
Abstract
This work reports on the nanoscale assembly of hybrid lanthanum oxide and silica structures, which form patterns of interfacial dense and porous networks. It was found that increasing the molar ratio of lanthanum nitrate to tetraethyl orthosilicate (TEOS) in an acid catalysed sol-gel process alters the expected microporous metal oxide silica structure to a predominantly mesoporous structure above a critical lanthanum concentration. This change manifests itself by the formation of a lanthanum silicate phase, which results from the reaction of lanthanum oxide nanoparticles with the silica matrix. This process converts the microporous silica into the denser silicate phase. Above a lanthanum to silica ratio of 0.15, the combination of growth and microporous silica consumption results in the formation of nanoscale hybrid lanthanum oxides, with the inter-nano-domain spacing forming mesoporous volume. As the size of these nano-domains increases with concentration, so does the mesoporous volume. The absence of lanthanum hydroxide (La(OH)3) suggests the formation of La2O3 surrounded by lanthanum silicate.
Similar content being viewed by others


Introduction
Silica is an extremely versatile material widely used for tailoring structures such as amorphous micro- and mesoporous structures1,2,3, crystalline materials such as zeolites4,5 and self-assembly materials such as mesoporous hollow spheres6 to name a few. The flexibility of silica derived sol-gel methods using colloidal precursors in tandem with further processing techniques has spurred the designing of novel silica structures. For instance, microporous silica structures are intrinsically linked to weakly branched fractal dimensions, which contain a high concentration of uncondensed silanol (Si-OH) species7,8,9. In addition, the use of organic templates10,11,12, metal oxides13,14, or both15, led to the manipulation of sol-gel chemistry and the production of a wide variety of porous structures in silica materials.
Embedding metal oxides into silica matrices to produce microporous structures has received the concerted attention of the scientific community, including nickel oxides16,17, cobalt oxides15,18,19, iron oxides20, palladium oxides21,22, binary palladium cobalt oxides23, as well as many others24,25,26,27. A feature of these reports is that the metal oxide remains homogeneously dispersed as nanoparticles in the silica matrices. In addition, Uhlmann et al.28 showed that the pore sizes of the interfaces of cobalt oxide and silica were around 3 Å, slightly smaller than the pore sizes (3–5 Å) in pure silica structures. In some formulations, even cobalt oxide to silica molar fractions as high as 1:1 still delivered microporous structures13. All these reports have limited the calcination temperature to no more than 630°C, to avoid the densification of the silica and/or formation of metal oxide silicates. Transition metals have been the preferred route in producing metal oxide silica porous structures.
Lanthanides have also been used to produce porous silica materials but to a lesser extent than transition metals. In some cases lanthanum was coated on nanoporous SBA-1529 or on silica layers30, whilst in other cases the combination of lanthanum niobate silica has formed pillared structures31 or lanthanum phosphate silica has produced mesoporous materials32. Célérier et al.33 produced mesoporous lanthanum silicate materials for use in solid oxide fuel cells (SOFC) due to their high ionic conductivity at low temperatures and their ability to form mesoporous materials which impose minimal resistance to fluidic species. In this instance, materials exhibited a large increase in the pore size distribution by forming a lanthanum silicate phase (La9.33Si6O26). However, no mechanism for the deviation in the pore size distribution was proposed, which is vital to achieve fine tuning of the system. The deviation from conventional micro- to mesoporous structures requires further understanding for metal doped silica materials, as in many cases the formation of metal oxide silicates require high calcination temperatures (>850°C)34.
The present study focuses on the effect of the concentration of lanthanum within the silica to determine the mechanism behind porosity alteration over the calcination process. Results of porosity measurements show the formation of mesoporous structures above a critical lanthanum concentration, which departs from results found with transition metal dopants. Characterisation of the chemical structure of the materials determined that the lanthanum forms nanoscale oxides, encased by a silicate layer within an amorphous silica matrix. A mechanistic model is proposed to explain the effects of material synthesis on the resultant structure.
Results and Discussion
In order to investigate the structural effect of lanthanum embedded in the silica matrix, xerogel samples were analysed via N2 adsorption. Figure 1a shows representative isotherms of the xerogel samples calcined at 500°C. The LaSi(0) xerogel (i.e. pure silica) shows a type I isotherm which is characteristic of microporous materials. It also displays the largest adsorbed volume of all the samples. The addition of small amounts of lanthanum (LaSi(0.03), (0.05)) results in a more right-angled isotherm at lower nitrogen partial pressures which corresponds to a shift to a narrower pore size distribution (PSD) at lower pore sizes, as seen in Figure 1b. This is likely due to the acidification of solution with the addition of lanthanum nitrate33,35, a classic method to increase the microporosity of silica in sol-gel theory. Furthermore, a substantial reduction in the volume of nitrogen adsorbed per xerogel mass is evident.

(a) N2 adsorption-desorption isotherms and (b) Pore size distribution calculated by density functional theory for the lanthanum silica xerogels calcined at 500°C.
The trend of decreasing micropore volume with increasing lanthanum content continues to higher lanthanum concentrations until a transition point is reached (La/Si > 0.15) whereby the isotherms shift to type IV; thus forming mesoporous networks. This is clearly demonstrated in Figure 1b, with microporous volume decreasing and mesoporous volume increasing proportionally to lanthanum concentration above La/Si = 0.15. Table 1 further emphasises the trends in the isotherms and pore size distributions, showing a loss in both the BET surface area and pore volume with increasing lanthanum concentration. This is true for all concentrations except for LaSi(50), whereby pore volume increases due to the large mesoporous volume that has formed.
Figure 2a shows the FTIR spectra of the dried lanthanum silica xerogels, with the typical silica (SiO2) vibrations located at 800, 1080 and 1220 cm−1 assigned to siloxane (Si-O-Si)36 and 960 cm−1 to silanol (Si-OH)37 bonds for non-doped silica, LaSi(0). The integration of lanthanum nitrate into the silica during the sol-gel synthesis results in the emergence of multiple sharp nitrate peaks at 1455, 1425, 1325, 1300, 1050, 1040, 870, 810, 780, 740, 715 cm−1 (Figure 2a). Upon calcination at 500°C, there is a significant reduction in the intensity of the nitrate peaks (Figure 2b), attributed to the partial decomposition of the lanthanum nitrate species38,39. The silanol peak at 960 cm−1 is no longer visible at 500°C for the LaSi(0) xerogels, becoming embedded as a shoulder in the broader siloxane peak from 1080 to 1220 cm−1. This is a direct result of the condensation reactions occurring during the heat treatment, a process which occurs by the forming of siloxane species at the expense of silanol groups.

Infrared spectra for xerogels with a lanthanum to silica molar ratio of 0 ≤ La/Si ≤ 0.5 heated to (a) 60°C, (b) 500°C and (c) LaSi(0.33) as a function of temperature.
The heat treatment of the LaSi(0.33) xerogel (Figure 2c) shows a significant effect in the reduction of nitrates, which decompose and leave the lanthanum silica matrix as the calcination temperature increases. Similarly, as the calcination temperature increases, the FTIR spectrum exhibits the emergence of a peak at 950 cm−1. This peak is distinctly different in shape to the traditional silanol peak at 960 cm−1, as can be seen by comparison with LaSi(0) in Figure 2b and emerges at temperatures above 200°C. Furthermore, this peak becomes more intense at higher lanthanum concentrations (Figure 2b). This peak has widely been reported to originate from the SiO44− ion, which comprises part of the lanthanum silicate species, a phase which forms at elevated temperatures due to the diffusion of La2O3 and silica33,40,41,42. As nitrates are decomposed during calcination, the thermodynamic propensity is for the formation of lanthanum oxides, thus suggesting the formation of metal oxide particles in silica, as reported elsewhere for cobalt and cobalt iron oxides13,19,43,44.
X-ray photoelectron spectroscopy (XPS) was carried out to gain a more thorough understanding of the chemical formation of the lanthanum silica xerogels. The XPS spectra in Figure 3 displays the O1s region of the xerogel for samples LaSi(0.15) to LaSi(0.50) calcined at 500°C. Deconvolution of the LaSi(0.15) spectra shows a large peak present at 532.8 eV, commonly associated with SiO241. As the concentration of lanthanum increases in the xerogels, the binding energy of the SiO2 peak decreases, consistent with results published elsewhere41. Deconvolution of the shoulder at lower binding energy shows a peak at approximately 529.8 eV which is assigned to La2O341,45,46,47,48. Furthermore, the deconvolution reveals the presence of a peak between the SiO2 and La2O3 species, at 530.8 eV, which has been extensively reported as lanthanum silicate41,45,47, thus confirming the results from the FTIR spectra in Figure 2b and c. Deconvolution of the lanthanum oxide and lanthanum silicate species within the O1s region matches well with deconvolution within the La3d region confirming the results.

Deconvoluted, baseline corrected XPS spectra of the O 1 s region in the xerogel with lanthanum concentration LaSi(0.33) calcined at 500°C.
TEM images of the xerogels were taken to investigate the presence of nano-domains composed of lanthanum oxide and lanthanum silicate. Figure 4a shows a representative image of the LaSi(0.33) xerogel. The presence of particulate aggregates can be seen, particularly in Figure 4b, which are similar to the nanoscale assembly that was reported by Célérier et al.33 for the formation of lanthanum silicates at high temperatures. Determination of the phases present in these nanoscale domains is difficult due to their amorphous nature (confirmed by XRD in Figure 5). Thus, to determine the phase of the various components in the TEM image, the LaSi(33) sample was heated to 1000°C to induce crystallinity. It can be seen in Figure 4c that the structure of the xerogel is very similar to that calcined at 500°C. High resolution TEM exhibits the presence of multiple crystalline phases (Figure 4d), those that are labelled are presented in Table 2. The red line bordering the dark areas indicates the interface between lanthanum oxide and lanthanum silicate, while the enclosed space between the dashed black line and the red line is amorphous and is proposed to be unreacted microporous silica.

Representative TEM image of LaSi(0.33) xerogel calcined at (a) 500°C and c) 1000°C. (b) and d) are high resolution images from within the boxes of a) and c) respectively.

XRD of LaSi(33) calcined at 1000°C.
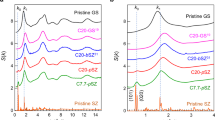
Figure 5 shows the XRD patterns of the LaSi(33) xerogel calcined at 500 and 1000°C. The xerogel calcined at 500°C exhibits a largely amorphous structure. Four broad peaks are present attributed to amorphous silica (2θ = 27°) and lanthanum oxide/lanthanum silicate crystallites (2θ = 13°, 43° and 64°). The 1000°C pattern shows numerous crystalline peaks, all of which were assigned to La2O3 (JCPDS cards 05-0602, 22-0369), La2SiO5 (JCPDS card 40-0234) and La9.33Si6O26 (JCPDS card 49-0443), with La2O3 and La9.33Si6O26 constituting the majority of the crystalline material. This corresponds to the TEM image shown in Figure 4d.
With further analysis of the XPS spectra (Figure 3), it is observed that the shoulder present in the LaSi(0.15) xerogel increases in intensity proportionally to the lanthanum concentration. However, the shape of the shoulder also changes, from a gradual tailing off in intensity at the lower binding energy side of the shoulder for the LaSi(0.15) sample to the formation of a distinct peak at low binding energies for the LaSi(0.50) sample. This deviation suggests a variation in the ratios of lanthanum oxide to lanthanum silicate. The ratio of these deconvoluted peak areas is displayed in Figure 6. It can be seen that the ratio of lanthanum oxide to lanthanum silicate increases linearly with lanthanum concentration. Assuming the formation of nano-domains containing both La2O3 and lanthanum silicate of a constant composition, this suggests that the average particle size is increasing proportional to the lanthanum concentration.

XPS deconvolution for 0.15 < La/Si < 0.50 samples calcined displaying the lanthanum oxide to lanthanum silicate intensity ratio at 500°C.
The TEM micrograph suggests the formation of lanthanum oxide nano-domains surrounded by lanthanum silicate. It is proposed that at low lanthanum concentrations (≤LaSi(0.15)) the reaction between La2O3 and silica converts microporous silica into a dense lanthanum silicate. This is supported by the emergence and growth of a peak at 950 cm−1 in the FTIR spectrum of lanthanum doped xerogels (Figure 2b) and the decrease in microporous volume of lanthanum doped xerogels (Figure 1a). In addition, studies have shown that transition metal oxides that are incorporated within microporous silica are accessible by ambient water molecules, due to the hydrophilic nature of the silica matrix18. Exposure of La2O3 to water results in a spontaneous reaction to produce lanthanum hydroxide (La(OH)3), a compound which does not appear within the infrared or XRD analyses as expected. This further indicates that the La2O3 phase must be fully enclosed within a dense silicate material at its interface, such that ambient moisture cannot contact the La2O3. Hence, these results strongly suggest the formation of a lanthanum nano-phase constituted by La2O3 encased by lanthanum silicate.
At high lanthanum concentrations (>LaSi(0.15)) it is proposed that the enhanced size of the nanoscale assembly of lanthanum increase their proximity. The reduction in inter-assembly spacing, in addition to the enhanced consumption of silica, results in the formation of nano-domains, as can be seen in Figure 4. As the lanthanum concentration increases, the nanoscale assembly of lanthanum silicates and oxides continues to increase in size, resulting in the expansion of the inter-nano-domain voids. This effect correlates well with the results in Figure 1b, with the mesoporous volume increasing proportionally to lanthanum concentration. The expansion of the porous network of lanthanum silicate materials over the calcination process has been shown previously by Célérier et al.33. This work shows that the underlying mechanism causing the expansion of the porous network is the formation and expansion of lanthanum nano-domains. This study thus provides a platform for porosity optimisation studies for lanthanum silica materials.
Methods
Synthesis of Lanthanum Silica Xerogels
The synthesis of lanthanum silica xerogels was carried out using an acid catalysed sol-gel procedure. Initially lanthanum nitrate hexahydrate (La(NO3)3*6H2O) was dissolved in 30 vol% aqueous hydrogen peroxide (H2O2). The solution was then diluted with absolute ethanol (EtOH) and water, stirred and cooled to 0°C in an ice bath. Finally tetraethyl orthosilicate (TEOS) was added drop wise and reacted for 3 hours before being dried for 96 hours at 60°C to form xerogels. The final molar ratio of all reagents was 9 H2O2 : x La(NO3)3*6H2O : 256 EtOH : 45.5 H2O : 4 TEOS with the lanthanum to silica ratio (La/Si) varied between 0 and 0.5. It should be noted that xerogels with the composition of La/Si = x will be denoted as LaSi(x) for the duration of this paper. Dried sols were crushed into a fine xerogel powder so that all the grain surfaces gave a good representation of the bulk sample. Finally the powders were calcined between 200 and 600°C in an air atmosphere.
Characterisation
Fourier transform infrared (FTIR) spectroscopy was performed using a PerkinElmer Spectrum 400 FT-IR/FT-FIR spectrometer with a diamond/ZnSe crystal. Data was collected between 4000–500 cm−1. Transmission electron microscopy (TEM) was performed using a JEOL 2100 transition electron microscope operating at 200 keV. Samples were prepared by suspending finely crushed xerogels in ethanol and depositing them on a hollow carbon copper grid. X-ray photoelectron spectroscopy (XPS) was performed after xerogel powders were re-crushed to minimise any contaminant formed on the surface layer. A Kratos Axis ULTRA X-ray Photoelectron Spectrometer incorporating a 165 mm hemispherical electron energy analyser was implemented to acquire the data. An Al Kα (1486.6 eV) monochromatic x-ray source was used at a 90° takeoff angle with 0.05 eV step size to obtain XP spectra. Charge compensation was performed by setting the adventitious C1s peak to a binding energy of 284.6 eV. Peak deconvolution of the high-resolution data was carried out using CasaXPS software with a Shirley baseline. Kratos library relative sensitivity factors (RSFs) were used to normalise atomic regions.
N2 adsorption-desorption isotherms were generated at 77K in a Micromeritics TriStar 3000 volumetric adsorption system. Before analysis, xerogels were degassed for 8 hours at 200°C in a Micromeritics VacPrep061 at a pressure of approximately 0.05 mbar. The pore size distribution (PSD) was calculated via density functional theory assuming cylindrical pores with an oxide surface.
References
Rajendra, V. & Brook, M. A. Controlled formation of macroporous or hollow silica particles in non-aqueous silicone dispersions. RSC Advances 3, 22229–22238 (2013).
Chen, X., Jiang, J., Yan, F., Tian, S. & Li, K. A novel low temperature vapor phase hydrolysis method for the production of nano-structured silica materials using silicon tetrachloride. RSC Advances 4, 8703–8710 (2014).
Ahmed, A., Clowes, R., Myers, P. & Zhang, H. Hierarchically porous silica monoliths with tuneable morphology, porosity and mechanical stability. J. Mater. Chem. 21, 5753–5763 (2011).
Keller, T. C., Isabettini, S., Verboekend, D., Rodrigues, E. G. & Pérez-Ramírez, J. Hierarchical high-silica zeolites as superior base catalysts. Chem. Sci. 5, 677–684 (2014).
Camblor, M. A., Corma, A. & Valencia, S. Spontaneous nucleation and growth of pure silica zeolite-β free of connectivity defects. Chem. Commun. 20, 2365–2366 (1996).
Yan, N. et al. Hollow porous SiO2 nanocubes towards high-performance anodes for lithium-ion batteries. Sci. Rep. 3, 1568; 10.1038/srep01568 (2013).
Brinker, C. J. & Scherer, G. W. Sol-gel Science: The Physics and Chemistry of Sol-gel Processing. (Academic Press, 1990).
Iler, R. K. The Chemistry of Silica. (Wiley-Interscience, 1979).
Diniz da Costa, J. C., Lu, G., Rudolph, V. & Lin, Y. Novel molecular sieve silica (MSS) membranes: characterisation and permeation of single-step and two-step sol-gel membranes. J. Memb. Sci. 198, 9–21 (2002).
Duke, M. C., Diniz da Costa, J. C., Do, D. D., Gray, P. G. & Lu, G. Q. Hydrothermally robust molecular sieve silica for wet gas separation. Adv. Funct. Mater. 16, 1215–1220 (2006).
Brinker, C. et al. Sol-gel strategies for controlled porosity inorganic materials. J. Memb. Sci. 94, 85–102 (1994).
Huo, C., Ouyang, J. & Yang, H. CuO nanoparticles encapsulated inside Al-MCM-41 mesoporous materials via direct synthetic route. Sci. Rep. 4, 3682; 10.1038/srep03682 (2014).
Yacou, C., Smart, S. & Diniz da Costa, J. C. Long term performance of cobalt oxide silica membrane module for high temperature H2 separation. Energ. Environ. Sci. 5, 5820–5832 (2011).
Zhang, N., Fu, X. & Xu, Y.-J. A facile and green approach to synthesize Pt@ CeO2 nanocomposite with tunable core-shell and yolk-shell structure and its application as a visible light photocatalyst. J. Mater. Chem. 21, 8152–8158 (2011).
Olguin, G., Yacou, C., Smart, S. & Diniz da Costa, J. C. Tailoring the oxidation state of cobalt through halide functionality in sol-gel silica. Sci.Rep. 3, 2249; 10.1038/srep02449 (2013).
Ikuhara, Y. H., Mori, H., Saito, T. & Iwamoto, Y. High temperature hydrogen adsorption properties of precursor derived nickel nanoparticle dispersed amorphous silica. J. Am. Ceram. Soc. 90, 546–552 (2007).
Kanezashi, M. & Asaeda, M. Hydrogen permeation characteristics and stability of Ni-doped silica membranes in steam at high temperature. J. Memb. Sci. 271, 86–93 (2006).
Uhlmann, D., Smart, S. & Diniz da Costa, J. C. High temperature steam investigation of cobalt oxide silica membranes for gas separation. Sep.Purif. Tech. 76, 171–178 (2010).
Igi, R., Yoshioka, T., Ikuhara, Y. H., Iwamoto, Y. & Tsuru, T. Characterization of Co doped silica for improved hydrothermal stability and application to hydrogen separation membranes at high temperatures. J. Am. Ceram. Soc. 91, 2975–2981 (2008).
Darmawan, A., Smart, S., Julbe, A. & Diniz da Costa, J. C. Iron oxide silica derived from sol-gel synthesis. Materials 4, 448–456 (2011).
Kanezashi, M., Sano, M., Yoshioka, T. & Tsuru, T. Extremely thin Pd–silica mixed-matrix membranes with nano-dispersion for improved hydrogen permeability. Chem. Commun. 46, 6171–6173 (2010).
Kanezashi, M., Fuchigami, D., Yoshioka, T. & Tsuru, T. Control of Pd dispersion in sol-gel derived amorphous silica membranes for hydrogen separation at high temperatures. J. Memb. Sci. 439, 78–86 (2013).
Ballinger, B., Motuzas, J., Smart, S. & Diniz da Costa, J. C. Palladium cobalt binary doping of molecular sieving silica membranes. J. Memb. Sci. 451, 185–191 (2014).
Fotou, G. P., Lin, Y. S. & Pratsinis, S. E. Hydrothermal stability of pure and modified microporous silica membranes. J. Mater. Sci. 30, 2803–2808 (1995).
Yoshida, K., Hirano, Y., Fujii, H., Tsuru, T. & Asaeda, M. Hydrothermal stability and performance of silica-zirconia membranes for hydrogen separation in hydrothermal conditions. J. Chem. Eng. Jpn. 34, 523–530 (2001).
Shen, S. C. & Kawi, S. Understanding of the effect of Al substitution on the hydrothermal stability of MCM-41. The J. Phys. Chem. B 103, 8870–8876 (1999).
Boffa, V., ten Elshof, J. E., Petukhov, A. V. & Blank, D. H. A. Microporous niobia–silica membrane with very low CO2 permeability. ChemSusChem 1, 437–443 (2008).
Uhlmann, D., Smart, S. & Diniz da Costa, J. C. H2S stability and separation performance of cobalt oxide silica membranes. J. Memb. Sci. 380, 48–54 (2011).
Yang, J. et al. A designed nanoporous material for phosphate removal with high efficiency. J. Mater. Chem. 21, 2489–2494 (2011).
Yang, J. et al. Rationally designed functional macroporous materials as new adsorbents for efficient phosphorus removal. J. Mater. Chem. 22, 9983–9990 (2012).
Guo, C.-X., Hou, W.-H., Guo, M., Yan, Q.-J. & Chen, Y. Synthesis of a new solid acid: silica pillared lanthanum niobatewith a supergallery. Chem. Commun. 8, 801–802 (1997).
Chai, Z., Suo, Q., Wang, H. & Wang, X. Mesoporous lanthanum phosphate nanostructures containing H 3 PO 4 as superior electrolyte for PEM fuel cells. RSC Advances 3, 21928–21935 (2013).
Célérier, S. et al. Synthesis of La9. 33Si6O26 Pore–solid nanoarchitectures via epoxide-driven sol–gel chemistry. Adv. Mater. 18, 615–618 (2006).
Esposito, S. et al. Cobalt–silicon mixed oxide nanocomposites by modified sol–gel method. J.Solid State Chem. 180, 3341–3350 (2007).
Biedermann, G. & Ciavatta, L. Studies on the hydrolysis of metal ions part 35: the hydrolysis of the lanthanum ion, La3+. Acta Chem. Scand. 15, 1347–1366 (1961).
Miller, C. R., Wang, D. K., Smart, S. & Diniz da Costa, J. C. Reversible redox effect on gas permeation of cobalt doped ethoxy polysiloxane (ES40) Membranes. Sci. Rep. 3, 1648; 10.1038/srep01648 (2013).
Duran, A., Serna, C., Fornes, V. & Fernandez Navarro, J. Structural considerations about SiO2 glasses prepared by sol-gel. J. Non-Cryst. Solids 82, 69–77 (1986).
Nyquist, R. A., Putzig, C. L. & Leugers, M. A. Infrared and Raman Spectral Atlas of Inorganic Compounds and Organic Salts. Vol. 3 (Academic Press, 1997).
Mentus, S., Jelić, D. & Grudić, V. Lanthanum nitrate decomposition by both temperature programmed heating and citrate gel combustion. J. Therm. Anal. Calorim. 90, 393–397 (2007).
Tao, S. & Irvine, J. T. S. Preparation and characterisation of apatite-type lanthanum silicates by a sol-gel process. Mater. Res. Bull. 36, 1245–1258 (2001).
Gougousi, T., Kelly, M. J., Terry, D. B. & Parsons, G. N. Properties of La-silicate high-K dielectric films formed by oxidation of La on silicon. J. Appl. Phys. 93, 1691 (2003).
Rodríguez-Reyna, E. et al. Structural, microstructural and vibrational characterization of apatite-type lanthanum silicates prepared by mechanical milling. J.Solid State Chem. 179, 522–531 (2006).
Cannas, C., Musinu, A., Peddis, D. & Piccaluga, G. Synthesis and characterization of CoFe2O4 nanoparticles dispersed in a silica matrix by a sol-gel autocombustion method. Chem. Mater. 18, 3835–3842 (2006).
Yan, X., Chen, J., Xue, Q. & Miele, P. Synthesis and magnetic properties of CoFe2O4 nanoparticles confined within mesoporous silica. Micropor. Mesopor. Mat. 135, 137–142 (2010).
Yamada, H., Shimizu, T. & Suzuki, E. Interface reaction of a silicon substrate and lanthanum oxide films deposited by metalorganic chemical vapor deposition. Jpn. J. Appl. Phys. 2 41, 368–370 (2002).
Uwamino, Y. & Ishizuka, H. X-ray photoelectron spectroscopy of rare-earth compounds. J. Electron Spectrosc. 34, 67–78 (1984).
Copel, M., Cartier, E. & Ross, F. Formation of a stratified lanthanum silicate dielectric by reaction with Si (001). Appl. Phys. Lett. 78, 1607 (2001).
Wong, H., Iwai, H., Kakushima, K., Yang, B. & Chu, P. XPS study of the bonding properties of lanthanum oxide/silicon interface with a trace amount of nitrogen incorporation. J. Electrochem. Soc. 157, G49 (2010).
Acknowledgements
The authors thank both Dr Barry Wood for his services towards XPS data acquisition, Dr Graeme Auchterlonie for the transmission electron microscopy (TEM) analysis and the Australian Research Council (ARC) for their financial support (DP110101185). J. C. Diniz da Costa acknowledges support via the ARC Future Fellowship Program (FT130100405).
Author information
Authors and Affiliations
Contributions
B.B. performed the experimental works, analysed results and wrote the manuscript. J.M., C.R.M., S.S. and J.C.D.d.C. assisted in the analyses of results. S.S. and J.C.D.d.C. reviewed the manuscript prior to submission.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Ballinger, B., Motuzas, J., Miller, C. et al. Nanoscale assembly of lanthanum silica with dense and porous interfacial structures. Sci Rep 5, 8210 (2015). https://doi.org/10.1038/srep08210
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08210
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
