
Abstract
The dipeptidyl peptidase 11 from Porphyromonas gingivalis (PgDPP11) belongs to the S46 family of serine peptidases and preferentially cleaves substrates with Asp/Glu at the P1 position. The molecular mechanism underlying the substrate specificity of PgDPP11, however, is unknown. Here, we report the crystal structure of PgDPP11. The enzyme contains a catalytic domain with a typical double β-barrel fold and a recently identified regulatory α-helical domain. Crystal structure analyses, docking studies and biochemical studies revealed that the side chain of Arg673 in the S1 subsite is essential for recognition of the Asp/Glu side chain at the P1 position of the bound substrate. Because S46 peptidases are not found in mammals and the Arg673 is conserved among DPP11s, we anticipate that DPP11s could be utilised as targets for antibiotics. In addition, the present structure analyses could be useful templates for the design of specific inhibitors of DPP11s from pathogenic organisms.
Similar content being viewed by others
Introduction
Periodontitis is a bacterially induced inflammatory disease that destroys the periodontal tissues, eventually leading to tooth loss1. Periodontitis is widely regarded as the second most common disease worldwide and chronic periodontitis affects approximately 750 million people as of 20102. Various studies have revealed that periodontitis is associated with systemic diseases such as diabetes and cardiovascular disease3, preterm and low-weight births4, Alzheimer’s disease5, cancers6, respiratory diseases7 and rheumatoid arthritis8. Porphyromonas gingivalis, a Gram-negative, anaerobic bacterium, is a major pathogen associated with the chronic form of periodontitis1,9. Because P. gingivalis is an asaccharolytic bacterium that gains its metabolic energy by fermenting amino acids10, P. gingivalis secretes various proteases/peptidases that are capable of digesting external proteins into peptides. The best-characterised proteases from P. gingivalis are two cysteine proteases, gingipains R (Rgp) and K (Kgp), that exhibit specificity for arginine and lysine, respectively11,12. Rgp and Kgp have been implicated as major virulence factors of P. gingivalis13,14,15 and inhibitors of these proteases have been reported to suppress the virulence of P. gingivalis16. P. gingivalis also produces other proteases/peptidases including collagenase17, dipeptidyl peptidase 4 (PgDPP4)18 and prolyl tripeptidyl-peptidase A (PgPTPA)19. P. gingivalis utilises di- and tripeptides, instead of single amino acids, as sources of carbon and energy20,21. Therefore, peptidases of P. gingivalis that provide di- and tripeptides are essential for the metabolism of the bacterium and much attention has been paid to dipeptidyl peptidases (DPPs) from P. gingivalis. In addition to DPP4, novel DPPs, DPP5 (PgDPP5), DPP7 (PgDPP7) and DPP11 (PgDPP11), have been identified from P. gingivalis22,23,24. The P. gingivalis peptidases PgPTPA, PgDPP4 and PgDPP5 have been classified as clan SC, family S9 in the MEROPS database25, while PgDPP7 and PgDPP11 have been assigned to another type of serine peptidase family, S46 in clan PA22,23. Whereas PgDPP7 exhibits a broad substrate specificity for both aliphatic and aromatic residues at the P1 position (NH2-P2-P1-P1’-P2’-…, where the P1-P1’ bond is the scissile bond), PgDPP11 exhibits a strict substrate specificity for acidic residues (Asp/Glu) at the P1 position. Because Asx (Asp and Asn) and Glx (Glu and Gln) are the most abundantly utilised amino acids in this bacterium20,21, PgDPP11 plays a critical role in the metabolism of P. gingivalis by degrading polypeptides carrying Asp and Glu.
The S46 peptidases are widely distributed in anaerobic Gram-negative species in the genera Bacteroides, Parabacteroides and Porphyromonas, but they are not found in mammals23,26. Therefore, the family S46 peptidases may represent ideal targets for novel antibiotics. Biochemical studies have revealed that Gly666 of PgDPP7 and Arg673 of PgDPP11 are crucial residues for the P1 specificity23,27 whereas Phe664 of PgDPP7 and Phe671 of PgDPP11 are involved in recognition of the P2 residue of the substrate28. Regarding the catalytic mechanism of the S46 peptidases, the serine protease catalytic triad29,30 has recently been identified for dipeptidyl aminopeptidase BII (DAP BII) from Pseudoxanthomonas mexicana WO2426. The catalytic triad of DAP BII is His86-Asp224-Ser657, corresponding to His89-Asp225-Ser648 of PgDPP7 and His85-Asp227-Ser655 of PgDPP11.
Recently, the first three-dimensional structure of a S46 peptidase has been determined, for DAP BII31,32. That study revealed that DAP BII is a homodimer and that each subunit contains a peptidase domain including a double β-barrel fold that is characteristic of the chymotrypsin superfamily33,34, as well as an unusual α-helical domain that regulates the exopeptidase activity of DAP BII. Peptide complex structures of DAP BII have revealed that the residues directly involved in recognition of the N-terminal amino group of the substrate peptide are Asn215, Trp216, Asn330 and Asp674 of DAP BII. The shapes of the S1 and S2 subsites of DAP BII are sufficiently deep and wide to accommodate any amino acids and are consistent with the non-specific peptide digestion profile of DAP BII35. The catalytic mechanism of DAP BII, possibly common among the S46 peptidases, has also been proposed based on the crystal structure analyses of a series of peptide complex structures of wild-type and mutant DAP BIIs. Although the overall structure, the molecular basis of the exopeptidase activity and the catalytic mechanism of the S46 peptidase have been revealed by the crystal structure analyses of DAP BII32, determinants for the substrate specificity of S46 peptidases at the atomic level remain to be fully elucidated.
In this study, we determined the crystal structure of PgDPP11. Crystal structure analyses, an amino acid sequence comparison, in silico docking studies and site-directed mutagenesis studies showed that Arg673 in PgDPP11 is essential for the Asp/Glu P1 specificity of PgDPP11. Because crystallisation trials of PgDPP11 complexed with oligopeptides containing Asp/Glu at the P1 position were not successful, we have implemented a different strategy to verify the involvement of Arg673 in substrate recognition. A mutant DAP BII (G675R), in which Gly675 was replaced with Arg to mimic the P1 specificity of PgDPP11, was prepared and successfully co-crystallised with a Leu-Glu dipeptide. The crystal structure of DAP BII (G675R) complexed with the Leu-Glu dipeptide revealed that the Arg675 in the mutant DAP BII is directly involved in the recognition of the Glu side chain of the bound dipeptide. This result suggests that the Arg side chain in the S1 subsite of S46 peptidases, e.g., Arg673 in PgDPP11 and Arg675 in the mutant DAP BII, is responsible for the Asp/Glu specificity of DPP11s in the S46 family.
Results
Overall structure of PgDPP11
The crystal structure of PgDPP11 was determined using multi-wavelength anomalous diffraction method at 2.5 Å resolution by analysing a Se-Met substituted PgDPP11 crystal (Fig. S1 and Table S1). The final model was obtained from a native data set using a space (the Japanese Experimental Module “Kibo” at the International Space Station)-grown crystal consisting of residues 22-720, 688 water molecules and a disulphide bridge (Cys69-Cys86). The final R and Rfree values are 0.188 and 0.226, respectively, at 1.66 Å resolution (Table S2). A protomer of PgDPP11 is situated in the asymmetric unit (Fig. 1A). Two protomers of PgDPP11 are related by a crystallographic twofold axis of the C2221 crystal and form a dimer (Fig. 1B). Dimerisation is also observed in the crystal structure of another S46 peptidase, DAP BII, which is the only other known crystal structure of this family of enzyme and was recently determined by our group32. The protruding β-hairpin dimerisation interface reported in DAP BII is also observed for PgDPP11. This type of inter-subunit interaction is found among several crystal forms of PgDPP11 (Table S1), indicating that such dimer formation is the natural state of PgDPP11. Each subunit contains a catalytic double β-barrel domain harbouring the Asp-His-Ser catalytic triad and an α-helical domain that caps the active site (Fig. 1A). The assignment of the secondary structural elements is provided in Fig. 2 and S2.

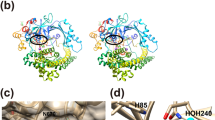
Three-dimensional structure of PgDPP11.
(A) Stereoview of the PgDPP11 subunit. Helices (1 to 33) and strands (N1 to N7 and C1 to C6 in the N- and C-terminal barrels, respectively) are coloured as in Fig. 2. (B) Dimeric structure of PgDPP11 in orthogonal views. The catalytic domain, helical domain and beta hairpin are respectively shown in blue, orange and green for one subunit and cyan, yellow and light green for the other subunit.

Amino acid sequences of clan PA S46 family peptidases.
The His-Asp-Ser catalytic triad (red), the N-terminal amino group recognition residues (blue) and candidates for the determinants of the P1 specificity of the S46 peptidases (green) are highlighted. Secondary structural elements are shown on top for PgDPP11. The abbreviations used (UniProt accession numbers) are as follows: PgDPP11 (B2RID1), Porphyromonas gingivalis DPP11; PeDPP11 (F8WQK8), Porphyromonas endodontalis DPP11; PgDPP7 (Q7MWU6), Porphyromonas gingivalis DPP7; PeDPP7 (C3JAQ3), Porphyromonas endodontalis DPP7; SpDPP11 (A4Y3F4), Shewanella putrefaciens DPP11; SmDPP7 (B4SLK2), Stenotrophomonas maltophilia DPP7 and PmDAPBII (V5YM14), Pseudoxanthomonas mexicana WO24 DAP BII.
The catalytic domain is composed of two non-contiguous stretches of PgDPP11 (residues 22-279 and 572-720) and contains a double β-barrel structure, which is a characteristic of the chymotrypsin superfamily, consisting of 17 α-helices (α1-α10 and α27-α33) and 15 β-strands (βN1-βN7 and βC1-βC6). The catalytic domain of the only structurally characterised S46 enzyme DAP BII32 can be superimposed on the catalytic domain of PgDPP11 (Fig. 3A). The serine peptidase catalytic triads, His85, Asp227 and Ser655 in PgDPP11 and His86, Asp224 and Ser657 in DAP BII26,32, are almost completely superimposable, with a root mean square (rms) deviation between the two structures of 0.94 Å for 306 structurally equivalent Cα atoms, which had 55% sequence identity for that region.

The catalytic and α-helical domains of PgDPP11.
(A) Superposition of the catalytic domains of PgDPP11 (blue) and DAP BII (yellow). The catalytic triads of these enzymes are shown in ball-and-stick format. (B) The protomers of PgDPP11 and DAP BII in orthogonal views. The superposition of each protomer was performed with respect to the structurally equivalent 306 Cα atom pairs of the respective catalytic domains.
The α-helical domain is inserted between strands C1 and C2 of the catalytic domain and spans residues 280-571. The domain consists of 16 helices packed into a helical bundle that caps the catalytic triad of the catalytic domain. No structural homologue of this domain is found in the DALI database36, except for the α-helical domain of DAP BII. Thus, the α-helical domain is absolutely restricted to S46 peptidases. A superposition of the α-helical domain of PgDPP11 with that of DAP BII shows that the helices 16 to 18 and 23 to 24 of PgDPP11 extend relatively far from the main bundle of the α-helical domain. In addition, a superposition of the overall structure of PgDPP11 with that of peptide-free DAP BII shows that the two domains have different relative orientations (Fig. 3B).
The catalytic triad of PgDPP11
The catalytic triad of PgDPP11, His85, Asp227 and Ser655, can be identified based on structural similarity with DAP BII26. The triad corresponds to His57, Asp102 and Ser195 in chymotrypsin29,30. Ser655 is located just before strand C4 in the C-terminal barrel, His85 is located in an insertion helix (helix 4) in the N-terminal barrel and Asp227 is located just before strand N7 in the N-terminal barrel. The hydroxyl group of Ser655 is exposed to solvent and hydrogen bonded to the imidazole group of His85 (OG(Ser655)---NE2(His85): 2.8 Å). One of the oxygen atoms of the carboxylate group of Asp227 forms a hydrogen bond with His85 and completes the catalytic triad (ND1(His85)---OD2(Asp227): 2.8 Å). As described above, the catalytic triad of PgDPP11 is spatially arranged in the same manner as that of DAP BII (Fig. 3A). The active site is located on one face of the double β-barrel catalytic domain and is covered by the α-helical domain. The putative oxyanion hole is formed by the backbone amide nitrogen atoms of Ser655 and Gly653. The respective corresponding residues in chymotrypsin are Ser195 and Gly19337,38.
The N-terminal amino group recognition residues and the S1 subsite of PgDPP11
PgDPP11 has 29% amino acid sequence identity with DAP BII (Fig. 2). The structural features of PgDPP11 revealed by the present study, including the double β-barrel catalytic domain, the α-helical regulatory domain and the protruding β-hairpin dimerisation interface, are conserved between the two enzymes (Figs 1 and 3). A noteworthy difference between PgDPP11 and DAP BII is their substrate specificities at the P1 position (NH2-P2-P1-P1’-P2’-…, where the P1-P1’ bond is the scissile bond). The active site of PgDPP11 involves N-terminal amino group recognition residues, S1 subsite, S2 subsite and catalytic triad described above.
The N-terminal amino group recognition residues of PgDPP11 are Asn218, Trp219 and Asp672 from the catalytic domain and Asn333 from the α-helical domain. The corresponding residues of DAP BII, revealed by X-ray crystallographic analyses, are Asn215, Trp216, Asp674 and Asn33032. For DAP BII, a large-scale conformational change, from open to closed, was observed upon peptide binding. The distance between the Cα atoms of Asn330 and catalytic Ser657 of DAP BII observed in the peptide-free, open conformation (PDB ID: 3WOJ) was 16.6 Å, whereas that observed in the dipeptide-bound, closed conformation (PDB ID: 3WOL) was 12.8 Å. The corresponding distance between the Cα atoms of Asn333 and Ser655 of PgDPP11 observed in the current peptide-free conformation was 15.5 Å. This suggests that the present structure of PgDPP11 corresponds to the open conformation of DAP BII and that the active site cleft of PgDPP11 would close upon peptide binding.
High-resolution diffraction data obtained from a space-grown crystal enabled us to identify two potassium ion-binding sites in the catalytic domain of PgDPP11 because the present crystallisation conditions contained 0.16 M tri-potassium citrate in the reservoir solution. One was found at the N-terminal amino group binding site and the other was found at the bottom of the S1 subsite. The former potassium ion was coordinated by the side chains of Asn218 (OD1(Asn218)---K: 2.8 Å) and Asp672 (OD1(Asp672)---K: 2.6 Å) and a water molecule (O(HOH)---K: 2.5 Å) and was also stabilised by a cation-pi interaction with the indole ring of Trp219 (Fig. 4A). Similar cation-pi interaction was observed between the indole ring of Trp216 in DAP BII and the N-terminal amino group of bound peptide (Fig. 4B)32. The relative arrangement of the side chains of Asn218, Trp219, Asp672 and Asn333 in PgDPP11 was significantly different from that of Asn215, Trp216, Asp674 and Asn330 in the peptide-bound DAP BII, in which conformational change (open to closed) occurred upon peptide binding. In contrast, the relative arrangement of the N-terminal amino group recognition residues of PgDPP11 was similar to that of peptide-free DAP BII. The only difference between the structures of the N-terminal amino group recognition residues of PgDPP11 and of peptide-free DAP BII is the conformations of Asn218 in PgDPP11 and Asn215 in DAP BII. The former residue is involved in binding potassium ion (Fig. 4A) and mimics a peptide-bound conformation as observed in the peptide-bound DAP BII, whereas the latter residue is flipped out from the N-terminal amino group binding site (Fig. 4C).

The active site of PgDPP11.
(A) Binding mode of the potassium ion in the amino group binding site of PgDPP11. (B) Super position of the amino group binding site of PgDPP11 (cyan) with that of Val-Tyr bound DAP BII (orange). (C) Super position of the active site of PgDPP11 (cyan) with that of the peptide-free DAP BII (pink). The PgDPP11 residue numbers of the catalytic triad and the Gly residue providing the NH group for the oxyanion hole, the N-terminal amino group recognition residues and candidates for the determinants of the strict P1 specificity are coloured red, blue and green, respectively. (D) The putative dipeptide binding mode of PgDPP11. The Leu-Asp dipeptide (orange) was docked into the active site of PgDPP11 (green). The molecular surface was generated for the closed conformation of wild-type DAP BII, the starting template of the docking study, to emphasise Arg side chains involved in the Asp recognition.
The putative S1 subsite of PgDPP11 is observed adjacent to the catalytic Ser655. The S1 subsite of PgDPP11 consists of His649, Thr650, Thr651, Gly652, Asn670, Arg673, Gly677, Gly680, Asp681 and Ser691 and is mainly composed of hydrophilic residues (Fig. 4C, cyan). In contrast, the S1 subsite of DAP BII32, is more hydrophobic, containing the residues Asp651, Ile652, Thr653, Gly654, Ala672, Gly675, Ser679, Ser682, Asn683 and Met693 (Fig. 4C, pink). Arg673 in PgDPP11, a crucial residue for the Asp/Glu specificity of PgDPP1123,27, corresponds to Gly675 of DAP BII and is located in the wall of subsite S1 (Fig. 4C). The present crystal structure analysis confirms the importance of this residue for the Asp/Glu specificity of PgDPP11. Our structure further suggests that Arg337 in the α-helical domain of PgDPP11 (corresponding to Asn334 of DAP BII, Fig. 2) may constitute another side wall of subsite S1 that would interact with the P1 residue of the substrate peptide when, upon substrate binding, PgDPP11 adopts a closed conformation as observed for DAP BII complexed with various peptides. To examine the roles of Arg337 and Arg673 during catalysis by PgDPP11, in silico docking and site-directed mutagenesis studies were performed.
The putative peptide-binding mode of PgDPP11
In silico docking of a Leu-Asp (LD) dipeptide into the active site of PgDPP11 suggested particular interactions between the bound dipeptide and the S1 subsite of PgDPP11 (Fig. 4D). The main chain of the docked dipeptide can bind to PgDPP11 in essentially the same mode as observed for various peptides in complexes with DAP BII32. A bifurcated hydrogen bond bridges the amino and carbonyl groups of the main chain of the N-terminal P2 residue with, respectively, the side-chain carbonyl and amide groups of Asn333 in the α-helical domain. The side chains of Asn218 and Asp672 are also involved in the recognition of the N-terminal amino group of the docked peptide. The indole ring of Trp219 is involved in a cation-pi interaction with the N-terminal amino group of the docked peptide. The NH group of the P1 residue of the bound peptide forms a hydrogen bond with the carbonyl oxygen of Phe671 (Fig. 4D). The carbonyl oxygen of the P1 residue is accommodated in the oxyanion hole formed by the catalytic Ser655 and Gly653 via hydrogen bonds to their main-chain NH groups.
The most remarkable observation in the in silico docking model is that inter-side-chain interactions between two Arg residues (Arg337 and Arg673) and two Asp residues (P1-Asp and Asp681) are possible. Thus the two positively charged Arg residues from the S1 and the two negatively charged Asp residues, one from the S1 subsite and the other from the P1 in the substrate, could mediate the S1-P1 interactions of the active site of PgDPP11. As described above, the S1 subsite of PgDPP11 consists primarily of hydrophilic residues (Fig. 4C). The enhanced hydrogen bonding capacity might contribute to the strict specificity for a negatively charged P1 residue in the substrate.
Site-directed mutagenesis studies of PgDPP11
To test the roles of Arg337 and Arg673 in substrate binding by PgDPP11, we replaced these residues with alanine, glycine, or asparagine and tested the enzymatic activity of the mutant proteins on synthetic substrates with Asp/Glu at the P1 position (Table 1). Mutation of Arg673 to Ala resulted in complete loss of activity, either by itself or in combination with the substitution of Arg337 with Ala. These results indicate that the Arg673 in PgDPP11 plays an important role in the recognition of the negatively charged residue of the substrate peptide at the P1 position. Interestingly, mutation of Arg673 to Gly resulted in drastically decreased but still significant activities against Asp/Glu substrates (0.017 U/mg and 0.0017 U/mg for Gly-Glu-pNA and Gly-Asp-pNA, respectively), as well as detectable activity against substrates harbouring hydrophobic residues at the P1 position (0.00038 U/mg and 0.00071 U/mg for Gly-Phe-pNA and Ala-Ala-pNA, respectively). The latter activities increased in a double mutant of PgDPP11 containing Arg673Gly and Arg337Asn (0.0062 U/mg and 0.0180 U/mg for Gly-Phe-pNA and Ala-Ala-pNA, respectively), which appears to be the result of an additive effect of the two mutations.
Contrary to our expectations, mutation of Arg337 in PgDPP11 (R337A, R337G and R337N in Table 1) led to a significant increase (or preservation), rather than decrease, of the enzymatic activity. For example, the specific activities of the Arg337Ala mutant of PgDPP11 against the synthetic substrates Gly-Glu-pNA (1.6 U/mg) and Gly-Asp-pNA (2.58 U/mg) are significantly higher than or equal to those of wild type PgDPP11 (0.77 U/mg and 2.66 U/mg for Gly-Glu-pNA and Gly-Asp-pNA, respectively). Similar results were obtained for the Arg337Asn mutant of PgDPP11 (3.01 U/mg and 4.6 U/mg for Gly-Glu-pNA and Gly-Asp-pNA, respectively). These data indicate that only Arg673, not Arg337, is essential for the Asp/Glu P1 specificity of PgDPP11.
Crystal structure analysis of a mutant DAP BII
To obtain structural evidence that the side chain of Arg673 in PgDPP11 is directly involved in the recognition of the Asp/Glu residue at the P1 position of substrate peptides, a huge number of crystallisation experiments by the canonical hanging-drop vapour diffusion method on the ground were performed to crystallise PgDPP11 in the presence of various peptides harbouring Asp/Glu at the P1 position. Furthermore, we also tried to crystallise the peptide-complex crystals of PgDPP11 by the counter-diffusion method in space. These attempts, however, were not successful. Therefore, as an alternative approach, Gly675 in DAP BII, which corresponds to Arg673 in PgDPP11, was mutated to Arg (G675R) and the mutant enzyme was tested on synthetic substrates with Asp/Glu at the P1 position. As shown in Table 1, the mutant DAP BII acquired activities of 0.028 U/mg for Gly-Glu-pNA and 0.0218 U/mg for Gly-Asp-pNA, which were not detectable in wild-type DAP BII. In addition, activities against synthetic substrates with hydrophobic residues at the P1 position were drastically reduced after substitution of Gly675 by Arg (0.0060 U/mg and 0.37 U/mg for Gly-Phe-pNA and Ala-Ala-pNA, respectively) compared with those of the wild-type DAP BII (3.3 U/mg and 9.5 U/mg for Gly-Phe-pNA and Ala-Ala-pNA, respectively).
The mutant DAP BII had acquired the enzymatic activity for synthetic substrates with Asp/Glu at P1 position (Table 1), allowing further analyses of their mode of interaction. The mutant DAP BII (G675R) was overexpressed and co-crystallised with a Leu-Glu dipeptide (Fig. S3). The structure of this complex was solved at 2.18 Å resolution and the refined structure was compared with recently described DAP BII structures32. As we had expected, the artificially introduced Arg675 side chain in the mutant DAP BII made direct contact with the side chain of the Glu residue of the bound dipeptide (NE(Arg675)---OE1(Glu): 2.9 Å) (Fig. 5A). The present crystallographic data on the mutant DAP BII clearly indicate that the Arg side chain in the S1 subsite of S46 peptidase, e.g., Arg673 in PgDPP11 and Arg675 in the mutant DAP BII, is responsible for the Asp/Glu specificity of these enzymes.

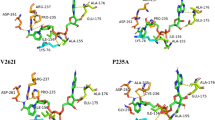
Crystal structure of DAP BII Gly675Arg mutant.
(A) Binding mode of the bound Leu-Glu dipeptide (pink) in the active site of the mutant DAP BII (yellow). (B-D) Superpositions of the crystal structure of Leu-Glu-bound mutant DAP BII (yellow) and (B) Val-Tyr (cyan)-bound wild-type DAP BII (orange), (C) peptide-free PgDPP11 (cyan) and (D) hypothetical model of Leu-Asp (orange)-bound PgDPP11 (green). Residue numbers are shown for DAP BII (A and B) and for PgDPP11 (C and D). The molecular surfaces were generated in the same way as in Fig. 4D.
Discussion
In this study, we have solved the crystal structure of PgDPP11, which specifically recognises an Asp/Glu residue at the P1 position of substrate peptides. To better understand this substrate specificity, we examined the role of the Arg673 in the S1 subsite. Arg673 is essential for specific recognition of Asp/Glu by PgDPP11 because substrates with either residue at the P1 position could no longer be converted after mutation of Arg673 (Table 1), which is consistent with previous results for PeDPP1123 and PgDPP1127. Interestingly, the Arg673 to Gly mutation, particularly when combined with the substitution of Arg337 to Asn, increased the reactivity of mutant PgDPP11 towards substrates with a hydrophobic residue at the P1 position (Table 1). These substitutions expand the S1 subsite of PgDPP11, which therefore partially mimics the active site of DAP BII that is specific for substrates with a hydrophobic residue at the P1 position. As reported for a Gly666Arg mutant of PgDPP727, we could change the specificity of DAP BII to substrates with a negatively charged residue at the P1 position after substitution of Gly675 by Arg. Arg673 of PgDPP11 is completely conserved in PgDPP11 type DPP11s (described later). Taken together, these phylogenetic data and the results of our site-directed mutagenesis studies on PgDPP11 and DAP BII strongly suggest that Arg673 in PgDPP11 is responsible for the recognition of the Asp/Glu residue at the P1 position of the substrate peptide.
To obtain structural evidence for a direct interaction between the side chain of Arg673 in PgDPP11 and the side chain of Asp/Glu at the P1 position of the substrate peptide, we tried to generate the crystals of PgDPP11 in complex with various peptides harbouring Asp/Glu at the P1 position. These attempts, however, were not successful. Because the Gly675Arg mutant DAP BII had acquired a preference for DPP11-type substrates (Table 1), we tried to crystallise the Gly675Arg mutant DAP BII complexed with peptides harbouring Asp/Glu at the P1 position. The crystal structure of the Gly675Arg mutant DAP BII reveals that the side chain of Arg675 is directly involved in an interaction with the side chain of Glu at the P1 position of the bound dipeptide (Fig. 5A). The recognition mode of the main chain of the bound dipeptide by the mutant DAP BII was quite similar to those observed for the wild type DAP BII complexed with dipeptides (Fig. 5B). The artificially introduced Arg675 is structurally equivalent to Arg673 in PgDPP11 (Fig. 5C, D). Thus, the present crystal structure analysis of the Gly675Arg mutant DAP BII provides the first structural evidence that the side chain of Arg residue in the S1 subsite of S46 peptidase is essential for recognition of the acidic side chain at the P1 position of the bound peptide. As an exceptional case, we must mention SpDPP11. In the case of SpDPP11, the architecture of the S1 subsite is somewhat different from that of PgDPP11. The residue corresponding to Arg673 in PgDPP11 is replaced by Ser684 in SpDPP11 (Fig. 2). To compensate for the lack of Arg residue at Ser684 in SpDPP11, the residue corresponding to Gly680 in PgDPP11, located at the bottom of the S1 subsite, is replaced by Lys691 in SpDPP11 (Fig. 2). The important role of Lys691 in hydrolysing the synthetic substrate harbouring the Asp/Glu at the P1 position was confirmed by site-directed mutagenesis studies27. In the case of SpDPP11-type DPP11s among the S46 family, harbouring Ser residue at the position corresponding to Arg673 in PgDPP11, Lys691 in SpDPP11 is well conserved (see below for details).
Among 1355 amino acid sequences of S46 peptidases found in 462 species (Fig. 6), 399 amino acid sequences are putatively assigned as DPP11. Among the 399 DPP11s, 347 DPP11s (PgDPP11-type DPP11s) have an Arg residue at the position equivalent to 673 in PgDPP11 and 52 DPP11s (SpDPP11-type DPP11s) have Ser and Lys residues at the positions equivalent to 673 and 680, respectively, in PgDPP11. If position 673 in a DPP11 is Arg, position 680 is Gly (49.86%), Ser (42.65%), or Asn (6.92%), whereas Lys and Arg never exist in any other positions of the S1 pocket. Therefore, the active site architectures of the two types of DPP11s are equivalent in that the S1 subsite of each has one positively charged side chain correlated with the Asp/Glu-specific P1 specificity of the enzyme. Taken together, the site-directed mutagenesis studies (Table 1), in silico docking studies (Fig. 4D) and crystallographic studies (Figs 4A and 5A) allow us to safely conclude that the side chain of Arg673 in PgDPP11 is responsible for recognition of the Asp/Glu side chain at the P1 position of the bound substrate. This conclusion is also applicable to the other DPP11s in the S46 family.

Amino acid sequence analyses of the S1 subsite of S46 peptidases
. Amino acid sequence variations were analysed for 1355 peptidases belonging to the S46 family. Residues located in the S1 subsite of PgDPP11 are shown in left. All: amino acid sequence variations of the 1355 peptidases for each position. G, R and S: analyses of sequence variations with respect to the nature of amino acid at the position equivalent to Arg673 in PgDPP11.
It is interesting to note that Arg673 in PgDPP11, located at the tip of strand C5, is structurally equivalent to Val216 in elastase29, a clan PA family S1 endopeptidase specific for small, uncharged residues (Fig. 7, left). These residues infill the S1 subsite of each enzyme and prevent the access of longer or bulky side chains at the P1 position of the substrate peptide. In contrast, the corresponding positions are occupied by Gly in DAP BII32 and chymotrypsin29, which can accommodate longer or bulky side chains as the P1 position of the substrate peptide (Fig. 7, right). Thus, S46 peptidases and S1 peptidases adopt structurally equivalent mechanisms in which the side chains located at the tip of strand C5 regulate the depth of their S1 subsite, although the peptide digestion patterns are quite different between these peptidase families: “exo” for S46 and “endo” for S1.

Schematic diagrams of the S1 subsites of clan PA peptidases.
The depths of the S1 subsites were determined by the nature and length of the side chain located at the tip of the strand C5. The P1 specificities/preferences are as follows: PgDPP11: negatively charged; DAP BII: aromatic (non-specific); Elastase: small uncharged; Chymotrypsin: aromatic.
Regarding the Arg337 located in the α-helical domain, although the Arg residue is also conserved in PeDPP11 (Fig. 2), the Arg residue is conserved only in 36 sequences (10.37%, Fig. 6) and conservative substitutions of Arg to Lys are observed in 140 sequences (40.35%). Unexpectedly, in the majority of PgDPP11-type DPP11s, that position holds an Asn residue (164 sequences, 47.26%). These data are consistent with the results of the present mutagenesis studies on the Arg337 in PgDPP11, which showed that the Arg337 in PgDPP11 is not essential for recognition of the Asp/Gly residue at the P1 position of the synthetic substrate (Table 1). For SpDPP11-type DPP11s, interestingly, the position is completely conserved as Lys (52 sequences, 100%). This indicates that the positively charged residue in the α-helical domain of SpDPP11-type DPP11s might contribute to some degree to recognition of the acidic side chain at the P1 position of the substrate peptide. In addition to the Arg/Gly difference at the wall of the S1 subsite, two hydrophilic residues, Thr650 (222 sequences, 63.98%) and Asn670 (225 sequences, 64.84%), are conserved among PgDPP11-type DPP11s, which prefer a hydrophilic Asp/Glu residue at the P1 position of the substrate peptide. However, two hydrophobic residues, Ile652 (785 sequences, 89.00%) and Ala672 (702 sequences, 79.59%), which correspond to Thr650 and Asn670 in PgDPP11, are conserved among DAP BII/DPP7s, which prefer a hydrophobic residue at the P1 position.
Because the S46 peptidases are not found in mammals, but do exist in some pathogenic organisms, such as P. gingivalis (a periodontal pathogen)1, Stenotrophomonas maltophilia (an opportunistic pathogen)39 and Xylella fastidiosa (a plant pathogen)40, the present structure of PgDPP11 is useful for drug design. Structural differences between PgDPP11 and human dipeptidyl peptidases (Table S3) support the potential. For example, recognition modes of the main-chain NH group of the P1 residue are clearly different between PgDPP11 (C = O group of Phe671) and DPP4 from Homo sapiens (HsDPP4)41 (none, because DPP4 prefers Pro residue, which lacks NH group). Actually, a DPP4 inhibitor, diprotin A (IC50 value of 0.3 μM against DPP4 from rat kidney)42, could not inhibit digestion of a synthetic substrate Gly-Asp-pNA by PgDPP11 at the inhibitor concentration of 0.3 mM (Suzuki et al., unpublished data). Another example is the difference in substrate specificities between PgDPP11 (P1-Asp/Glu) and HsDPPI (non-specific). The S1 subsite of PgDPP11 forms a pocket to accommodate the P1 side chain, whereas the S1 subsite of HsDPPI shows a character of a surface rather than a pocket and the P1 side chain is exposed to the solvent43,44. Therefore, a PgDPP11 inhibitor could be an anti-periodontitis drug because the enzyme plays a critical role in the metabolism of P. gingivalis by degrading polypeptides carrying Asp and Glu20,21 and the inhibitor is likely to have little effect on the activities of human dipeptidyl peptidases. The active site architecture of PgDPP11 would be a good target for designing specific inhibitors against DPP11s of the S46 peptidases because positively charged Arg673 in PgDPP11 is highly conserved among the DPP11s. In general, several factors, such as shape, electrostatics, flexibility, hydration and allostery, are important for improving the selectivity in inhibitor design45. Among these factors, electrostatic complementarity is often useful for designing selective inhibitors. A representative example is the electrostatic interaction between the Arg side chains of an influenza neuraminidase and the carboxylate group of ZANAMIVIR46. Thus, the Arg side chain in the S1 subsite of DPP11s would be a good pharmacophore for a DPP11 selective inhibitor harbouring a negatively charged group.
Two potassium ion-binding sites were identified in the catalytic domain of PgDPP11 based on a high-resolution diffraction data obtained from a space-grown crystal. One site was meaningful because the site corresponds to the N-terminal amino group binding site (Fig. 4A). Before obtaining the high-resolution data (1.66 Å) from the space-grown crystal, the potassium ions were tentatively assigned as water molecules for data from a ground-grown crystal (1.96 Å), although weak residual electron densities (positive peaks in |Fo|-|Fc| map) were observed around the water molecules. When the high-resolution data were available, stronger residual electron densities were observed around the water molecules and the temperature factors of the water molecules were substantially lower than the surrounding atoms. The water molecules were replaced with potassium ions and then the residual densities disappeared and the temperature factors of the potassium ions were comparable to the surrounding atoms. Not only the resolution (1.66 Å vs. 1.96 Å), but also the average mosaicity (0.182 vs. 0.392) and other statistics are improved in the space grown crystal (Table S1), such improvements are frequently observed in space grown crystals47. As a result, a finer electron density map was obtained. Because PgDPP11 crystals obtained in space were of better quality than those obtained on the ground and they enabled us more precise structure analyses, we consider that crystallisation experiments under microgravity in this case lead us to more articulate understanding.
In this study, we present the crystal structure of PgDPP11, a member of the S46 peptidase family. The crystal structure analyses, in silico docking studies and site-directed mutagenesis studies clearly explain the molecular basis of the Asp/Glu specificity of PgDPP11, which is determined by the conserved Arg residue in the S1 subsite. We anticipate that the present structural analyses will support the design of specific inhibitors of DPP11s produced by pathogenic organisms, which require these enzymes for survival.
Methods
Overexpression and purification of PgDPP11
PgDPP11 was expressed and purified as described elsewhere48. A synthetic gene coding for full-length PgDPP11 (residues 1-720), codon-optimised for expression in E. coli, was purchased from Genscript (Piscataway, USA) and cloned into the pET22b expression plasmid (Merck, Darmstadt, Germany). The mature PgDPP11 was composed of 699 amino acids (residues 22 to 720), with a theoretical molecular weight of 79618.27 and an isoelectric point of 5.87. E. coli BL21 Gold(DE3) cells (Agilent Technologies, Santa Clara, USA) transformed with the pET22b-PgDPP11 expression plasmid (Merck) were grown in 2xYT media at 298 K to an OD600 of 0.6. Overproduction of PgDPP11 was performed using the Overnight Express Autoinduction System 1 (Merck). Thereafter, the cells were harvested by centrifugation at 8,000 × g. Cells were disrupted using BugBuster Protein Extraction Reagent (Merck). The cell extract was obtained by centrifuging the lysate at 27,000 × g for 30 min. PgDPP11 was purified by precipitation with 35 to 70% ammonium sulfate and hydrophobic column chromatography using a HiPrep 16/10 Butyl column (GE Healthcare, Little Chalfont, UK). The eluate was desalted using a HiPrep 26/10 desalting column (GE Healthcare) and finally subjected to anion-exchange column chromatography using a Mono Q 5/50 GL column (GE Healthcare). The fractions containing PgDPP11 were pooled, buffer-exchanged to 80 mM Tris/HCl pH 8.5 using a Vivaspin 20 concentrator (GE Healthcare) and concentrated to 10 mg/ml using the concentrator. Se-Met-substituted PgDPP11 was expressed using the Overnight Express Autoinduction System 2 (Merck). The Se-Met derivative was purified in a manner similar to that used for wild-type PgDPP11 described above.
Crystallisation
To obtain peptide-free PgDPP11 crystals48, the samples were crystallised using the hanging-drop method, in which 1 μl of protein solution (10 mg/ml PgDPP11 in 80 mM Tris-HCl, pH 8.5) was mixed with the same volume of reservoir solution (20% (v/v) glycerol, 16% (m/v) PEG8000 and 0.16 M tri-potassium citrate) and incubated at 293 K. The drops were suspended over 200 μl of reservoir solution in 48-well plates. Peptide-free crystals were also obtained using a counter-diffusion crystallisation method49 under a microgravity environment in the Japanese Experimental Module “Kibo” at the International Space Station (ISS)47.
X-ray data collection
Because the crystallisation conditions of PgDPP11 described above included 20% (v/v) glycerol in the reservoir solution, X-ray data could be collected under cryogenic conditions without the need for any additional cryoprotectant. Crystals obtained from the hanging-drop method were directly mounted in nylon loops and flash-cooled in a cold nitrogen gas stream at 100 K immediately before data collection. Data were collected by the rotation method at 100 K using ADSC Quantum CCD detectors with synchrotron radiation sources at the Photon Factory. The Laue group and unit-cell parameters were determined using the XDS software package50. The resulting cell parameters and data-collection statistics are summarised in Table S1.Table S1.
Structure determination
Initially, we attempted to solve the structure of PgDPP11 by the molecular replacement method. The structures of DAP BII complexed with or without a peptide32, having approximately 29% sequence identity with PgDPP11, were used as a search model. However, these attempts failed. Subsequently, we prepared Se-Met substituted PgDPP11. Initial phases were determined for the peptide-free Se-Met PgDPP11 using the multi-wavelength anomalous diffraction (MAD) method. Se-Met-substituted PgDPP11 was crystallised in the space group P212121. Se-MAD phases were determined using the program SHARP/autoSHARP51. Automatic model building and refinement were performed using the programs ARP/wARP52 and REFMAC553 and further iterative manual model building and refinement were performed using the programs Coot54 and REFMAC5. The Se-Met-substituted PgDPP11 was refined at 2.5 Å resolution and the resulting model was used as the starting model for the structural refinement of wild-type PgDPP11. The refinement statistics of PgDPP11 are summarised in Table S2. Least-squares comparisons of the molecular models were conducted using the program UCSF Chimera55. Multiple sequence alignment of S46 peptidases was performed using the program ClustalW56. Figures were produced using UCSF Chimera.
Docking study of PgDPP11
A starting template for the closed conformation of PgDPP11 was constructed by superimposing the crystal structure of the catalytic and α-helical domains of peptide-free PgDPP11 obtained by the present study onto that of the catalytic and α-helical domains, respectively, of the crystal structure of the Val-Tyr bound DAP BII32. Then, the closed conformation model of PgDPP11 was further optimised with the program MODELLER57 by using the starting template described above and the amino acid sequence of PgDPP11. Docking studies were performed using the Schrodinger modelling package. The standard protein preparation protocol was used involving (1) addition of missing hydrogen atoms to the starting template, (2) assignment of an ionisable state to each charged groups and (3) energy minimisation to optimise all hydrogen-bonding networks. The docking grid of 20 × 20 × 20 Å3 was generated based on the observed docking site of dipeptide in DAP BII. The Val-Asp dipeptide molecule was docked into the active site of PgDPP11. The docking reliability was supported by the fact that this protocol successfully re-docked the Val-Tyr dipeptide into the active site of DAP BII as observed in the crystal structure.
Enzymatic activity assay of wild-type and mutant PgDPP11s
Arg337 and Arg673 of PgDPP11, which were initially assumed to be involved in the recognition of the P1 residue of the substrate peptide, were individually mutated to Ala, Gly, or Asn. The mutants were expressed using the pET system and purified using the method described above for wild-type PgDPP11. The enzymatic activities of the wild-type and mutant enzymes were measured based on the hydrolysis of synthetic substrates; Gly-Phe-p-nitroanilide (Gly-Phe-pNA), Ala-Ala-pNA, Gly-Glu-pNA and Gly-Asp-pNA. The reaction mixture, composed of 100 μl 3 mM synthetic substrate, 100 μl appropriately diluted enzyme solution, 500 μl 100 mM sodium phosphate buffer (pH 7.0) containing 10 mM EDTA and 300 μl water, was incubated at 310 K for 20 min. The reaction was stopped by the addition of 50 μl of 100% trichloroacetic acid and centrifuged at 20,000 × g for 5 min; the extent of hydrolysis was measured by detecting the absorbance at 385 nm. The activity of the incubation mixtures was measured at a final substrate concentration of 0.3 mM and expressed as the mean of three different experiments. The relative activities of the mutant PgDPP11s are summarised in Table 1. kcat and Km values could not be obtained due to the low activity of the mutant enzymes.
Enzymatic activity assay of a mutant DAP BII
Gly675 of DAP BII, which is located at the S1 subsite and structurally equivalent to Arg673 in PgDPP11, was mutated to Arg (G675R). The mutant DAP BII was expressed and purified as described for wild type DAP BII32. The enzymatic activities of the wild-type and mutant DAP BIIs were measured as described for PgDPP11 above, except for the reaction buffer: 500 μl 100 mM Tris-HCl buffer (pH 8.0).
Crystallographic analysis of a mutant DAP BII
Crystallisation of G675R mutant DAP BII in the presence of a Leu-Glu dipeptide was performed as described for wild type DAP BII32. Briefly, the samples were crystallised using the hanging-drop method, in which 1 μl of peptide complex solution (10 mg/ml DAP BII and 3 mM Leu-Glu dipeptide in 80 mM Tris-HCl, pH 8.5) was mixed with the same volume of reservoir solution (18% (w/v) PEG 8000, 20% (v/v) glycerol, 2 mM zinc chloride and 160 mM magnesium acetate) and incubated at 293 K. The drops were suspended over 500 μl of reservoir solution in 24-well plates. X-ray data collection was performed at the Photon Factory. Phase determination was performed by the molecular replacement method using the dipeptide (Val-Tyr) complex of wild-type DAP BII as the starting model.
Additional Information
How to cite this article: Sakamoto, Y. et al. Structural and mutational analyses of dipeptidyl peptidase 11 from Porphyromonas gingivalis reveal the molecular basis for strict substrate specificity. Sci. Rep. 5, 11151; doi: 10.1038/srep11151 (2015).
Accession codes: Atomic coordinates for the reported structures have been deposited with the Protein Data Bank under accession codes 4Y01 (Se-Met PgDPP11, P212121), 4Y04 (WT PgDPP11, space-grown crystal, C2221), 4Y02 (WT PgDPP11, ground-grown crystal, C2221), 4XZY (WT PgDPP11, P212121) and 4Y06 (G675R mutant DAP BII, Leu-Glu complex).
References
Bostanci, N. & Belibasakis, G. N. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS. Microbiol. Lett. 333, 1–9 (2012).
Vos, T. et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2163–2196 (2012).
Humphrey, L. L., Fu, R., Buckley, D. I., Freeman, M. & Helfand, M. Periodontal disease and coronary heart disease incidence: a systematic review and meta-analysis. J. Gen. Intern. Med. 23, 2079–2086 (2008).
Dasanayake, A. P., Gennaro, S., Hendricks-Munoz, K. D. & Chhun, N. Maternal periodontal disease, pregnancy and neonatal outcomes. MCN. Am. J. Matern. Child. Nurs. 33, 45–49 (2008).
Kamer, A. R. et al. Inflammation and Alzheimer’s disease: possible role of periodontal diseases. Alzheimer’s Dement. 4, 242–250 (2008).
Hujoel, P. P., Drangsholt, M., Spiekerman, C. & Weiss, N. S. An exploration of the periodontitis-cancer association. Ann. Epidemiol. 13, 312–316 (2003).
Renvert, S. Destructive periodontal disease in relation to diabetes mellitus, cardiovascular diseases, osteoporosis and respiratory diseases. Oral. Health. Prev. Dent. 1. Suppl. 1, 341–357; discussison 358 (2003).
Detert, J., Pischon, N., Burmester, G. R. & Buttgereit, F. The association between rheumatoid arthritis and periodontal disease. Arthritis Res. Ther. 12, 218 (2010).
Holt, S. C., Ebersole, J., Felton, J., Brunsvold, M. & Kornman, K. S. Implantation of Bacteroides gingivalis in nonhuman primates initiates progression of periodontitis. Science 239, 55–57 (1988).
Shah, H. & Williams, D. Catabolism of aspartate and asparagine by Bacteroides intermedius and Bacteroides gingivalis. Curr. Microbiol. 15, 313–318 (1987).
Chen, Z., Potempa, J., Polanowski, A., Wikstrom, M. & Travis, J. Purification and characterization of a 50-kDa cysteine proteinase (gingipain) from Porphyromonas gingivalis. J. Biol. Chem. 267, 18896–18901 (1992).
Okamoto, K. et al. Structural characterization of argingipain, a novel arginine-specific cysteine proteinase as a major periodontal pathogenic factor from Porphyromonas gingivalis. Arch. Biochem. Biophys. 316, 917–925 (1995).
Nakayama, K., Kadowaki, T., Okamoto, K. & Yamamoto, K. Construction and characterization of arginine-specific cysteine protease (Arg-gingipain)-deficient mutants of Porphyromonas gingivalis. Evidence for significant contribution of Arg-gingipain to virulence. J. Biol. Chem. 270, 23619–23626 (1995).
Lamont, R. J. & Jenkinson, H. F. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62, 1244–1263 (1998).
Shi, Y. et al. Genetic analyses of protelolysis, hemoglobin binding and hemagglutination of Porphyromonas gingivalis. Construction of mutants with a combination of rgpA, rgpB, kgp and hagA. J. Biol. Chem. 274, 17955–17960 (1999).
Kadowaki, T. et al. Suppression of pathogenicity of Porphyromonas gingivalis by newly developed gingipain inhibitors. Mol. Pharmacol. 66, 1599–1606 (2004).
Mayrand, D. & Grenier, D. Detection of collagenase activity in oral bacteria. Can. J. Microbiol. 31, 134–138 (1985).
Grenier, D. & McBride, B. C. Isolation of a membrane-associated Bacteroides gingivalis glycylprolyl protease. Infect Immun. 55, 3131–3136 (1987).
Banbula, A. et al. Prolyl tripeptidyl peptidase from Porphyromonas gingivalis. A novel enzyme with possible pathological implications for the development of periodontitis. J. Biol. Chem. 274, 9246–9252 (1999).
Takahashi, N., Sato, T. & Yamada, T. Metabolic pathways for cytotoxic end product formation from glutamate- and aspartate-containing peptides by Porphyromonas gingivalis. J. Bacteriol. 182, 4704–4710 (2000).
Takahashi, N. & Sato, T. Preferential utilization of dipeptides by Porphyromonas gingivalis. J. Dent. Res. 80, 1425–1429 (2001).
Banbula, A. et al. Porphyromonas gingivalis DPP-7 represents a novel type of dipeptidylpeptidase. J. Biol. Chem. 276, 6299–6305 (2001).
Ohara-Nemoto, Y. et al. Asp- and Glu-specific novel dipeptidyl peptidase 11 of Porphyromonas gingivalis ensures utilization of proteinaceous energy sources. J. Biol. Chem. 286, 38115–38127 (2011).
Ohara-Nemoto, Y. et al. Identification and characterization of prokaryotic dipeptidyl-peptidase 5 from Porphyromonas gingivalis. J. Biol. Chem. 289, 5436–5448 (2014).
Rawlings, N. D., Barrett, A. J. & Bateman, A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40, D343–350 (2012).
Suzuki, Y. et al. Identification of the catalytic triad of family S46 exopeptidases, closely related to clan PA endopeptidases. Sci. Rep. 4, 4292 (2014).
Rouf, S. M. et al. Discrimination based on Gly and Arg/Ser at position 673 between dipeptidyl-peptidase (DPP) 7 and DPP11, widely distributed DPPs in pathogenic and environmental gram-negative bacteria. Biochimie 95, 824–832 (2013).
Rouf, S. M. et al. Phenylalanine 664 of dipeptidyl peptidase (DPP) 7 and Phenylalanine 671 of DPP11 mediate preference for P2-position hydrophobic residues of a substrate. FEBS Open Biol. 3, 177–183 (2013).
Kraut, J. Serine proteases: structure and mechanism of catalysis. Annu. Rev. Biochem. 46, 331–358 (1977).
Polgar, L. The catalytic triad of serine peptidases. Cell Mol. Life Sci. 62, 2161–2172 (2005).
Sakamoto, Y. et al. Crystallization and preliminary X-ray crystallographic studies of dipeptidyl aminopeptidase BII from Pseudoxanthomonas mexicana WO24. Acta Crystallogr. F Struct. Biol. Commun. 70, 221–224 (2014).
Sakamoto, Y. et al. S46 peptidases are the first exopeptidases to be members of clan PA. Sci. Rep. 4, 4977 (2014).
Matthews, B. W., Sigler, P. B., Henderson, R. & Blow, D. M. Three-dimensional structure of tosyl-alpha-chymotrypsin. Nature 214, 652–656 (1967).
Bode, W. & Schwager, P. The refined crystal structure of bovine beta-trypsin at 1.8 Å resolution. II. Crystallographic refinement, calcium binding site, benzamidine binding site and active site at pH 7.0. J. Mol. Biol. 98, 693–717 (1975).
Ogasawara, W., Kobayashi, G., Okada, H. & Morikawa, Y. Two types of novel dipeptidyl aminopeptidases from Pseudomonas sp. strain WO24. J. Bacteriol. 178, 6288–6295 (1996).
Holm, L. & Rosenstrom, P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 (2010).
Henderson, R. Structure of crystalline alpha-chymotrypsin: IV. The structure of indoleacryloyl-alpha-chymotrypsin and its relevance to the hydrolytic mechanism of the enzyme. J. Mol. Biol. 54, 341–354 (1970).
Robertus, J. D., Kraut, J., Alden, R. A. & Birktoft, J. J. Subtilisin; a stereochemical mechanism involving transition-state stabilization. Biochemistry 11, 4293–4303 (1972).
Brooke, J. S. Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin. Microbiol. Rev. 25, 2–41 (2012).
Lambais, M. R., Goldman, M. H., Camargo, L. E. & Goldman, G. H. A genomic approach to the understanding of Xylella fastidiosa pathogenicity. Curr. Opin. Microbiol. 3, 459–462 (2000).
Rasmussen, H. B., Branner, S., Wiberg, F. C. & Wagtmann, N. Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog. Nat. Struct. Biol. 10, 19–25 (2003).
Umezawa, H. et al. Diprotins A and B, inhibitors of dipeptidyl peptidase IV, produced by bacteria. J. Antibiot. 37, 422–425 (1984).
Turk, D. et al. Structure of human dipeptidyl peptidase I (cathepsin C): exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 20, 6570–6582 (2001).
Molgaard, A. et al. The crystal structure of human dipeptidyl peptidase I (cathepsin C) in complex with the inhibitor Gly-Phe-CHN2. Biochem. J. 401, 645–650 (2007).
Huggins, D. J., Sherman, W. & Tidor, B. Rational approaches to improving selectivity in drug design. J. Med. Chem. 55, 1424–1444 (2012).
Varghese, J. N., Epa, V. C. & Colman, P. M. Three-dimensional structure of the complex of 4-guanidino-Neu5Ac2en and influenza virus neuraminidase. Protein. Sci. 4, 1081–1087 (1995).
Takahashi, S. et al. JAXA protein crystallization in space: ongoing improvements for growing high-quality crystals. J. Synchrotron Radiat. 20, 968–973 (2013).
Sakamoto, Y. et al. Crystallization and preliminary X-ray crystallographic studies of dipeptidyl peptidase 11 from Porphyromonas gingivalis. Acta Crystallogr. F Struct. Biol. Commun. 71, 206–210 (2015).
Garcia-Ruiz, J. M. & Morena, A. Investigations on protein crystal growth by the gel acupuncture method. Acta Crystallogr. D Biol. Crystallogr. 50, 484–490 (1994).
Kabsch, W. X D S Acta Crystallogr . D Biol. Crystallogr. 66, 125–132 (2010).
Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M. & Paciorek, W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr. D Biol. Crystallogr. 59, 2023–2030 (2003).
Morris, R. J., Perrakis, A. & Lamzin, V. S. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 374, 229–244 (2003).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Pettersen, E. F. et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Sali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993).
Acknowledgements
We thank Drs. Y. Yamada and N. Matsugaki of the Photon Factory and Dr. A. Higashiura, K. Hasegawa and E. Yamashita of SPring-8 for their help with data collection at the synchrotron facilities. This study was supported in part by the Platform for Drug Discovery, Informatics and Structural Life Science (to N.T.), the Program for the Strategic Research Foundation at Private Universities from the MEXT of Japan (to N.T. and Y.Sa) and a Grant-in-Aid for Scientific Research(C) (to Y.Sa.). This study was also supported in part by “High-Quality Protein Crystal Growth Experiment on JEM” promoted by JAXA (Japan Aerospace Exploration Agency) (to Y.Sa.). The Russian Spacecraft “Progress” and “Soyuz” provided by the Russian Federal Space Agency were used for space transportation. Some of the space crystallisation technology was developed by ESA (the European Space Agency) and the University of Granada.
Author information
Authors and Affiliations
Contributions
Y.Sa. crystallised the protein, collected X-ray data, analysed the data and wrote the paper; Y.Su. cloned the construct, performed biochemical analyses and wrote the paper; I.I., C.T., S.R. and M.F. crystallised the protein, collected X-ray data and analysed the data; K.I., H.T., M.Y. and K.O. supported the crystallisation of the protein under microgravity; H.G., T.N. designed the study and analysed the data; W.O. and N.T. designed the study, analysed the data and wrote the paper. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Sakamoto, Y., Suzuki, Y., Iizuka, I. et al. Structural and mutational analyses of dipeptidyl peptidase 11 from Porphyromonas gingivalis reveal the molecular basis for strict substrate specificity. Sci Rep 5, 11151 (2015). https://doi.org/10.1038/srep11151
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11151
This article is cited by
-
Immunoelectron Microscopic Analysis of Dipeptidyl-Peptidases and Dipeptide Transporter Involved in Nutrient Acquisition in Porphyromonas gingivalis
Current Microbiology (2023)
-
Structural basis for an exceptionally strong preference for asparagine residue at the S2 subsite of Stenotrophomonas maltophilia dipeptidyl peptidase 7
Scientific Reports (2021)
-
Fragment-based discovery of the first nonpeptidyl inhibitor of an S46 family peptidase
Scientific Reports (2019)
-
Bacterial protease uses distinct thermodynamic signatures for substrate recognition
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
