Mater. Today 6, 24–33 (2003).
Satterthwaite, C. & Toepke, I. Superconductivity of Hydrides and Deuterides of Thorium. Phys. Rev. Lett. 25, 741–743 (1970).
Íñiguez, J., Yildirim, T., Udovic, T., Sulic, M. & Jensen, C. Structure and hydrogen dynamics of pure and Ti-doped sodium alanate. Phys. Rev. B 70, 060101 (2004).
Sandrock, G., Gross, K. & Thomas, G. Effect of Ti-catalyst content on the reversible hydrogen storage properties of the sodium alanates. J. Alloy. Compd. 339, 299–308 (2002).
Yamanaka, S., Miyake, M. & Katsura, M. Study on the hydrogen solubility in zirconium alloys. J. Nucl. Mater. 247, 315–321 (1997).
Yamanaka, S. et al. Characteristics of zirconium hydride and deuteride. J. Alloy. Compd. 330, 99–104 (2002).
Konashi, K., Ikeshoji, T., Kawazoe, Y. & Matsui, H. A molecular dynamics study of thermal conductivity of zirconium hydride. J. Alloy. Compd. 356-357, 279–282 (2003).
Kenji, K. et al. in Proceedings of the 2006 international congress on advances in nuclear power plants-ICAPP'06 (2006).
Gao, G., Bergara, A., Liu, G. & Ma, Y. Pressure induced phase transitions in TiH2. J. Appl. Phys. 113, 103512 (2013).
Huang, X. et al. Structural stability and compressive behavior of ZrH2under hydrostatic pressure and nonhydrostatic pressure. RSC Adv. 4, 46780–46786 (2014).
Quijano, R. & de Coss, R. Electronic structure and energetics of the tetragonal distortion for TiH2, ZrH2 and HfH2: A first-principles study. Phys. Rev. B 80, 184103 (2009).
Born M. & Huang K. Dynamical Theory of Crystal Lattice, Oxford University Press, Oxford (1954).
Allen, P. B. & Dynes, R. Transition temperature of strong-coupled superconductors reanalyzed. Phys. Rev. B 12, 905 (1975).
Oganov, A. R. & Glass, C. W. Crystal structure prediction using ab initio evolutionary techniques: principles and applications. J. Chem. Phys. 124, 244704 (2006).
Oganov, A. R., Lyakhov, A. O. & Valle, M. How Evolutionary Crystal Structure Prediction Works–and Why. Acc. Chem. Res. 44, 227–237 (2011).
Lyakhov, A. O., Oganov, A. R., Stokes, H. T. & Zhu, Q. New developments in evolutionary structure prediction algorithm USPEX. Comput. Phys. Commun. 184, 1172–1182 (2013).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Bader, R. F. Atoms in molecules. Accounts Chem. Res. 18, 9–15 (1985).
Henkelman, G., Arnaldsson, A. & Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comp. Mater. Sci. 36, 354–360 (2006).
Tang, W., Sanville, E. & Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Mat. 21, 084204 (2009).
Parlinski, K., Li, Z. & Kawazoe, Y. First-principles determination of the soft mode in cubic ZrO 2. Phys. Rev. Lett. 78, 4063 (1997).
Togo, A., Oba, F. & Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 78, 134106 (2008).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Mat. 21, 395502 (2009).
Acknowledgements
This work was supported by the National Basic Research Program of China (No. 2011CB808200), Program for Changjiang Scholars and Innovative Research Team in University (No. IRT1132), National Natural Science Foundation of China (Nos. 51032001, 11074090, 11204100, 10979001, 51025206, 11104102 and 11404134), National Found for Fostering Talents of basic Science (No. J1103202), China Postdoctoral Science Foundation (2012M511326, 2013T60314 and 2014M561279) Parts of calculations were performed in the High Performance Computing Center (HPCC) of Jilin University.
Author information
Authors and Affiliations
Contributions
T.C. initiated the project. Y.L. performed the first principle calculations and prepared all figures. Y.L., X.H., D.D. and T.C. analyzed the data and wrote the manuscript text. F.T., H.L., D.L., Z.Z., X.S., H.Y., H.Z. and B.L. reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liu, Y., Huang, X., Duan, D. et al. First-principles study on the structural and electronic properties of metallic HfH2 under pressure. Sci Rep 5, 11381 (2015). https://doi.org/10.1038/srep11381
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11381
This article is cited by
-
Strong correlation between electronic bonding network and critical temperature in hydrogen-based superconductors
Nature Communications (2021)
-
Structural, mechanical and electronic properties and hardness of ionic vanadium dihydrides under pressure from first-principles computations
Scientific Reports (2020)
-
Fungal Hybrid B heme peroxidases – unique fusions of a heme peroxidase domain with a carbohydrate-binding domain
Scientific Reports (2017)
-
Pressure dependence of electronic structure and superconductivity of the MnX (X = N, P, As, Sb)
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
