
Abstract
Inhomogeneity in the ground state is an intriguing, emergent phenomenon in magnetism. Recently, it has been observed in the magnetostructural channel of the geometrically frustrated α-NaMnO2, for the first time in the absence of active charge degrees of freedom. Here we report an in-depth numerical and local-probe experimental study of the isostructural sister compound CuMnO2 that emphasizes and provides an explanation for the crucial differences between the two systems. The experimentally verified, much more homogeneous, ground state of the stoichiometric CuMnO2 is attributed to the reduced magnetoelastic competition between the counteracting magnetic-exchange and elastic-energy contributions. The comparison of the two systems additionally highlights the role of disorder and allows the understanding of the puzzling phenomenon of phase separation in uniform antiferromagnets.
Similar content being viewed by others


Introduction
Although phase separation in a uniform system is a widespread phenomenon in diverse fields of matter1,2,3, ranging from biological systems4,5,6, to soft matter7,8 and strongly correlated electron systems9,10,11,12,13,14,15, in magnetism the microscopic pattering has been, until recently, almost exclusively limited to thin ferromagnetic (FM) films16,17. In this case, such a pattering is a trade-off between minimizing the exchange and the dipolar energies. It thus represents one possible manifestation of a general requirement of multiple competing phases that can lead to inhomogeneous states. Lately, it has become increasingly apparent that a similar competition between energetically nearly equivalent phases is also responsible for phase separation in geometrically frustrated spin systems18,19,20,21 that are generically characterized by ground-state degeneracy22. However, the balance between the competing phases in these systems is generally much more delicate and, therefore, poorly understood.
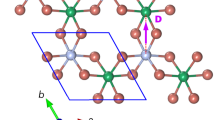
Recently, the spatially anisotropic triangular antiferromagnet α-NaMnO2, with dominant intrachain (J1) and geometrically frustrated interchain (J2) antiferromagnetic (AFM) exchange interactions (inset in Figure 1a), has been highlighted as a paradigm of a phase-separated ground state in the absence of active charge degrees of freedom18. Its AFM order that sets in below the Néel temperature TN = 45 K is accompanied by a simultaneous structural deformation23. This was initially suggested as being a phase transition from the high-temperature monoclinic (C2/m) to the low-temperature triclinic ( ) crystal structure23. However, more detailed, recent experiments have shown that the magnetic order fails to drive this improper ferroelastic transition to completion18. Instead, an intricate magnetostructurally inhomogeneous state on the nano-scale has been discovered below the TN. Such a state was suggested to be an unforeseen consequence of the subtle interplay between the geometrical frustration and the competing structural phases18.
) crystal structure23. However, more detailed, recent experiments have shown that the magnetic order fails to drive this improper ferroelastic transition to completion18. Instead, an intricate magnetostructurally inhomogeneous state on the nano-scale has been discovered below the TN. Such a state was suggested to be an unforeseen consequence of the subtle interplay between the geometrical frustration and the competing structural phases18.

Determination of J's and TN.
(a) The magnetic susceptibility χ = M/H (M is the magnetization and H is the applied magnetic field) of CuMnO2, measured by a SQUID magnetometer in a field of μ0H = 0.1 T. The solid and dashed lines denote the best FTLM fits with the average and the scaled curves, respectively (see Methods for details). The former yields the exchange-coupling constants J1 = 53.5 K, J2/J1 = 0.25 and the latter J1 = 52.1 K, J2/J1 = 0.29. Inset shows the spatially anisotropic triangular spin lattice of the CuMnO2 in the monoclinic setting, with intrachain J1 (thick bonds) and interchain J2 (thin bonds) exchange constants. (b) The temperature dependence of the staggered susceptibility χs multiplied by J1 for spin-2 chains (adopted from Ref. 34). The solid line is a guide to the eye. The Néel transition temperatures TN = 1.40J1 = 74 K in CuMnO2 and TN = 0.87J1 = 57 K in α-NaMnO2 are predicted (dashed lines) by equation (2).
In order to fully understand this novel phenomenon, further theoretical studies and experimental investigations of related compounds are of paramount importance. In this respect, a comparison with the crystallographically24 and magnetically25 analogous sister compound CuMnO2, known as the mineral crednerite, is particularly relevant. Here, in contrast to α-NaMnO2, the emergent magnetic order below TN = 65 K is believed to lift the macroscopic degeneracy in the spin space completely, by inducing the monoclinic-to-triclinic structural phase transition26. This spin-induced phase transition is witnessed by the splitting of several families of nuclear Bragg reflections18,27. It was suggested to reflect the strong magnetoelastic (ME) coupling that allows for the development of shear strain at a low energy cost27. Interestingly, the strain is significantly enhanced28 in the off-stoichiometric29 Cu1+xMn1−xO2, where TN is reduced and the structural transition temperature is further suppressed with increasing x even for small doping levels28,30,31. Moreover, in off-stoichiometric samples the interlayer ordering changes from AFM, observed in stoichiometric CuMnO2, to FM28,30,31, which was attributed to a partial substitution of Cu2+ for Mn3+ that should effectively change the interlayer exchange coupling from AFM to FM32. On the other hand, this implies that CuMnO2 may be very close to an electronic instability, possibly of a similar kind to that found in stoichiometric α-NaMnO2.
Obtaining in-depth information about the magnetic and structural properties of CuMnO2 on the local scale should clarify the differences with respect to the isostructural α-NaMnO2. Such knowledge would also help to address the pending issue of the microscopic origin of the phase-separation phenomenon in geometrically frustrated magnets. The most obvious ambiguities arise from questions like: why is the TN enhanced in CuMnO2 compared to α-NaMnO2, despite the theoretically predicted sizably smaller exchange interactions in the former compound33; what is the role of the ME coupling in establishing the structural distortion below the TN, what is the role of disorder; and ultimately, why does the structural phase transition appear to be fully developed in CuMnO2, while in α-NaMnO2 it only manifests in a phase-separated state. Here, we answer these questions by combining numerical calculations with local-probe experimental investigations. First, we determine the dominant intralayer exchange interactions by modelling the magnetic susceptibility via exact-diagonalization calculations. We demonstrate that the difference in the TN for the two compounds can be understood via the mismatch of the two non-equivalent interchain exchange interactions, i.e., by the different extent of the frustration present in the two compounds, while the ME contribution is negligible in this respect. Moreover, we provide the first experimental microscopic insight into the magnetism of CuMnO2 via 63,65Cu nuclear magnetic resonance (NMR) and nuclear quadrupolar resonance (NQR) measurements, as well as complementary muon spin relaxation (μSR) measurements. These experiments clearly reveal that the ground state is more homogeneous in the Cu case than in the Na case and suggest that the stoichiometric CuMnO2 is on the verge of a phase-separation instability. Finally, our ab-initio calculations suggest that the more homogeneous state of the stoichiometric CuMnO2 originates from an enhanced energy difference (when compared to α-NaMnO2) between the two competing phases, born out of the magnetic-exchange and the elastic-energy changes below the TN.
Results
Determination of the dominant exchange interactions and the TN
In order to understand the apparently significantly different properties of the two isostructural compounds, CuMnO2 and α-NaMnO2, a proper determination of the dominant terms in the Hamiltonian is crucial. Therefore, we applied numerical finite-temperature Lanczos method (FTLM) simulations and density-functional theory (DFT) calculations. The former were aimed at quantifying the two main magnetic exchange interactions (J1 and J2) of the isotropic Heisenberg model on the spatially anisotropic two-dimensional (2D) triangular lattice (inset in Figure 1a),

Here, the first sum runs over the (stronger) intrachain bonds in one direction, while the second sum runs over the (weaker) interchain bonds in the other two directions on the triangular lattice of spins S = 2.
The temperature-dependent magnetic susceptibility χ(T) of this model was calculated for various m × n spin clusters (see Methods). However, even for the largest reachable cluster sizes (7 × 2), some finite-size effects remain present at low T. With the use of results from many clusters (m = 2–7) these effects can, however, be reduced and the value of χ at the thermodynamic limit is thus approached. Both, the average susceptibility curve from the two largest-size clusters (that gives a better approximation than the individual clusters; see Methods) and the susceptibility curve obtained in an approach similar to finite-size scaling (see Methods) fit the experimental data above the TN very well (Figure 1a). A disagreement with the experimental data below the TN, on the other hand, is expected, because the 2D Heisenberg model cannot account for a finite TN. Both approaches yield very similar exchange-coupling constants, which we estimate to be J1 = 53 K and J2/J1 = 0.27(2). These are in very good agreement with recent ab-initio predictions33, J1 = 56 K and  , which, in principle, could be erroneous due to the unknown on-site repulsion32.
, which, in principle, could be erroneous due to the unknown on-site repulsion32.
Our calculations thus confirm that the exchange interactions are indeed reduced for CuMnO2 compared to α-NaMnO2, where35 J1 = 65 K and J2/J1 = 0.44. Despite this fact, in CuMnO2 the TN is increased with respect to that found in α-NaMnO2 by more than 40%. So far, this has been attributed to a difference in the interlayer coupling26 J′, which, however, is rather small32,33 and can, therefore, only slightly affect the TN on 2D Heisenberg lattices36. Furthermore, the amount of frustration reflected in the J2/J1 ratio should directly influence the TN, as the frustration is known to suppress the spin correlations37. In α-NaMnO2 and CuMnO2 the intrachain exchange coupling is dominant. Therefore, these compounds can be regarded as systems of coupled spin chains, which is manifested in the one-dimensional character of the magnetic excitations in α-NaMnO238. For such systems the TN can be determined with the use of a random-phase approximation39,40. In this approach the interchain coupling is treated at the mean-field level, whereas the intrachain interactions are treated exactly. For isotropic interchain coupling in a non-frustrated lattice the TN is determined by the condition zJ2χs(TN) = 1 (Ref. 36,39,40,41), where χs(T) is the chain's staggered susceptibility. Within this approach, we generalize the above condition for TN to include the two interchain constants (J2a > J2b) pertinent to the triclinic phase of CuMnO2 and α-NaMnO2, as well as the interlayer coupling J′;

Here, z = z′ = 2 corresponds to the number of neighbouring coupled chains and planes, respectively while J2b adopts the minus sign because it frustrates the AFM order dictated by the larger J2a. The constant k renormalizes the coordination numbers and is reduced from unity41,42 because of quantum effects36. As (J2a − J2b)/J1 = 0.083 and 0.035 in CuMnO2 and α-NaMnO2, respectively33, while J′/J1 is expected to be an order of magnitude smaller32,33, we estimate the TN by neglecting J′ in equation (2) and by taking k = 0.7, which is appropriate for quasi-one dimensional cases (see Fig. 2 in Ref. 36). This gives TN = 74 K in CuMnO2 and TN = 57 K in α-NaMnO2 (see Figure 1b), which are in good agreement with the experimental values of 65 K and 45 K, respectively. We note that for anisotropic interchain couplings the constant k is expected to be further reduced and, ultimately, for J′ → 0 also k → 0 (Ref. 41,42), leading to TN → 0, which is consistent with the Mermin-Wagner theorem (no long-range order in the 2D Heisenberg model at finite T). However, it has been shown43 that for quasi-2D systems the dependence of the TN and, in turn also of k, on J′ is sub-logarithmic. Therefore, for the exchange-coupling constants related to CuMnO2 and α-NaMnO2, k will be somewhat, but not drastically, reduced from the value 0.7, which is perfectly in line with the small theoretical overestimates of the TN.

DFT calculations.
(a) The calculated difference in the total-energy density for the three different magnetoelastic components. The solid lines are linear fits for the xx and yy components and a quadratic fit for the xy component. (b) The ab-initio calculated total energy of the relaxed monoclinic (m) and triclinic (t) structures of the CuMnO2 as a function of the volume for the antiferromagnetic (AFM) and non-magnetic (NM) cases. The insets zoom at the regions around the local minima of the relaxed structures. The global minimum of the energy is set to zero and the corresponding volume of the triclinic structure V0 is used for volume normalization.
This analysis reveals that the Néel transition is predominantly determined by the Heisenberg Hamiltonian of equation (1). Moreover, the ferrodistortive structural transition accompanying the magnetic ordering and leading to the splitting of the two interchain exchange constants (J2a, J2b) in the triclinic phase is needed to ensure a finite TN. The extent to which frustration is relieved in the triclinic phase of the CuMnO2 elevates its ordering temperature above the ordering temperature in α-NaMnO2. Other factors, such as the interlayer coupling, the magnetic anisotropy and the ME coupling, can, at best, only slightly shift the TN.
Total-energy change at the TN
Having established that the magnetic ordering at the TN is predominantly set by the 2D Heisenberg Hamiltonian and the tendency of both systems to remove magnetic degeneracy in the ground state by lattice deformation, the question that arises is what is the microscopic origin of such a complex transformation. In this respect, the ME coupling has been suggested as being the key factor23,26,27 in both α-NaMnO2 and CuMnO2. However, the ME coupling has been shown to be insubstantial in the former case18 and thus needs to be evaluated also in the CuMnO2. The total-energy change at the TN, associated solely with the magnetoelasticity, arises from the coupling terms44  between the strain-tensor components
between the strain-tensor components  and the magnetization-direction vector m = (mx, my, mz). The strength of the ME coupling and consequently the corresponding contribution to the total energy, is proportional to the ME-coupling coefficients bij (i, j = x, y, z). The coefficients bxx = 2.3 MJ/m3, byy = 1.6 MJ/m3 and bxy = 3.4 MJ/m3 are determined as linear terms in the calculated dependence of the total-energy-density change Δfij on the strain (see Methods), which is shown in Figure 2a. The ME energy gain is the largest for the shear-strain component
and the magnetization-direction vector m = (mx, my, mz). The strength of the ME coupling and consequently the corresponding contribution to the total energy, is proportional to the ME-coupling coefficients bij (i, j = x, y, z). The coefficients bxx = 2.3 MJ/m3, byy = 1.6 MJ/m3 and bxy = 3.4 MJ/m3 are determined as linear terms in the calculated dependence of the total-energy-density change Δfij on the strain (see Methods), which is shown in Figure 2a. The ME energy gain is the largest for the shear-strain component  , which is associated with the monoclinic-to-triclinic deformation. However, for the experimental strain
, which is associated with the monoclinic-to-triclinic deformation. However, for the experimental strain  (see Methods) the magnetoelastic energy change amounts to only 2.7 μeV per triclinic unit cell, which is very similar to the value of 2.5 μeV found in α-NaMnO2.
(see Methods) the magnetoelastic energy change amounts to only 2.7 μeV per triclinic unit cell, which is very similar to the value of 2.5 μeV found in α-NaMnO2.
The contribution of the ME coupling to the total energy change at the TN is thus negligible in both compounds. Therefore, the complex phase transition at the TN has to reflect changes in the magnetic-exchange and elastic energies18. Our ab-initio calculations of both relevant contributions to the total energy in the CuMnO2 for the non-spin-polarized case, relevant to non-magnetically ordered structures, reveal that the monoclinic structure is energetically lower than the triclinic one, although only by ~1 meV per 4 formula units (f.u.); see Figure 2b. This is in-line with the C2/m crystal symmetry found experimentally at room temperature. However, once the magnetic order sets in, the total energy of the triclinic structure is lowered below that of the monoclinic structure by about 4 meV per 4 f.u. This change of ~1.25 meV per Mn3+ ion is mainly a consequence of the exchange-energy decrease during the structural phase transition, associated with the removal of the degenerate magnetic states due to the interchain frustration. The resulting splitting of the interchain exchange constants by J2a − J2b = 0.4 meV (Ref. 33) releases S2(J2a − J2b) = 1.6 meV of energy per Mn that is slightly larger than the total-energy change at the TN, as it is partially spent to compensate for the elastic-energy increase in the triclinic phase.
The calculated total-energy difference below the TN of 1 meV per f.u. in CuMnO2 between the two structures that is about 3-times above the calculation error bar (see Methods), is markedly larger than in the α-NaMnO2, where the calculated difference was below the calculation error bar;  per f.u.45. However, in absolute terms this difference is small, even in the CuMnO2, so that a competition between the near-degenerate monoclinic and triclinic structures is expected for both compounds. Experimental local-probe magnetic techniques are then essential for highlighting possible differences between the two systems.
per f.u.45. However, in absolute terms this difference is small, even in the CuMnO2, so that a competition between the near-degenerate monoclinic and triclinic structures is expected for both compounds. Experimental local-probe magnetic techniques are then essential for highlighting possible differences between the two systems.
NMR/NQR insight to the magnetism
Information about the magnetic properties of CuMnO2 on the local scale are revealed in the NQR/NMR experiments via the hyperfine (hf) coupling Ahf of the electronic and the 63,65Cu nuclear magnetic moments. Moreover, the quadrupolar splitting in the electric-field gradient (EFG) provides information about the material's structural properties. The NQR spectra measured in zero field correspond to a single line for each copper isotope46, while the powder NMR spectra are structured (see Methods for details). Our simultaneous fit of the NQR and NMR data at 80 K (Figure 3a) yields a hf coupling constant 63Ahf = 2.3(1) T/μB that is significantly larger than the coupling constant 23Ahf = 0.11(1) T/μB found18 in α-NaMnO2. Since Ahf scales with the orbital overlap, the charge transfer from the Mn3+ ions to the interlayer cations (Cu+ or Na+) is much larger in the CuMnO2 than in the α-NaMnO2. This implies a stronger J′ in the former compound and thus is in line with the somewhat better agreement between the experimental and the predicted TN, as the renormalization factor k in equation (2) is closer to the used value of 0.7.

NMR results.
(a) 80-K 63,65Cu NQR and NMR (inset) spectra of CuMnO2. The solid lines represent a simultaneous NMR/NQR fit (see Methods for details), assuming a Gaussian distribution of the NQR frequencies  , that yields the quadrupolar frequency 63νQ = 27.0(1) MHz, the asymmetry parameter η = 0.20(5), the isotropic hf shift Khf = 1.6(1)% that is much larger than the dipolar shift Kd = 0.11% and the individual line widths 63δ = 0.17(1) MHz, 65δ = 0.14(1) MHz. The dashed lines show the center of the NQR lines and the reference NMR frequencies corresponding to a zero magnetic shift. The 63Cu NQR spectrum at 4.6 K is added for comparison. (b) The temperature dependence of the 63Cu NQR line width 63δ and the line position 63νc. The inset highlights the hf paths through the O2− sites that provide the coupling of each Cu nuclei with six surrounding Mn3+ magnetic moments (arrows), ordered with the magnetic wave vector27
, that yields the quadrupolar frequency 63νQ = 27.0(1) MHz, the asymmetry parameter η = 0.20(5), the isotropic hf shift Khf = 1.6(1)% that is much larger than the dipolar shift Kd = 0.11% and the individual line widths 63δ = 0.17(1) MHz, 65δ = 0.14(1) MHz. The dashed lines show the center of the NQR lines and the reference NMR frequencies corresponding to a zero magnetic shift. The 63Cu NQR spectrum at 4.6 K is added for comparison. (b) The temperature dependence of the 63Cu NQR line width 63δ and the line position 63νc. The inset highlights the hf paths through the O2− sites that provide the coupling of each Cu nuclei with six surrounding Mn3+ magnetic moments (arrows), ordered with the magnetic wave vector27  . (c) Comparison of the temperature-dependent 63Cu NQR/NMR spin-lattice relaxation rate 1/T1 in the CuMnO2 and 23Na NMR 1/T1 in the α-NaMnO2 (Ref. 18). The latter is normalized by the squared ratio of the hf coupling constants. The error bars represent the standard deviation of the fit parameters.
. (c) Comparison of the temperature-dependent 63Cu NQR/NMR spin-lattice relaxation rate 1/T1 in the CuMnO2 and 23Na NMR 1/T1 in the α-NaMnO2 (Ref. 18). The latter is normalized by the squared ratio of the hf coupling constants. The error bars represent the standard deviation of the fit parameters.
The width δ of the NQR spectra at 80 K, amounting to 0.16 MHz/27.2 MHz = 0.6% of the line-position value νc already in the paramagnetic phase (Figure 3b), is rather large. The line widths 63δ > 65δ reveal spectral broadening being in accordance with the quadrupole moments 63Q > 65Q and contradicting the gyromagnetic ratios 63γ < 65γ. Therefore, sizeable structural distortions of the local environments must be present. The temperature dependence of both νc and δ shows a pronounced sudden increase below the TN (Figure 3b), clearly marking the phase transition. The anomaly in νc at the TN is attributed to the structural transformation of the CuMnO2 sample, directly affecting the quadrupolar frequency νQ. Namely, static internal magnetic fields below the TN cause a symmetric broadening/splitting of the NQR line so that its center of gravity is unaffected46. On the other hand, the pronounced increase of δ by a factor of ~2 at the TN, exceeding the change of νc by several orders of magnitude, can only be magnetic in origin.
We must emphasize that the existence of the NQR signal below the TN is unexpected. Namely, in the frame of the homogeneously ordered magnetic phase27 with  the Cu nuclei would experience extremely large internal magnetic fields. Although the Cu site is a structural center of inversion, the magnetic order breaks this symmetry, as the spins at ±r from a given Cu site are FM ordered (see the inset in Figure 3b), in contrast to the α-NaMnO2, where the order of the two corresponding spins is AFM18. Such a spin configuration in CuMnO2 yields a large local hf field
the Cu nuclei would experience extremely large internal magnetic fields. Although the Cu site is a structural center of inversion, the magnetic order breaks this symmetry, as the spins at ±r from a given Cu site are FM ordered (see the inset in Figure 3b), in contrast to the α-NaMnO2, where the order of the two corresponding spins is AFM18. Such a spin configuration in CuMnO2 yields a large local hf field  (μ = 3.05 μB is the size of the ordered26 Mn3+ moment). This field leads to extremely broad NQR spectra,
(μ = 3.05 μB is the size of the ordered26 Mn3+ moment). This field leads to extremely broad NQR spectra,  , being two orders of magnitude broader than the experimental ones. Indeed, the NQR signal below the TN corresponds to a minority fraction of all the 63Cu nuclei, while a majority of the signal is lost at the TN due to the onset of large internal fields. Namely, the Boltzman-corrected intensity of the NQR signal at 4.6 K, when further corrected for nuclear relaxation effects, is smaller than the intensity at 80 K by a factor of ~17. This reveals that, unexpectedly, about 6% of all the Cu sites in our sample experience small or no internal magnetic fields and do not correspond to the reported homogeneous magnetic phase. We note that the AFM order of the moments positioned symmetrically with respect to the Cu site, or the absence of any order, result in a zero static local magnetic field at the Cu site and would explain the NQR-observable sites below the TN. Since this minority signal exhibits clear anomalies at the TN (Figure 3b) it is obviously well coupled to the bulk that undergoes the magnetostructural transition. This is confirmed by the temperature dependence of the spin-lattice relaxation rate, 1/T1, that shows a maximum at the TN due to critical spin fluctuations47 related to the magnetic instability of the bulk. However, in contrast to the monotonic decrease found in the Na-based compound below the TN, in the CuMnO2, another clear maximum in both the NMR and NQR 1/T1 is observed at around 10 K. This reveals an, as yet, unobserved instability that could be either magnetic or structural in its nature.
, being two orders of magnitude broader than the experimental ones. Indeed, the NQR signal below the TN corresponds to a minority fraction of all the 63Cu nuclei, while a majority of the signal is lost at the TN due to the onset of large internal fields. Namely, the Boltzman-corrected intensity of the NQR signal at 4.6 K, when further corrected for nuclear relaxation effects, is smaller than the intensity at 80 K by a factor of ~17. This reveals that, unexpectedly, about 6% of all the Cu sites in our sample experience small or no internal magnetic fields and do not correspond to the reported homogeneous magnetic phase. We note that the AFM order of the moments positioned symmetrically with respect to the Cu site, or the absence of any order, result in a zero static local magnetic field at the Cu site and would explain the NQR-observable sites below the TN. Since this minority signal exhibits clear anomalies at the TN (Figure 3b) it is obviously well coupled to the bulk that undergoes the magnetostructural transition. This is confirmed by the temperature dependence of the spin-lattice relaxation rate, 1/T1, that shows a maximum at the TN due to critical spin fluctuations47 related to the magnetic instability of the bulk. However, in contrast to the monotonic decrease found in the Na-based compound below the TN, in the CuMnO2, another clear maximum in both the NMR and NQR 1/T1 is observed at around 10 K. This reveals an, as yet, unobserved instability that could be either magnetic or structural in its nature.
Probing the magnetic disorder with μSR
In order to provide more insight into the magnetic state in the CuMnO2 that is, according to the unexpected minority NQR signal, apparently not as homogeneous as inferred from previous bulk measurements, we resorted to the μSR local-probe technique. Moreover, this technique reveals details about the low-temperature anomaly in the NMR/NQR relaxation at 10 K. In contrast to NQR/NMR, which is limited because the intrinsic signal disappears below the TN, the μSR measurements can assess the magnetic properties of the entire CuMnO2 sample also below the TN. This time a hf/dipolar coupling between the electronic magnetic moments and the muon magnetic moment is utilized after a muon stops in the sample. The resulting local magnetic field Bμ at the muon site affects the μSR asymmetry A(t) that is proportional to the muon polarization precessing in Bμ.
In CuMnO2, the weak-transverse-field (wTF) experiment that effectively keeps track of the temperature-dependent ordered part of the sample by measuring the amplitude of the oscillating μSR signal48, reveals that the fraction of the muons detecting large frozen internal fields starts growing already below 80 K (Figure 4a); i.e., far above the TN, which can be attributed to developing short-range spin correlations48. Below the TN, the whole sample becomes magnetically ordered within only a few kelvins, leaving no room for a non-frozen fraction above the experimental error bar of a few percent. Similar information is obtained from the zero-field (ZF) μSR, where the initial asymmetry strongly decreases around the TN and at low temperatures reaches 1/3 of its high-temperature value (inset in Figure 4b). Such a reduction is characteristic of the establishment of strong static internal fields in powder samples. Statistically, in 1/3 of all cases the muon magnetic moment is aligned parallel to the Bμ and therefore exhibits no precession, while rapid oscillations of the asymmetry in other cases diminish the μSR signal on a coarse time scale.

μSR results.
(a) The temperature-dependent magnetically ordered volume fraction [1 − A0(T)/A0(120 K)] of the CuMnO2, derived from the wTF μSR asymmetry AwTF data (inset); solid lines are fits to the model  , where γμ = 2π × 135.5 MHz/T is the muon gyromagnetic ratio, BwTF is the transverse applied magnetic field and λT the transverse muon relaxation rate. The A0(T) term corresponds to muons experiencing no sizeable static internal magnetic field, while the C(T) term describes those muons that reside at sites with large static fields (
, where γμ = 2π × 135.5 MHz/T is the muon gyromagnetic ratio, BwTF is the transverse applied magnetic field and λT the transverse muon relaxation rate. The A0(T) term corresponds to muons experiencing no sizeable static internal magnetic field, while the C(T) term describes those muons that reside at sites with large static fields ( ). (b) The longitudinal muon relaxation rate derived from the ZF muon asymmetry AZF data (inset); solid lines are fits to the model
). (b) The longitudinal muon relaxation rate derived from the ZF muon asymmetry AZF data (inset); solid lines are fits to the model  , where the initial asymmetry
, where the initial asymmetry  is temperature dependent to account for the disappearance of the oscillating component below the TN. (c) μSR asymmetry of CuMnO2 (upper panel) and α-NaMnO2 (lower panel; adopted from Ref. 18) at 5 K. The solid and dashed lines represent the corresponding fits to the “two-component” model of equation (3) and a model with only one oscillating component, respectively. Fitting to the CuMnO2 data yields χ2 = 0.71 and 1.71 for the former and the latter models, respectively. The error bars represent the standard deviation of the fit parameters. For the muon asymmetry data the latter are set by the square root of the total number of detected positrons.
is temperature dependent to account for the disappearance of the oscillating component below the TN. (c) μSR asymmetry of CuMnO2 (upper panel) and α-NaMnO2 (lower panel; adopted from Ref. 18) at 5 K. The solid and dashed lines represent the corresponding fits to the “two-component” model of equation (3) and a model with only one oscillating component, respectively. Fitting to the CuMnO2 data yields χ2 = 0.71 and 1.71 for the former and the latter models, respectively. The error bars represent the standard deviation of the fit parameters. For the muon asymmetry data the latter are set by the square root of the total number of detected positrons.
A detailed look at the ZF relaxation curves below TN (Figure 4c) also allows for the detection of the quickly-oscillating component. Similar to the α-NaMnO2 case18, the two-component model

fits well with the experimental data. Here, fj denotes temperature-independent probabilities that the muons stop at either of the two magnetically non-equivalent stopping sites j. The preferential site is occupied in 70(5)% cases. The internal field at this site is only slightly higher than at the second site (0.59 and 0.54 T at the first and the second site, respectively, at 5 K); however, a fit with only a single oscillating component (the dashed line in Figure 4c) results in a much poorer agreement with the data. The damping rate of the oscillations λT,j that is due to the finite width of the local-field distributions18 in CuMnO2 is reduced by a factor of ~3 when compared to the α-NaMnO2 (Figure 4c), indicating more homogeneous magnetism.
In the ZF experiment, the magnetic phase transition at the TN is expressed as a maximum of the longitudinal muon-relaxation rate λL, like in the NQR/NMR relaxation experiments. Moreover, the second maximum observed in the NQR/NMR experiments at 10 K is also found in the μSR. Since the ZF μSR signal corresponds to the total volume of the sample and the muons are only sensitive to magnetism, this reveals that the low-temperature anomaly is of magnetic origin and is intrinsic to the CuMnO2 system.
Discussion
The FTLM and DFT numerical calculations provide a solid basis for addressing the experimentally observed similarities and differences between the CuMnO2 and the α-NaMnO2. Considering the latter calculations in the magnetically ordered state, the triclinic phase is energetically preferred in both compounds. With increasing temperature, the staggered susceptibility decreases and this leads to a finite TN. Above the TN the exchange-energy gain associated with the magnetically ordered state disappears, which in turn leads to a structural transformation to the monoclinic phase that is energetically preferred in the non-magnetic state. The isotropic Heisenberg Hamiltonian of the spatially anisotropic triangular lattice is dominantly responsible for elevating the TN in the CuMnO2 with respect to the α-NaMnO2, while the magnetoelastic and the interlayer couplings play a less important role.
On the other hand, our local-probe experiments on the CuMnO2 revealed some subtle, yet profound, features that should be carefully considered in the attempt to understand the presence/absence of nano-scale phase separation in the spatially anisotropic triangular lattice. Both, the NQR/NMR and the μSR investigations demonstrated that CuMnO2 undergoes a magnetostructural phase transition at TN = 65 K almost completely. The minority NQR component (~6%) that remains present below the TN can be explained by regions where the interlayer magnetic ordering is FM instead of being AFM, as the latter causes the disappearance of the NQR signal due to large local fields. The NQR signal exhibits a magnetic anomaly around 10 K, which is expressed by the increased relaxation rates of the NQR/NMR as well as the μSR. Since μSR, on the other hand, detects a bulk magnetic signal, the small NQR component is apparently coupled to the bulk magnetic phase. This is further confirmed by the line position and the width of the NQR spectra, changing considerably at the TN. The coupling with the bulk phase can then be regarded in the context of the nano-scale phase inhomogeneity. A comparison of the ZF μSR asymmetry curves of the CuMnO2 and the α-NaMnO2 is quite informative in this respect. The notably reduced damping of the oscillations in the ordered phase of the former compound provides evidence of much narrower field distributions and hence less disorder. This conclusion is also in line with the number of the interlayer cation (Cu+ and Na+) sites experiencing internal fields that do not comply with the symmetry of the bulk magnetic order, which in the CuMnO2 is decreased to 6%, from the 30% found18 in the α-NaMnO2.
The magnetostructurally inhomogeneous ground state of the α-NaMnO2 on the nano-scale has previously been attributed to the combined effects of geometrical frustration and near-degenerate monoclinic and triclinic structural phases18. We believe that the key factor controlling such an inhomogeneity is the difference in the total energy of the two competing phases in the magnetically ordered state. This difference is notably larger in the CuMnO2 (1 meV per f.u.) than in the α-NaMnO2, where it is below the computational error bar (<0.5 meV per f.u.45). In the latter compound, an infinitesimal quenched disorder, locally favouring one phase over the other, can then be held responsible for triggering the phase separation. Similar effects are suppressed in the stoichiometric CuMnO2, but would become enhanced for larger deviations from perfect system uniformity. Indeed, enhanced strain, acting as a precursor of the monoclinic-to-triclinic structural phase transition, has been observed18,23 in the high-temperature monoclinic phase in stoichiometric α-NaMnO2, while in the CuMnO2 a Cu-Mn off-stoichiometry is required to produce such a strain28. Moreover, the diffuse magnetic scattering characteristic of 2D correlated regions that coexist with sharp magnetic Bragg peaks (one of the signatures of the inhomogeneity18 found in the α-NaMnO2) is also found26,30,31 in the CuMnO2. However, in contrast to the α-NaMnO2, where it persists to low temperatures, in stoichiometric CuMnO2 it gradually gives way to the 3D ordered phase below the TN. Interestingly though, in off-stoichiometric samples30 the volume fraction of the 2D-correlated phase shows no decrease below the TN, implying that the 2D-ordered regions keep competing with the 3D order at low temperatures. The total-energy difference of the competing phases below the magnetostructural transition, reflecting the interplay of the magnetic-exchange and the elastic energies, then seems to determine the amount of disorder required to stabilize the inhomogeneous ground state on a geometrically frustrated triangular lattice. Systems with near-degenerate competing phases can be locally perturbed more easily. Such an inhomogeneity may, therefore, be a more general feature of geometrically frustrated magnets.
Methods
Finite-temperature Lanczos method simulations
Calculations of the spin susceptibility for the S = 2 Heisenberg model on the anisotropic triangular lattice (equation (1)), were performed with the finite-temperature Lanczos method (FTLM)49,50 and were used to determine the leading exchange couplings J1 and J2 in the CuMnO2. Within the FTLM finite-size clusters are diagonalized in a similar manner as for the standard exact diagonalization Lanczos method (at T = 0) and additional random vector averaging over the R vectors is employed to determine the properties at T > 0. Typically, R ~ 10 suffices for the largest systems and the lowest T, while smaller systems require a larger R. The limitations of the method are mainly set by finite-size effects, which are the largest at low T and determine the lowest reachable T(~J1). In order to reduce the finite-size effects we used periodic boundary conditions, adjusted cluster shapes, the largest reachable cluster sizes (up to N = 14 sites) and additional approximations for the values in the thermodynamic limit.
The temperature-dependent magnetic susceptibility χ(T) of the model given by equation (1) was calculated previously in Ref. 35 for α-NaMnO2. It was shown that in the regime of interest, elongated spin clusters are the most appropriate. In particular, if a cluster has m independent spins in the J1 direction and n spins in the J2 directions, it was realized that due to J2 < J1 and two competing J2 bonds, χ does not depend on n for n > 2 (see Fig. 2 in Ref. 35). This fact allows us to reduce the finite-size effects by using a larger m. We note that due to the alternating behaviour of χ with m (see Figure 5), which originates in periodic boundary conditions and antiferromagnetic spin-spin correlations, the average susceptibility curve from the two largest-size clusters (m = 6 and 7) is a better approximation than the m = 7 curve.

The finite-size effects in the FTLM calculation.
The calculated susceptibility on various m × n clusters for the optimal parameters J1 = 53 K and J2/J1 = 0.27. The curve averaging the 6 × 2 and 7 × 2 data and the scaled curve are also shown.
Scaling-like approximation for the susceptibility
The results for several different sizes of finite clusters and their systematics, shown in Figure 5, allow a scaling-like analysis to obtain a better approximation of χ in the thermodynamic limit. Typical scaling analyses use scaling functions of the form χ(N) = a + b/N and additional higher terms when needed; e.g., c/N2. Since our calculations are limited to rather small maximum system sizes by S = 2, we also use the results from small systems (starting with m = 2). Consequently, such scaling functions are not appropriate. In particular, at higher T (see, for example, T > 5J1 in Figure 5) χ has already converged with N for systems with m ≥ 5, while for m < 5 notable finite-size effects are seen. Therefore, the scaling function should be close to a constant for 1/N smaller than some value, while at larger 1/N, the scaling function should allow for a stronger N dependence. For these reasons we use a generalized scaling function of the form χ(N) = a + b [exp(c/N) − 1], which corresponds to typically used functions in the limit of small 1/N. In order to also capture the alternating component of χ with m (Figure 5) we add, in a similar fashion, the term b1 [exp(c1/N) − 1] (−1)N/2. Such a scaling function also gives a correct (converged with N) result for high T, while typical scaling functions fail in this respect. We have performed such a scaling for each T separately. However, since we are limited to small systems with notable finite-size effects at low  and since the scaling function has many parameters, the result of such an analysis should not be taken as a strict thermodynamic limit. Rather, it should be regarded as a next approximation of it, compared to the result from simpler averaging of the two largest-cluster curves.
and since the scaling function has many parameters, the result of such an analysis should not be taken as a strict thermodynamic limit. Rather, it should be regarded as a next approximation of it, compared to the result from simpler averaging of the two largest-cluster curves.
Density-functional theory calculations
The calculations of the total energies and the magneto-elastic (ME) coupling coefficients were performed within the framework of the density-functional theory (DFT) and the generalized-gradient approximation (GGA)51 for the exchange-correlation contribution by applying the Quantum Espresso code52. The electron-ion interactions were described by the Vanderbilt ultrasoft potentials53 including the spin-orbit coupling for the Mn atoms. The plane-wave cut-off parameters were set to 585 eV and 4678 eV for the expansion of the wave functions and the potential, respectively. In order to take into account the proper antiferromagnetic ordering, the 1 × 2 × 2 supercells of the monoclinic and the triclinic structures were used. The calculations of the total energies as a function of the unit-cell volume for the different types of magnetic ordering were carried out by using 4 × 8 × 2 reciprocal vectors in the full Brillouin zone (BZ) for the Methfessel-Paxton sampling54 integration. The criterion for the self consistency was the total-energy difference between two subsequent iterations being less than 10−8 Ry. The monoclinic phase was further optimized by minimizing the total energy and the inter-atomic forces with respect to the lattice parameters and the atomic positions. The resulting structure served as the zero-strain reference for the calculations of the ME coefficients that are based on the evaluation of the total-energy differences of the order of <10−4 Ry, which is also the accuracy for the determination of the total-energy differences between the monoclinic and triclinic phases in Figure 2b, calculated per 4 f.u. The tests yielded 8 × 16 × 4 reciprocal vectors in the full BZ to be enough for well-converged results.
Determination of the magnetoelastic coupling
The magnetoelastic coupling constants bij are calculated from the associated magnetoelastic energy density. This contains the products  of the strain-tensor components
of the strain-tensor components  and the components mi of the normalized magnetization. The form of the magnetoelastic energy density is determined by the symmetry of a particular system44. The magnetism of the CuMnO2 and the α-NaMnO2 is essentially two-dimensional; therefore, only the terms with the lateral strain-tensor components are important. For the monoclinic symmetry, these include
and the components mi of the normalized magnetization. The form of the magnetoelastic energy density is determined by the symmetry of a particular system44. The magnetism of the CuMnO2 and the α-NaMnO2 is essentially two-dimensional; therefore, only the terms with the lateral strain-tensor components are important. For the monoclinic symmetry, these include

Individual magnetoelastic terms are then determined by specifically choosing strain components and magnetization directions and calculating the total-energy density  , from
, from

The above total-energy differences are calculated ab-initio as a function of  for relaxed crystal structures. In CuMnO2, Δfxx and Δfyy change linearly with increasing strain at least up to
for relaxed crystal structures. In CuMnO2, Δfxx and Δfyy change linearly with increasing strain at least up to  , while an additional quadratic term is observed in Δfxy (Figure 2a). The experimental strain value
, while an additional quadratic term is observed in Δfxy (Figure 2a). The experimental strain value  that is obtained by calculating the relative shift of the Mn2+ ions in the triclinic structure, when compared to the monoclinic structure (based on high-resolution synchrotron XRD data analysis18), is an order of magnitude lower. Therefore, the linear term is dominant for all three contributions and allows the extraction of the three magnetoelastic constants bxx = 2.3 MJ/m3, byy = 1.6 MJ/m3 and bxy = 3.4 MJ/m3.
that is obtained by calculating the relative shift of the Mn2+ ions in the triclinic structure, when compared to the monoclinic structure (based on high-resolution synchrotron XRD data analysis18), is an order of magnitude lower. Therefore, the linear term is dominant for all three contributions and allows the extraction of the three magnetoelastic constants bxx = 2.3 MJ/m3, byy = 1.6 MJ/m3 and bxy = 3.4 MJ/m3.
Nuclear magnetic/quadrupolar resonance
63,65Cu (I = 3/2) NMR/NQR measurements were performed on a high-quality powder sample with the same phase purity and stoichiometry as in the study presented in Ref. 27. The NMR/NQR spectra and the spin-lattice relaxation were measured between 4.6 K and 120 K in a magnetic field of 8.9 T (NMR) and in zero magnetic field (NQR) on a custom-built spectrometer. Frequency sweeping and a solid-echo pulse sequence were used for recording the spectra, while a saturation recovery method was used for measuring the spin-lattice relaxation. Typical π/2-pulse lengths were 3.5 μs and 6 μs in the NMR and NQR experiments, respectively. The reference NMR Larmor frequencies of 63ν0 = 100.728 MHz and 65ν0 = 107.908 MHz were determined with a 0.1 M NaCl-solution reference by taking into account the gyromagnetic ratios 23γ = 2π × 11.261 MHz/T, 63γ = 2π × 11.295 MHz/T and 65γ = 2π × 12.089 MHz/T.
The NQR spectrum of each isotope is particularly simple, as it is given by a single line46 at  , with the ratio of the quadrupolar frequencies 63νQ/65νQ = 1.08 fixed by the corresponding quadrupolar moments and the EFG tensor
, with the ratio of the quadrupolar frequencies 63νQ/65νQ = 1.08 fixed by the corresponding quadrupolar moments and the EFG tensor  asymmetry parameter being η = (Vxx − Vyy)/Vzz. The NMR spectrum is more complicated, because the applied magnetic field B0 breaks the symmetry in the spin space. The central-transition (−1/2
asymmetry parameter being η = (Vxx − Vyy)/Vzz. The NMR spectrum is more complicated, because the applied magnetic field B0 breaks the symmetry in the spin space. The central-transition (−1/2  1/2) powder NMR line adopts a characteristic structure because of the angular-dependent NMR shift K from the reference frequencies ν0 = γB0/2π,
1/2) powder NMR line adopts a characteristic structure because of the angular-dependent NMR shift K from the reference frequencies ν0 = γB0/2π,  . In analogy18 to the α-NaMnO2, we take the hf shift
. In analogy18 to the α-NaMnO2, we take the hf shift  (
( is the reduced Planck constant) to be isotropic, while the dipolar contribution Kd and the quadrupolar46 shift 63,65KQ can be accurately calculated. The former has a uniaxial symmetry and is calculated18 (Kd = 0.11% is the dominant eigenvalue) by taking into consideration all the Mn3+ paramagnetic spins around a given Cu site within a sphere large enough to ensure convergence.
is the reduced Planck constant) to be isotropic, while the dipolar contribution Kd and the quadrupolar46 shift 63,65KQ can be accurately calculated. The former has a uniaxial symmetry and is calculated18 (Kd = 0.11% is the dominant eigenvalue) by taking into consideration all the Mn3+ paramagnetic spins around a given Cu site within a sphere large enough to ensure convergence.
A homogeneous life-time broadening of the NQR spectra is negligible. The spin-spin relaxation time 63T2 = 46 μs at 80 K yields 63δh = 6.9 kHz, which is much smaller than the spectral width. The spin-lattice relaxation is of magnetic origin. We find the isotopic effect 65T1/63T1 = 0.86 that is in accordance with magnetic relaxation dictating  .
.
Muon spin relaxation
The μSR investigation was carried out on the General Purpose Surface muon (GPS) instrument at the Paul Scherrer Institute, Villigen, using the same powder sample as in the NMR/NQR experiments. Zero-field (ZF) and weak-transverse-field (wTF) measurements in a 3 mT magnetic field were performed in the temperature range between 5 and 120 K. The veto mode was utilized to minimize the background signal. The ZF μSR measurements below the TN revealed that each muon stops at one of the two possible non-equivalent stopping sites, like was observed18 in α-NaMnO2.
References
Seul, M. & Andelman, D. Domain shapes and patterns: the phenomenology of modulated phases. Science 267, 476–483 (1995).
Malescio, G. & Pellicane, G. Stripe phases from isotropic repulsive interactions. Nat. Mater. 2, 97–100 (2003).
Dagotto, E. Complexity in strongly correlated electronic systems. Science 309, 257–262 (2005).
Seifert, U. Configurations of fluid membranes and vesicles. Adv. Phys. 46, 13–137 (1997).
Baumgart, T., Hess, S. T. & Webb, W. W. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature 425, 821–824 (2003).
Kondo, S. & Miura, T. Reaction-diffusion model as a framework for understanding biological pattern formation. Science 329, 1616–1620 (2010).
Maclennan, J. & Seul, M. Novel stripe textures in nonchiral hexatic liquid-crystal films. Phys. Rev. Lett. 69, 2082 (1992).
Harrison, C. Mechanisms of ordering in striped patterns. Science 290, 1558–1560 (2000).
Tranquada, J. M., Sternlieb, B. J., Axe, J. D., Nakamura, Y. & Uchida, S. Evidence for stripe correlations of spins and holes in copper oxide superconductors. Nature 375, 561–563 (1995).
Dagotto, E., Hotta, T. & Moreo, A. Colossal magnetoresistant materials: the key role of phase separation. Phys. Rep. 344, 1–153 (2001).
Roger, M. et al. Patterning of sodium ions and the control of electrons in sodium cobaltate. Nature 445, 631–634 (2007).
Vojta, M. Lattice symmetry breaking in cuprate superconductors: stripes, nematics and superconductivity. Adv. Phys. 58, 699–820 (2009).
Park, J. T. et al. Electronic phase separation in the slightly underdoped iron pnictide superconductor Ba1−xKxFe2As2 . Phys. Rev. Lett. 102, 117006 (2009).
Bauer, E. D. et al. Electronic inhomogeneity in a Kondo lattice. Proc. Natl. Acad. Sci. USA 108, 6857–6861 (2011).
Seo, S. et al. Disorder in quantum critical superconductors. Nat. Phys. 10, 120–125 (2013).
De'Bell, K., MacIsaac, A. B. & Whitehead, J. P. Dipolar effects in magnetic thin films and quasi-two-dimensional systems. Rev. Mod. Phys. 72, 225 (2000).
Portmann, O., Vaterlaus, A. & Pescia, D. An inverse transition of magnetic domain patterns in ultrathin films. Nature 422, 701–704 (2003).
Zorko, A., Adamopoulos, O., Komelj, M., Arčon, D. & Lappas, A. Frustration-induced nanometre-scale inhomogeneity in a triangular antiferromagnet. Nat. Commun. 5, 3222 (2014).
de Groot, J. et al. Competing ferri- and antiferromagnetic phases in geometrically frustrated LuFe2O4 . Phys. Rev. Lett. 108, 037206 (2012).
Kamiya, Y. & Batista, C. D. Formation of magnetic microphases in Ca3Co2O6 . Phys. Rev. Lett. 109, 067204 (2012).
Nakajima, S. et al. Microscopic phase separation in triangular-lattice quantum spin magnet κ – (BEDT-TTF)2Cu2(CN)3 probed by muon spin relaxation. J. Phys. Soc. Jpn. 81, 063706 (2012).
Lacroix, C., Mendels, P. & Mila, F. (eds.) Introduction to Frustrated Magnetism (Springer-Verlag, Heidelberg, 2011).
Giot, M. et al. Magnetoelastic coupling and symmetry breaking in the frustrated antiferromagnet α-NaMnO2 . Phys. Rev. Lett. 99, 247211 (2007).
Kondrashev, I. D. The crystal structure and composition of crednerite. Sov. Phys. Crystallogr. 3, 703–706 (1959).
Doumerc, J.-P. et al. Magnetic properties of the crednerite CuMnO2 . Eur. J. Solid State Inorg. Chem. 31, 705 (1994).
Damay, F. et al. Spin-lattice coupling induced phase transition in the S = 2 frustrated antiferromagnet CuMnO2 . Phys. Rev. B 80, 094410 (2009).
Vecchini, C. et al. Magnetoelastic coupling in the frustrated antiferromagnetic triangular lattice CuMnO2 . Phys. Rev. B 82, 094404 (2010).
Poienar, M. et al. Substitution effect on the interplane coupling in crednerite: the Cu1.04Mn0.96O2 case. Chem. Mater. 23, 85–94 (2011).
Trari, M. et al. Preparation and physical properties of the solid solutions Cu1+xMn1−xO2 (0 ≤ x ≤ 0.2). J. Solid State Chem. 178, 2751–2758 (2005).
Garlea, V. O., Savici, A. T. & Jin, R. Tuning the magnetic ground state of a triangular lattice system Cu(Mn1−xCux)O2 . Phys. Rev. B 83, 172407 (2011).
Terada, N. et al. Magnetic correlations and the influence of atomic disorder in frustrated isosceles triangular lattice antiferromagnet CuMnO2 . Phys. Rev. B 84, 064432 (2011).
Ushakov, A. V., Streltsov, S. V. & Khomskii, D. I. Orbital structure and magnetic ordering in stoichiometric and doped crednerite CuMnO2 . Phys. Rev. B 89, 024406 (2014).
Jia, T. et al. Magnetic frustration in α-NaMnO2 and CuMnO2 . J. Appl. Phys. 109, 07E102 (2011).
Kim, Y. J., Greven, M., Wiese, U.-J. & Birgeneau, R. J. Monte-carlo study of correlations in quantum spin chains at non-zero temperature. Eur. Phys. J. B 4, 291–297 (1998).
Zorko, A. et al. Magnetic interactions in α-NaMnO2: Quantum spin-2 system on a spatially anisotropic two-dimensional triangular lattice. Phys. Rev. B 77, 024412 (2008).
Yasuda, C. et al. Néel temperature of quasi-low-dimensional Heisenberg antiferromagnets. Phys. Rev. Lett. 94, 217201 (2005).
Zheng, W., Singh, R. R. P., McKenzie, R. H. & Coldea, R. Temperature dependence of the magnetic susceptibility for triangular-lattice antiferromagnets with spatially anisotropic exchange constants. Phys. Rev. B 71, 134422 (2005).
Stock, C. et al. One-dimensional magnetic fluctuations in the spin-2 triangular lattice α-NaMnO2 . Phys. Rev. Lett. 103, 077202 (2009).
Scalapino, D. J., Imry, Y. & Pincus, P. Generalized Ginzburg-Landau theory of pseudo-one-dimensional systems. Phys. Rev. B 11, 2042–2048 (1975).
Schulz, H. J. Dynamics of coupled quantum spin chains. Phys. Rev. Lett. 77, 2790–2793 (1996).
Irkhin, V. Y. & Katanin, A. A. Calculation of Neel temperature for S = 1/2 Heisenberg quasi-one-dimensional antiferromagnets. Phys. Rev. B 61, 6757 (2000).
Bocquet, M. Finite-temperature perturbation theory for quasi-one-dimensional spin- Heisenberg antiferromagnets. Phys. Rev. B 65, 184415 (2002).
Siurakshina, L., Ihle, D. & Hayn, R. Theory of magnetic order in the three-dimensional spatially anisotropic Heisenberg model. Phys. Rev. B 61, 14601 (2000).
du Trémolet de Lacheisserie, E. Magnetostriction: Theory and Application of Magnetoelasticity (CRC Press, Boca Raton, 1993).
Ouyang, Z. W. & Wang, B. First-principles study of structural distortions in frustrated antiferromagnet α-NaMnO2 . Phys. Rev. B 82, 064405 (2010).
Abragam, A. Principles of Nuclear Magnetism (Oxford University Press, Oxford, 1961).
Moriya, T. Nuclear magnetic resonance in antiferromagnets. Prog. Theor. Phys. 16, 23–44 (1956).
Yaouanc, A. & Dalmas de Réotier, P. Muon Spin Rotation, Relaxation and Resonance (Oxford University Press, Oxford, 2011).
Jaklič, J. & Prelovšek, P. Finite-temperature properties of doped antiferromagnets. Adv. Phys. 49, 1–92 (2000).
Prelovšek, P. & Bonča, J. Strongly Correlated Systems - Numerical Methods. Springer Series in Solid–State Sciences 176 (Springer, Berlin, 2013).
Perdew, J., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 21, 395502 (2009).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, R7892 (1990).
Methfessel, M. & Paxton, A. T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 40, 3616 (1989).
Acknowledgements
We acknowledge fruitful discussions with S. El Shawish. This work has been supported in part by the Slovenian Research Agency Program P1-0125. A.L. acknowledges financial support in the framework of the KRIPIS action, PROENYL research project No. MIS-448305 (2013SE01380034) that was funded by the General Secretariat for Research and Technology, Ministry of Education, Greece and the European Regional Development Fund (Sectoral Operational Programme: Competitiveness and Entrepreneurship, NSRF 2007–2013)/European Commission. The μSR results are based on experiments performed at the Swiss Muon Source (SμS), Paul Scherrer Institute, Villigen, Switzerland.
Author information
Authors and Affiliations
Contributions
A.Z., D.A. and A.L. designed and supervised the project. The FTLM simulations were performed by J.K., while the ab-initio calculations were carried out by M.K. The samples were synthesized and characterized by O.A.. The μSR experiments were performed by A.Z. and H.L. The NMR/NQR experiments were conducted and analysed by A.Z., who also wrote the paper. All authors contributed to the interpretation of the data, discussed the results and reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zorko, A., Kokalj, J., Komelj, M. et al. Magnetic inhomogeneity on a triangular lattice: the magnetic-exchange versus the elastic energy and the role of disorder. Sci Rep 5, 9272 (2015). https://doi.org/10.1038/srep09272
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09272
This article is cited by
-
Development of short and long-range magnetic order in the double perovskite based frustrated triangular lattice antiferromagnet Ba\(_{2}\)MnTeO\(_{6}\)
Scientific Reports (2021)
-
High-temperature short-range order in Mn3RhSi
Communications Materials (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
