
Abstract
The hierarchically macro/micro-porous silicon–carbon–nitrogen (Si–C–N) hybrid material was presented with novel functionalities of totally selective and highly efficient adsorption for organic dyes. The hybrid material was conveniently generated by the pyrolysis of commercial polysilazane precursors using polydivinylbenzene microspheres as sacrificial templates. Owing to the Van der Waals force between sp2-hybridized carbon domains and triphenyl structure of dyes and electrostatic interaction between dyes and Si-C-N matrix, it exhibites high adsorption capacity and good regeneration and recycling ability for the dyes with triphenyl structure, such as methyl blue (MB), acid fuchsin (AF), basic fuchsin and malachite green. The adsorption process is determined by both surface adsorption and intraparticle diffusion. According to the Langmuir model, the adsorption capacity is 1327.7 mg·g−1 and 1084.5 mg·g−1 for MB and AF, respectively, which is much higher than that of many other adsorbents. On the contrary, the hybrid materials do not adsorb the dyes with azo benzene structures, such as methyl orange, methyl red and congro red. Thus, the hierarchically porous Si–C–N hybrid material from a facile and low cost polymer-derived strategy provides a new perspective and possesses a significant potential in the treatment of wastewater with complex organic pollutants.
Similar content being viewed by others
Introduction
The pyrolysis of preceramic precursors has recently attracted considerable attention in manufacturing ceramics with tailored compositions (Si–C,1 Si–O–C,2 Si–C–N,3 or Si–B–C–N4) and diverse morphologies (bulk materials5, fibers6,7, coatings8, tubes9, foams10,11 and porous components12,13) because of the advantages of precursor molecules designed in combination with the shaping processes. Among the polymer-derived ceramics (PDCs), the ternary silicon–carbon–nitrogen (Si–C–N) system has been intensively investigated to generate amorphous or crystalline non-oxide Si–C–N ceramics due to its exceptionally high temperature resistance14, oxidation resistance15 and mechanical strength16, therefore, it can be used even in some harsh chemical conditions, such as high temperature, extensive wear and corrosive media.
For PDCs, the pyrolysis temperature significantly determines the nature of the resulting structures. At a temperature above 800°C, the ceramics with dense and low porous structures can be generated17. When a moderate pyrolysis temperature of 400–800°C was employed, the emission of volatile species of precursor resulted in porous structures in an organic–inorganic hybrid state. High specific surface area (SSA) makes them suitable for catalytic supporters, adsorption and removal of pollutants18,19,20 For example, the foamed creamers from preceramic precursors, fillers and blow agent by pyrolysis at a moderate temperature of 600°C can be used as an alternative material for activated carbons in hydrocarbon adsorption18.
Moreover, when the hybrid materials are required to achieve multifunctionality, the hierarchical porosity is often desirable. For example, the macro/micro-porous structure can effectively improve the catalysis, adsorption, or separation performance of microporous materials. In general, the macropore channels provide chemical and mechanical stability, low pressure drop and high external mass transfer rate due to the interconnected pore structures21. However, the micro/meso-porous structures would achieve the functionality for a given application because of large surface area. Herein, extensive research efforts have been devoted to prepare porous materials with tailored hierarchical pore structures. By using polysiloxanes and platinic acid as precursors and polystyrene beads as templates, Adam et al.22 obtained the hierarchically ordered macro/micro Si–C–O–(Pt) foamed ceramics with high specific surface and potential in adsorption and catalyst application.
Now, various dyes effluents obtained from paper, food, plastic, textile and cosmetic fields pose a serious threat to the environment due to their deep coloration, low biodegradability and carcinogenicity23,24. Disposal of dyes from wastewater has become a significant issue. The adsorption25, biological treatment26, chemical coagulation27 and photocatalytic degradation28 have been investigated for the removal of dyes from the solution. Adsorption has shown an effective and promising perspective because of its simplicity and flexibility in design and operation29,30. Numerous adsorbents have been employed for removal of common dyes31, such as metal oxide microspheres32, carbon-based materials33 and graphenes34. These methods are unfavorable in practical application because of low absorption efficiency, long absorption time and high costs. Polymer-derived Si–C–N materials possess many distinctive advantages, such as electrochemical properties and chemical inertness. However, insignificant attention has been paid to their potential application involving removal of dyes. Notably, the Si–C–N systems possess significant potential in the field of advanced ceramics. However, the Si–C–N systems have never been employed for the separation of pollutants from wastewater. This study provides a new perspective, that by a controlled pyrolysis, it was possible to produce high SSA Si–C–N hybrid materials, which exhibited excellent adsorption performance towards dye pollutants.
In this contribution, the hierarchically macro/micro-porous Si–C–N hybrid materials were prepared by pyrolysis of polysilazane (PSZ) at a low temperature using sacrificial fillers of polydivinylbenzene microspheres (PDVB) as schematically illustrated in Figure 1. Interestingly, the porous hybrid material exhibited highly efficient and totally selective adsorption properties for triphenylmethane dyes. Thus, the Si–C–N hybrid material, which can be mass-produced at a low cost, possesses promising potential as a novel adsorbent for the separation of organic dyes illustrated in Figure 2 (methyl blue (MB), acid fuchsin (AF), basic fuchsin (BF), methyl violet (MV), malachite green (MG), methyl orange (MO), congro red (CR) and methyl red (MR)) from aqueous solution.

Schematic illustration of preparation of hierarchically porous silicon–carbon–nitrogen hybrid materials from a pyrolysis of polysilazane and nickelocene by using polydivinylbenzene (PDVB) microspheres.

The molecular structures of representative dyes, i.e. methyl blue (MB), acid fuchsin (AF), malachite green (MG), methyl violet (MV), methyl red (MR), methyl orange (MO) and congo red (CR).
Results
Pyrolysis Behavior of Polymeric Precursors
For the preparation of macro/micro-porous hybrid Si–C–N materials, the linear PSZ composed of vinyl groups and silicon hydrogen bonds (Si–H) was employed because it was one of the low-cost and general polymeric precursors for PDCs. The molecular structure of PSZ was characterized by 1H NMR and gel permeation chromatography as presented in Figures S1–2 in Supporting Information. Similar to the well-known HT-180035, the molar ratio of silicon hydrogen bonds and vinyl groups on PSZ mainchain is 4:1 with a Mn of 800 g·mol−1.
Figure 3 shows the pyrolysis behaviors of P1 (pure PDVB sacrificial filler), P2 (NiCp2 containing polysilazane) and P3 (polysilazane with NiCp2 and sacrificial filler) under nitrogen by simultaneous TGA and DSC. From the TGA curves in Figure 2a, the PDVB undergoes a dramatic thermolytic degradation in the temperature range from 430 to 500°C, in correspondence with a significant exothermic peak as presented in DSC curve (Figure 3b). The weight loss is about 80% at 1,000°C, indicating that the cross-linked PDVB microspheres retain a few residues after pyrolysis. Subsequently, for the P2 (PSZ:NiCp2 = 1:0.04), a weight loss of 15% is observed within the temperature range from 450 to 800°C. Above 800°C, the TGA curve remains almost constant until 1,200°C. The sharp endothermic peak at around 170°C is related to the cross-linking reaction involving radical polymerization of vinyl groups and hydrosilylation between vinyl group and silicon hydrogen bond36. In terms of P3 (PSZ:PDVB:NiCp2 = 1:1:0.04), a remarkable weight loss is observed from 430 to 800°C, which is attributed to the decomposition of PDVB and chain scission of PSZ detected by simultaneous mass spectrometry.

Thermograms of P1 (pure sacrificial filler), P2 (NiCp2 containing polysilazane) and P3 (polysilazane containing NiCp2 and sacrificial filler) obtained from simultaneous analysis by TGA/DSC under an argon atmosphere at a scanning rate of 10 K min−1, (a) TGA curves, (b) DSC curves.
Figure 4 shows the analysis of simultaneous TGA and mass spectrometry of P3. For the comparison, the analysis was also performed for P0 (native polysilazane), P1 (pure sacrificial filler) and P2 (NiCp2 containing polysilazane) as presented in Figure S3. There exist two peaks for P3, i.e., peaks maximum at 430 and 600°C. According to the thermalgrams of P1 and P2, the first set of peaks for P3 are mainly corresponding to the decomposition products of PDVB (H2, m/z = 2, hydrocarbons CHx, x = 1–4, m/z = 14–16, NH3, m/z = 17, CHxCHy, m/z = 25, 26 and 28) as well as other oligomer fragments (m/z = 38–74). However, another group of peaks is mainly attributed to cleavage of PSZ (H2, m/z = 2, hydrocarbons CHx, x = 1–4, m/z = 132–16, NH3, m/z = 17, CHxCHy, m/z = 25, 26 and 28).

The simultaneous TGA and mass spectrometry of P3 (PSZ:PDVB:NiCp2 = 1:1:4).
From the thermogram in Figure S3c for P2, the evolution of CH4 or hydrocarbons in the temperature range of 400–900°C is ascribed to the decomposition of Si–CH3 groups and cleavage of C–C bonds. The H2 evolution is ascribed to the dehydrocoupling reaction of Si–H/Si–H or Si–H/–NH– groups. Besides, the emission of NH3 is attributed to transamination reactions. Notably, for P2 and P3 containing NiCp2, the evolution of NH3 in correspondence with transamination reactions becomes weaker, even cannot be detected in comparison to the native PSZ in Figure S3a. Thus, the incorporation of nickel reduced the production of NH3 from the transamination reactions and more elements are reserved, which is in agreement with the TGA curve shown in Figure 3a.
Microstructures of Hybrid Materials
The powder XRD patterns for porous hybrid materials (PSZ:PDVB:NiCp2 = 1:1:0.04) pyrolyzed at different temperature are displayed in Figure 5a. Pyrolysis at low temperature (600 and 750°C) leads to materials with complete amorphous structures. When the pyrolysis temperature is increased to 1,100°C, distinct crystalline peak is still not observed, indicating the good structural stability of porous Si–C–N materials. The broad diffraction peak at 2θ = 26° indicates the presence of numerous free carbons derived from the residues of PDVB and PSZ. A main feature of PDCs is the possibility to incorporate free carbon into the matrix, which is favorable for the structural stability against crystallization and high-temperature resistance to oxidation37. For example, Mera et al.38 reported that the crystallization depended on the carbon content for different precursors and the sample with the highest carbon content was less likely to form the β-SiC phase. Herein, it could be concluded that although the incorporation of Ni was helpful for the formation of crystalline phase, the presence of carbon could render the amorphous characteristic at higher temperatures39. Besides, the addition of nickel could catalyze the formation of conjugated carbons (C = C, sp2) similar to polyaromatic-like conjugated structure36,40, which might be helpful for the interaction between the porous Si–C–N material and the organic dyes.

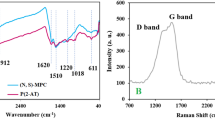
(a) Powder XRD patterns of the Si–C–N porous materials obtained from P3 (PSZ:PDVB:NiCp2 = 1:1:4) at 600, 750 and 1100°C, (b) Raman spectrum of Si–C–N porous materials obtained from P1 (PSZ:PDVB:NiCp2 = :2:0.04) pyrolyzed at 600°C.
Raman spectrum is one of powerful tools to further characterize free carbons. For the carbons with low crystallinity, Raman spectra mainly consist of a strong “sp3-like”resonant contribution and sp2-like doublet41. From Figure 5b, the C1 exhibits a broad peak at 1334 cm−1 as well as a sharp absorption peak at 1600 cm−1, corresponding to carbon vibration of the so called D and G band, respectively. The D band at 1334 cm−1 originates from sp3-bonded carbons and the G band at 1600 cm−1 is attributed to the E2g vibration mode of all the sp2 bonds of graphite42. Thus the Roman spectra clearly give a verification of sp2-bonded carbons in Si-C-N hybrid materials.
Composition of Hybrid Materials
The detailed structure of porous Si–C–N hybrid material (PSZ:PDVB:NiCp2 = 1:2:0.04) pyrolyzed at 600°C was evaluated by XPS as presented in Figure 6 and the according atomic composition was listed in Table 1. Obviously, the material is mainly composed of silicon, carbon, nitrogen and nickel elements, although the peak corresponding to Ni2p is not observed because of its lower content. The Si2p peak is deconvoluted into the C–Si–O, N–Si–N, N–Si–O and Si–Ox peaks43,44. The C1s peak is decomposed into the C–Si, C = C, C–C, C–O–H and C = O peaks45,46,47 and the N1s peak is deconvoluted into the Si–N–C, Si–Nx and Si–N–O peaks48. The excess content of oxygen in the 5–10 nm surface of porous material measured by XPS is mainly due to the O2 absorption by powder sample before measurement. Simultaneously, a little oxygen might be obtained from the polysilazane precursors employed in the preparation.

XPS spectra of porous Si–C–N hybrid material from precursor (PSZ:PDVB:NiCp2 = 1:2:0.04) pyrolyzed at 600°C (C1).
Hierarchical Pores of Hybrid Materials
For the preparation of porous material by sacrifice filler template, the porous structure is in correspondence with the template size. Furthermore, the content of template material also has a considerable influence on the subsequent structures in terms of interconnectivity, porosity and wall thickness. The pyrolyzed porous structures were obtained by PSZ precursors with varied percentage of PDVB. The pyrolysis temperature was maintained at 750°C. The SEM images in Figure 7 exhibit that the porous materials possess an expected regular morphology comprised of pores. It can be clearly seen that the isolated pores are distributed uniformly in matrix when the content of filler template is relatively low. When the content of microsphere fillers is increased, the distance between the isolated pores becomes smaller and the non-contact microspheres tend to come in contact with each other to form a highly porous component. Furthermore, when the content of microsphere fillers is increased to 200%, the interconnected cellular chunks are produced and the volume of voids in the matrix increases continuously.

SEM images of Si–C–N porous materials from precursors with a PSZ:PDVB:NiCp2 of (a) 1:0.3:0.04, (b) 1:0.5:0.04, (c) 1:1:0.04, (d) 1:2:0.04.
When the content of PDVB was relatively low, the PSZ tended to form the continuous phase and the microspheres were separately dispersed in the matrix to form the morphology similar to sea–island structure, where the size of pores in matrix was mainly 1 μm similar to the sacrificial filler. However, with increased amount of PDVB, more and more PDVB microspheres acquired access to make a contact with each other to form a sea–sea structure. In this case, the voids between microspheres were filled by PSZ and pyrolysis fragments of PSZ were dispersed uniformly to constitute the interconnected cellular structure. Therefore, the mass transfer resistance caused by micropores during the diffusion process could be significantly reduced by the presence of interconnected macropores.
It is well-known that the maximum pyrolysis temperature plays a critical role in determining the nature of the resultant PDCs, in particular, the SSA and pore size distribution49. In order to investigate the influence of pyrolysis temperature on microstructure of the materials, two types of samples under different pyrolysis condition were prepared to determine the specific surface area by N2 absorption–desorption measurement, i.e. C1 (PSZ:PDVB:NiCp2 = 1:2:0.04, 600°C) and C2 (PSZ:PDVB:NiCp2 = 1:2:0.04, 750°C). As shown in Figure 8a, the adsorption–desorption curve of C1 demonstrates a steep increase at very low relative pressures, which is a typical characteristic of microporous material with type I isotherm. Subsequently, a slow increase in the adsorption amount occurs during the relative pressure range of 0.4–0.8, indicating the presence of a few mesopores. Thus, the adsorption can be considered as multilayer adsorption and capillary condensation in these mesopores by nitrogen molecules when P/P0 is below and above 0.4, respectively. The calculated BET surface area and pore volume for C1 are 251.6 m2·g−1 and 0.09 cm3·g−1, respectively.

Nitrogen adsorption–desorption isotherms and the corresponding pore size distribution (PSD) calculated using adsorption branch of the BJH algorithm for (a) C1 and (b) C2.
On the contrary, for C2 pyrolyzed at 750°C, the nitrogen adsorption–desorption isotherm changes from type I to type II isotherm. As shown in Figure 8b, almost no adsorption occurs at the low pressure range, indicating the disappearance of numerous micropores. With the increased P/P0, the amount of nitrogen adsorbed increases very slowly. Moreover, the adsorption–desorption curves coincide well and almost no hysteresis loop can be observed, implying the absence of mesopores. The calculated BET surface area for C2 reduces drastically to 3.2 m2·g−1 in comparison to C1. Moreover, for both C1 and C2, the adsorption capacity still increases when the relative pressure is above 0.8, indicating the presence of large pores produced by the pyrolysis of PDVB. According to the literature50, the polymer to ceramic transformation, in general, occurs in the temperature range of 500–800°C accompanied by shrinkage and density increase. The TGA and mass spectrometry curve of precursor (PSZ:NiCp2 = 1:0.04) shown in Figure 4 displays that the ion current density reaches maximum at about 600°C confirmed by the decomposed organic moieties of the preceramic polymer (mainly CHx and CHxCHy), which contributes to numerous micropores in the matrix. However, the build-up of porosity is transient because it can be eliminated at an elevated temperature to achieve the ceramization process51. The BET results in combination with those from SEM analyses clearly indicate the presence of hierarchical macro/micropore structures in Si–C–N material at a low pyrolysis temperature of 600°C. Herein, the C1 with high BET surface area was selected as the target material to investigate the adsorption performance for organic dyes.
Adsorption Kinetics
From the viewpoint of adsorption kinetics, the adsorption is a heterogeneous process that involves physical, chemical and electrostatic interactions. The adsorption rate of dyes depends on the contact time of the solid and liquid as well as on diffusion process. The temporal evolution of UV spectra for two dyes of MB and AF on C1 dyes is presented in Figure 9 and the adsorption kinetic curves are presented in Figure 10. The C1 exhibits an excellent adsorption performance for two dyes. In general, the removal efficiency of 83.5% and 70.1% can be achieved within the first 60 min for MB and AF, respectively, with the initial concentration of 200 mg·L−1. The adsorption removal of two dyes was rapid at the initial period (10 min) and then became slow with the increased contact time until equilibrium. The removal rate of pollutants is initially rapid, however, it gradually decreases with increased time until it reaches equilibrium. This phenomenon is ascribed to the fact that a large number of vacant adsorption sites on C1 are available at the initial adsorption stage and after a lapse of time, the remaining vacant sites are difficult to occupy because of repulsive forces between the dye solute molecules on the solid and bulk phases52.

The temporal evolution of UV spectral of two dye solution in the presence of Si–C–N porous material (a) MB (b) AF.

(a) Adsorption kinetics curves and (b) pseudo-second-order kinetic plots for the adsorption of two dyes on C1 (initial concentration: 200 mg L−1 for MB and AF, dosage of C1: 666 mg L−1).
To study the transient adsorption behavior of dyes, two common chemical reaction kinetic models, i.e., pseudo-first-order (PF) and pseudo-second-order (PS) kinetic models, were used to analyze the adsorption process. The linearized pseudo-first-order kinetic model is expressed as follows53.

where Qe and Qt are the amounts of dye adsorbed (mg·g−1) at an equilibrium and time t (min), respectively. The K1 is the rate constant of the pseudo-first-order kinetic model (1/min). The linear form of pseudo-second-order kinetic model is given by the following equation.

where K2 (g·mg−1 ·min−1) is the rate constant for the pseudo-second-order reaction. Figure 10b shows the fitting data to the pseudo-second-order kinetic model. Accordingly, the kinetic parameters and the correlation coefficients R2 are determined by nonlinear regression and are listed in Table 2. The value of R2 clearly indicate that pseudo-second-order kinetic model achieves a better fitting of the experimental data for two dyes and the calculated Qe values (Qe,cal) are close to the experimental values (Qe,exp). Therefore, the pseudo-first-second kinetic model is more appropriate to characterize the adsorption behaviors of two dyes onto C1 compared to the pseudo-first-order model (see Figure S4) and the adsorption rate depends on the amount of solute at equilibrium54.
To obtain a deep understanding on the characteristics of the adsorption process, the pore-diffusion (intraparticle diffusion) model presented by Webber was also applied to fit the experimental data55. The intraparticle diffusion model can be expressed by the following equation.

where Kt is the intraparticle diffusion rate constant (g·mg−1·min0.5) and C is a constant providing the information about the thickness of the boundary layer. In Figure 11, the plots of Qt vs. t1/2 show at least two linear segments in correspondence with different stages in adsorption. For two linear plots, the Qt vs. t1/2 have intercept value far from zero, which indicates that intraparticle diffusion is not the rate-controlling step of the adsorption process and the external mass transfer may be significant in the rate-controlling step. The first segment is attributed to the outer diffusion due to the mass transfer from the dye solution to the outer surface of the adsorbent (film diffusion or boundary layer diffusion). The second portion indicates a steady adsorption step, corresponding to the diffusion of adsorbate from the adsorbent exterior to the pores or capillaries of the adsorbent internal structure (inner diffusion)56. Thus, the presence of multilinearity and the boundary layer thickness indicate that the adsorption of two dyes on C1 is a complex process and both the surface adsorption and intraparticle diffusion occur during the adsorption process57.

Pore-diffusion model plots for the adsorption of MB and AF on C1.
Adsorption Isotherm
Figure 12a shows the adsorption isotherms of the two dyes on C1 at 25°C. The results reveal that the porous material exhibits excellent adsorption ability toward two dyes. For example, the removal percentage can be as high as 99.2% and 97.2% when the initial concentrations were 300 mg L−1 for MB and 200 mg·L−1 for AF, respectively. In the low concentration range, the equilibrium adsorption capacities for all the dyes increase linearly with the increased initial concentrations. However, the linear growth of the adsorption capacities is retarded at higher initial concentrations because numerous dye molecules found difficulty in accessing vacant sites of the adsorbent, most of which have been occupied. The isotherms are all L-type, which reveals that the porous material has high affinity toward these dyes.

(a) Adsorption isotherms of two dyes on C1 and (b) Fitting plots of Langmuir isotherm models for two dyes on C1.
The Langmuir and Freundlich isotherm models were adopted to fit the isotherm data and the fitting parameters are listed in Table 3. The Langmuir model is based on the assumption that the adsorption process is a monolayer adsorption on a homogeneous adsorbent surface58. These binding sites are finite and have the same affinity for adsorbate. Besides, there is no interaction between adsorbed molecules. The linearized Langmuir equation is expressed as follows.

where Ce (mg·L−1) is the equilibrium dye concentration in liquid phase, Qe (mg·g−1) is the equilibrium adsorption capacity, Qm (mg·g−1) is maximum adsorption capacity and KL (L·mg−1) is the affinity parameter. The fitting experimental data of Langmuir isotherm model are shown in Figure 12b indicating that the fitting results give high linearity with correlation coefficient of 0.99975 for MB and 0.99984 for AF. Moreover, it suggests that the adsorption of two dyes on C1 is monolayer in nature and the surface is homogeneous. The calculated maximum adsorption capacity according to Langmuir isotherm model is 1327.7 mg·g−1 for MB and 1084.5 mg·g−1 for AF. The adsorption capacity of two dyes onto C1 is much higher than that of many other adsorbents, such as magnetic chitosan grafted with graphene oxide and organo-bentonite (compared to adsorption for MB), modified cellulose and carbon nanotube-graphene hybrid aerogel (compared to adsorption for AF). The values indicating the comparison of adsorption capacities for MB and AF onto various adsorbents are listed in Table 4. Although the adsorption capacity for MB is not so high compared to barium phosphate nano-flake and β-cyclodextrin chitosan nanoparticles; the facile preparation process, good recyclability and low cost of Si–C–N hybrid materials are more favorable in wastewater treatment59,60,61,62,63,64,65,66.
In contrast, the Freundlich isotherm model is, in general, used to describe the adsorption on a heterogeneous surface with interaction between adsorbed molecules and is not restricted to the formation of a monolayer67. The linearized expression of this model is given by equation (7).

where Qe is the amount of dye adsorbed per gram of adsorbent (mg·g−1), Ce is the equilibrium dye concentration in solution (mg·L−1), KF ((mg/g)·(L/mg)−1) is the Freundlich constant and 1/n is the heterogeneity factor. The fitting plots of Freundlich isotherm model for the absorption of dyes on C1 are shown in Figure S5. The Freundlich model fails to describe the experimental data, which indicates the presence of homogeneous adsorption sites on the porous Si–C–N hybrid material.
Besides, the thermodynamic parameters were calculated to predict the process of adsorption by free energy of adsorption (ΔG0), which is expressed as follows68.

where K0 is the equilibrium constant at a certain temperature and R is the gas constant. For an adsorption reaction, K0 is defined as follows.

where as is the activity of adsorbed dye, ae is the activity of dye in solution at equilibrium, Qe is the amount of dye adsorbed by per unit mass of adsorbent (mg·g−1), vs is the activity coefficient of the adsorbed dye and ve is the activity coefficient of dye in solution. The concentration of dye in the solution decreases and reaches zero; therefore, K0 can be obtained by plotting Ln(Qe/Ce) vs. Qe and extrapolating Qe to zero, as shown in Figure 13. The value of R2 obtained from a linear regression analysis is 0.90267 and 0.86085 for MB and AF, respectively. The value of K0 from the straight line intercept with the vertical axis is 6.09 for MB and 5.32 for AF. Accordingly, the ΔG0 values are −4.48 kJ·mol−1 for MB and −4.14 kJ·mol−1 for AF. It confirms that the adsorption of dyes onto C1 is spontaneous and thermodynamically favorable.

Plots of Ln(Qe/Ce) vs. Qe for the calculation of thermodynamic parameters.
Selective adsorption, regeneration and recycling properties
The selective adsorption experiments were performed on several other dyes with different molecular structures as presented in Figure 14 and Figure 15. The results exhibit that the porous Si–C–N hybrid material also shows totally selective adsorption property, i.e. high adsorption capacity for MG, MV and BF and no adsorption for MO, MR and CR. It seems to be that all dyes with bulky triphenyl structure can be absorbed by C1, however, the dyes with azo benzene (MO, MR, CR) cannot be absorbed. The repeatability is also important for an adsorbent for its feasible and practical application. The hybrid material could be easily regenerated by sintering because of its resistance to high temperature. In this study, MB was used as the model dye to investigate the recyclability of porous Si–C–N hybrid material. Figure 16 shows the removal percentages of MB in five recycle runs using regenerated C1. It was observed that the removal efficiency was 96.94 and 96.11% in the first two runs and decreased slightly to 94.33% and 93.56% in the fourth and fifth runs, respectively. The good repeatability was attributed to the stability of sp2 hybridized carbons as well as good structural stability of amorphous Si-C-N framework at the temperature below 600°C, which ensured enough adsorption sites for five recycling process to treat the low concentration dye solution (C0 = 200 mg·L−1). Moreover, Figure S6 shows the XPS analysis performed on the sample C1 recycled after five runs (C1-five). Compared to the original C1, the surface elemental composition of C1-five underwent some modifications. The results of the calculations revealed that the relative content of silicon and nitrogen decreased, however the relative content of carbon and oxygen increased dramatically compared to the composition of C1. However, the adsorption efficiency did not change significantly, indicating the good adsorption properties. Therefore, it was expected that this material was promising in the practical application of adsorption of dyes from wastewater.

Adsorption behaviors of C1 for different dyes (a) basic fuchsin, (b) methyl violet, (c) malachite green, (d) methyl orange, (e) congo red and (f) methyl red.

Schematic illustration of the dyes selective adsorption of Si–C–N porous materials.

Removal percentages of MB in five recycle runs using regenerated C1 (initial concentration: 200 mg·L−1).
Discussion
First, we conclude that the porous Si–C–N hybrid material selectively adsorb the dyes with triphenyl structure because of the Van der Waals force between the sp2-hybridized carbon domain in adsorbent (proved by Raman spectrum and FTIR) and the triphenyl structure of dyes. Herein, the semiemperical quantumchemical method AM1 (Austin Model #1) was adopted to determine Van der Waals forces between sp2-hybridized carbon domains in Si–C–N hybrid material and dyes although it seems rough for the quantitative canculation69. According to the results of AM1 modeling in Figure 17 and Figure S7, the atomic distance between sp2 carbon clusters of porous Si–C–N hybrid material and MB is between 3.84 to 4.49Å, which is within the typical range of Van der Waals distance (3–4Å). However for MO, the atomic distance between carbon clusters and AF is between 5.50–5.87Å, which is beyond the typical range of Van der Waals distance. So, it is believed that the adsorption sites on porous Si–C–N hybrid material may be related to the presence of sp2carbons that are proved by Raman spectra in Figure 5b. Thus, the Van der Waals forces between carbon atoms (sp2) in porous hybrid material and the triphenyl structure of dyes may be the main driving force for the adsorption of MB, AF, MG, MV, or BF. On the other hand, the electrostatic interaction between the Si-NH-Si sites in porous Si–C–N hybrid material and amino groups in dyes should also be considered. The presence of -NH, -NH2, N+ in MB, NH and NH2 in AF, MG and MV, is beneficial to the electrostatic interaction to Si–C–N hybrid material. While the presence of electron-donating groups, such as -N = N- in MO, MR, CR, is hindered to the electrostatic interaction to Si–C–N hybrid material as illustrated in Figure 17.

The suggested selective adsorption mechanism, (a) the interaction between sp2 carbon domain and triphenyl structure of MB, (b) the interaction between sp2carbon domain and azo-benzene ring structure of MO calculated according to AM1, (c) suggested selective adsorption mechanism of dyes for porous Si–C–N hybrid material.
The FTIR spectroscopy and XPS analyses were performed to gain insights into the adsorption mechanism. The analyses of FTIR spectra of C1, MB/AF and MB/AF adsorbed C1 (MB-C1/AF-C1) are presented in Figure S8. For C1, the symmetric deformation vibration corresponding to Si–CH3 at 1268 cm−1 is still present. Similarly, the peak at 2924 cm−1 related to the C–H asymmetric stretching vibrations (C (sp3) –H band) is faintly visible, indicating the presence of saturated groups. An intense and broad peak at 3440 cm−1 indicates the retention of N–H bond which is consistent with the mass spectrometry analysis. Importantly, the broad peaks at 1624 and 1404 cm−1 are ascribed to the conjugated bands of free carbons (C = C, sp2), which is indicative of a polyaromatic-like conjugation structure. Besides, the intense peak at 1032 cm−1 is attributed to amorphous Si–NH–Si phase, which is also important for the adsorption of dyes. After absorption of MB on C1 (C1-MB), the characteristic peaks of MB, such as the ring stretch at 1577 cm−1 and three peaks at 1122, 1037 and 616 cm−1 corresponding to sulfonate groups, are recorded in the spectrum of the adsorptive adduct, indicating that MB has been anchored to the surface of C1. In the case of AF adsorbed C1 (C1-AF), the characteristic peaks of AF, such as the peaks at 1625 and 1584 cm−1, respectively, related to NH2 and aromatic rings of AF and the two peaks at 1037 and 629 cm−1 derived from sulfonate groups of AF, are recorded in the spectrum of the adsorptive adduct, which indicates that AF has been attached to the surface of C1. There are no other new peaks observed for C1-MB and C1-AF, thus, indicating that it is a physical adsorption. Figure 18 shows the nitrogen adsorption isotherms, demonstrating that the specific surface area of the porous material dramatically reduces to 3.88 m2·g−1 and 2.19 m2·g−1 for C1-MB and C1-AF, respectively, which indicates that most micropores have been filled with MB and AF molecules after the adsorption. Figure S9 exhibits that the carbon content of C1-MB and C1-AF increase significantly compared to C1 after adsorption, indicating the adsorption of a large number of dye molecules (MB and AF) on C1.

Nitrogen adsorption–desorption isotherms of (a) C1-MB and (b) C1-AF.
Especially, the MAS Solid-state NMR was employed to give an experimental evidence for the adsorption mechanism. From the 13C NMR spectra (Figure 19a), there is an intense peak in the range of 120-150 ppm for C1, which is assigned to sp2-hybridized carbon atoms of Si-C-N materials71,70. In addition, the peak at δ 1 ppm is ascribed to sp3-hybridized carbon atoms. For the pure MB, in addition to the broad signal in the range of δ 77–160 ppm assigned to the carbon atoms on the aromatic rings, a small peak at δ 178 ppm may be due to the carbonyl groups caused by air environment. For the C1 after adsorption of MB (C1-MB), the peak of sp2-hybridized carbon atoms at δ 120–150 ppm is significantly higher than that of sp3-hybridized carbon atoms at δ 1 ppm, indicating that numerous MB molecules have been adsorbed to the surface of porous C1. In order to have a clear study on the interaction of sp2-hybridized carbons between C1 and MB, the sp2-hybridization carbon atoms peaks of three samples were fitted using Gaussian peaks as shown in Figure 19b. Compared with pure C1 and MB, although no obvious chemical shift in the range of 120–150 ppm changes can be observed for C1-MB, the peak at δ 153.5 ppm derived from the central carbon atoms attached to triphenyl structure of MB was disappeared. It demonstrates that the chemical environmental of the carbon atoms attached to triphenyl structure of MB is changed because of the stack of MB molecules on surface of porous C1. Therefore, the chemical shift those carbons atoms changes towards high-field after adsorption. So the FTIR, XPS and MAS Solid-state NMR measurements mentioned above demonstrate that Van der Waals force between sp2-hybridized carbon atoms and electrostatic interaction might be the main reason for the highly effective and totally selective adsorption functionality of Si–C–N hybrid materials.

The MAS Solid-state 13C NMR spectra of MB, C1 and C1-MB (a) and the sp2-hybridization carbon atoms peaks fitted using Gaussian peaks (b).
Conclusions
The hierarchically porous Si–C–N hybrid material was prepared by the pyrolysis of polysilazane precursor using PDVB microspheres as sacrificial templates at a lower temperature. The porous Si–C–N hybrid material with high specific surface and interconnected macropores exhibited excellent removal capability for dyes with triphenyl structures due to the Van der Waals force between sp2-hybridized carbon domains or clusters in porous material and triphenyl structure of dyes. Besides, the hybrid material possessed the selective absorption nature toward dyes with triphenyl structure compared to those dyes with azo benzene structures. Langmuir isotherm model showed a better fit with adsorption data than Freundlich isotherm model and the calculated adsorption capacity were 1327.7 mg·g−1 for MB and 1084.5 mg·g−1 for AF. The adsorption capacity was much higher than that of several other reported adsorbents, such as magnetic chitosan grafted with graphene oxide and organo-bentonite (compared to adsorption for MB), modified cellulose and carbon nanotube-graphene hybrid aerogel (compared to adsorption for AF). This hierarchically macro/micro hybrid Si–C–N material obtained by a facile and low cost pyrolysis of commercial polymeric precursors exhibited a significant potential in removing organic pollutants from wastewater.
Methods
Materials
The PSZ (The Institute of Chemistry, Chinese Academy of Sciences) was used as-received without further purification. The molar ratio of vinyl groups and silicon hydrogen bonds in main chains was 1:4 according to the NMR integral. Nickelocene (NiCp2) was purchased from Strem Chemicals (China). Anhydrous tetrahydrofuran (THF) was freshly distilled under reflux using sodium/benzophenone prior to its use. PDVB microspheres of nominal size 1–1.5 μm were synthesized according to the method reported in the literature72. All the dyes (MB, AF, BF, MV, MG, MO, CR and MR) were supplied by Kermel Chemical Reagent Co., Ltd. (Tianjin, China).
Preparation of Hybrid Materials
The PSZ, PDVB microspheres and NiCp2 were mixed by standard Schlenk techniques because PSZ was relatively sensitive to moisture and oxygen. PSZ (2 g), PDVB (0.6 g) and NiCp2 (0.08 g) were dissolved in anhydrous THF (20 mL). The solution was stirred at room temperature for one day to obtain homogeneous suspension. Hybrid materials with different compositions were prepared by varying the ratio of PSZ, PDVB and NiCp2 (1:0.3:0.04, 1:0.5:0.04, 1:1:0.04, 1:2:0.04). Subsequently, the solvent was evaporated under high vacuum using liquid nitrogen trap to make the precursors available as dry powder. Then, the as-prepared dry green powders were transferred into the vertical tube furnace (GSL-1700X, Kejing New Mater. Ltd., China) for cross-linking and pyrolysis under argon atmosphere. Three pyrolysis profiles were developed as follows: first, the precursors were heated to 170°C for cross-linking (holding time: 1 h), pyrolyzed at 600°C for 4 h and then cooled down to ambient temperature. Second, the precursors were heated from room temperature to 170°C for cross-linking (holding time: 1 h), decomposed at 550°C for 2 h, pyrolyzed at 750°C for 4 h and then cooled down to ambient temperature. Third, the precursors were heated from room temperature to 170°C for cross-linking (holding time: 1 h), decomposed at 550°C for 2 h, pyrolzed at 1100°C for 4 h and then cooled down to ambient temperature. The heating and cooling rates for all the experiments were 5 K min−1.
Characterization
The pyrolysis process of precursors was investigated by thermogravimetric (TGA) and differential scanning calorimetry (DSC) analyses by a simultaneous thermoanalyzer STA 449 F3 coupled with a quadrupole mass spectrometer QMS 403 C Aëolos (Netzsch GmbH & Co., Selb, Germany) with a heating rate of 10 K min−1 under argon atmosphere (a gas flow of 50 mL min−1) in a temperature range between 40 and 1,300°C. Fourier Transform Infrared Spectroscopy (FTIR) was recorded on a Nicolet ISTO spectrometer (Varian, Palo Alto, California, USA) over a range from 4,000 to 400 cm−1. Raman spectroscopy study was carried out using a Raman Microprobe Instrument (Renishaw, UK) with 514.5 nm Ar+ laser excitation. The MAS Solid-state NMR measurement was performed on a NMR spectrometer (Bruker Anance III-400, Germany). The resonance for 13C was 79.46. All spectra were acquired using magic angle spinning (MAS 54.74) technique with a rotation frequency of 6 kHz for 13C. All samples were measured with a crosspolarization sequence (CP). The recycle delay used was 3 s for 13C and the acquisition time was 42.3 ms for 13C. The adamantine was used as chemical reference for 13C NMR (38.5 ppm from tetra-methylsilane). Powder X-ray diffraction (XRD) measurements were conducted on X′Pert Pro Powder diffractometer (Cu Kα radiation, 40 kV, 40 mA) (PANalytical B.V., Netherlands) using crushed ceramic powders. The X’Celerator scientific RTMS detection unit was used for detection. X-ray photoelectron spectroscopy (XPS) core level spectra were measured using a monochromatic Al K R X-ray source. The analyzer was operated at 23.5 eV pass energy and the analyzed area was 800 μm in diameter. Binding energies were referenced to the adventitious hydrocarbon C1s line at 285.0 eV and the curve fitting of the XPS spectra was performed using the least-squares method. The analysis by scanning electron microscopy (SEM) was performed by a VEGA3 XMH instrument (Tescan Co., Czech Republic). The samples were measured before sputtering a thin layer (1–2 nm) of gold. Pore volume and surface area analysis of the selected porous materials were carried out according to Brunauer–Emmett–Teller (BET) method by nitrogen adsorption–desorption isotherm analysis at −195.8°C (77.35 K, TriStar II 3020, Micromeritics Instrument Co., USA). The powdered samples were degassed at 150°C for 24 h prior to measurement. Specific surface area was calculated using the BET equation. Pore size distributions were derived from the desorption branch of the isotherms using Barrett–Joyner–Halenda (BJH) method.
Dye Adsorption Behavior
The batch mode adsorption was carried out to study the adsorption kinetics and adsorption capacities for dyes, including MB and AF. Dye concentration was determined by measuring at maximum UV absorbance (λmax = 590 nm for MB, λmax = 545 nm for AF). The calibration curves were plotted between absorbance and concentration of dyes to obtain the absorbance-concentration profile based on Beer–Lambert's law (see Table S1–S2 and Figure S10 in Supporting Information). For the kinetic study, solid adsorbent (20 mg) was dispersed in solution (30 mL) with the initial concentration of 200 mg·L−1 for MB and AF, respectively. The mixture was continuously allowed to stir in the magnetic stirrer with a speed of 300 rpm at a constant temperature of 25°C. At different time intervals, around 3 mL of suspension was extracted and centrifuged at 4,000 rpm for 5 min for subsequent analysis. For the equilibrium experiment, solid adsorbent (5 mg) was dispersed in dye solution (10 mL) with a certain initial concentration in the range of 300–1200 mg·L−1 for MB and 200–900 mg·L−1 for AF, respectively. The mixture was allowed to stir continuously for 24 h at ambient temperature until the equilibrium was reached. All the adsorption experiments were conducted twice and only the mean values were reported. The maximum deviation for the duplicates was usually less than 5%. The dye concentration in the clear supernatant was determined by a XinMao Ultraviolet–Visible (UV–Vis) spectrophotometer (UV-7504 PC). Besides, the dye solution with high concentration was measured by dilution. The adsorption percentage R (%) and the adsorption capacity Qe (mg·g−1) were calculated by eqs 8 and 9, respectively.


where C0 (mg L−1) is the initial concentration, Ce (mg L−1) is the equilibrium concentration after adsorption. V (L) is the solution volume and m (g) is the mass of the adsorbent. The molecular structures and characteristic properties of representative dyes investigated are shown in Figure 2 and listed in Table S3, respectively.
The Regeneration and Recycling
The recyclability of porous Si–C–N hybrid material was investigated by performing repeated adsorption and regeneration using MB as the model dye. During the adsorption step, porous Si–C–N material (20 mg) was added into MB aqueous solution (40 mL) with an initial concentration of 100 mg·L−1. The suspension was stirred at 300 rpm for 24 h and then the solid was separated via centrifugation and freeze-dried overnight. The absorbance in the supernatant was determined by UV spectrophotometer. Subsequently, the dried powder was sintered at 400°C for 2 h in vertical tube furnace under nitrogen atmosphere to remove the dye. Notably, there was a loss of about 20% porous material after sintering which was attributed to the mass loss caused by centrifugation. To eliminate the effect of mass loss, a few parallel batches of adsorption/regeneration were conducted at the same time and under the same conditions. After each run, the regenerated porous material from these parallel batches was combined and the same amount of adsorbent was extracted from this mixture for the next run.
References
Nghiem, Q. D., Asthana, A., Sung, I-K. & Kim, D-P. Fabrication of Porous SiC-Based Ceramic Microchannels via Pyrolysis of Templated Preceramic Polymers. J. Mater. Res. 21, 1543–1549 (2006).
Biasetto, L., Francis, A., Palade, P., Principi, G. & Colombo, P. Polymer-Derived Microcellular SiOC Foams with Magnetic Functionality. J. Mater. Sci. 43, 4119–4126 (2008).
Sung, I-K., Christian, I. K., Mitchell, M., Kim, D. P. & Kenis, P. J. A. Tailored Macroporous SiCN and SiC Structures for High-Temperature Fuel Reforming. Adv. Funct. Mater. 15, 1336–1342 (2005).
Yang, H., Deschatelets, P., Brittain, S. T. & Whitesides, G. M. Fabrication of High Performance Ceramic Microstructures from a Polymeric Precursor Using Soft Lithography. Adv. Mater. 13, 54–57 (2001).
Harshe, R., Balan, C. & Riedel, R. Amorphous Si(Al)OC Ceramic from Polysiloxanes: Bulk Ceramic Processing, Crystallization Behavior and Applications. J. Eur. Ceram. Soc. 24, 3471–3482 (2004).
Wideman, T. & Sneddon, L. G. Dipentylamine-Modified Polyborazylene: a New, Melt-Spinnable Polymeric Precursor to Boron Nitride Ceramic Fibers. Chem. Mater. 8, 3–5 (1996).
Guron, M. M. et al. Preceramic Polymer Blends as Precursors for Boron-Carbide/Silicon-Carbide Composite Ceramics and Ceramic Fibers. Chem. Mater. 21, 1708–1715 (2009).
Torrey, J. D. et al. Composite Polymer Derived Ceramic System for Oxidizing Environments. J. Mater. Sci. 41, 4617–4622 (2006).
Melcher, R., Cromme, P., Scheffler, M. & Greil, P. Centrifugal Casting of Thin-Walled Ceramic Tubes from Preceramic Polymers. J. Am. Ceram. Soc. 86, 1211–1213 (2003).
Colombo, P. & Bernardo, E. Macro-and Micro-Cellular Porous Ceramics from Preceramic Polymers. Compos. Sci. Technol. 63, 2353–2359 (2003).
Nedunchezhian, S., Sujith, R. & Kumar, R. Processing and Characterization of Polymer Precursor Derived Silicon Oxycarbide Ceramic Foams and Compacts. J. Adv. Ceram. 2, 318–324 (2013).
Prasad, R. M. et al. Thermal Decomposition of Carbon-Rich Polymer-Derived Silicon Carbonitrides Leading to Ceramics with High Specific Surface Area and Tunable Micro- and Mesoporosity. J. Eur. Ceram. Soc. 32, 477–484 (2012).
Nghiem, Q. D. & Kim, D. P. Direct Preparation of High Surface Area Mesoporous SiC-Based Ceramic by Pyrolysis of a Self-Assembled Polycarbosilane-Block-Polystyrene Diblock Copolymer. Chem. Mater. 20, 3735–3739 (2008).
Kleebe, H. J., Störmer, H., Trassl, S. & Ziegler, G. Thermal Stability of SiCN Ceramics Studied by Spectroscopy and Electron Microscopy. Appl. Organomet. Chem. 15, 858–866 (2001).
Bharadwaj, L., Fan, Y., Zhang, L., Jiang, D. & An, L. Oxidation Behavior of a Fully Dense Polymer-Derived Amorphous Silicon Carbonitride Ceramic. J. Am. Ceram. Soc. 87, 483–486 (2004).
Shah, S. R. & Raj, R. Mechanical Properties of a Fully Dense Polymer Derived Ceramic Made by a Novel Pressure Casting Process. Acta Mater. 50, 4093–4103 (2002).
Francis, A., Ionescu, E., Fasel, C. & Riedel, R. Crystallization Behavior and Controlling Mechanism of Iron-Containing Si-C-N Ceramics. Inorg. Chem. 48, 10078–10083 (2009).
Wilhelm, M., Soltmann, C., Koch, D. & Grathwohl, G. Ceramers-Functional Materials for Adsorption Techniques. J. Eur. Ceram. Soc. 25, 271–276 (2005).
Adam, M., Wilhelm, M. & Grathwohl, G. Polysiloxane Derived Hybrid Ceramics with Nanodispersed Pt. Micropor. Mesopor. Mater. 151, 195–200 (2012).
Wilhelm, M., Adam, M., Bäumer, M. & Grathwohl, G. Synthesis and Properties of Porous Hybrid Materials Containing Metallic Nanoparticles. Adv. Eng. Mater. 10, 241–245 (2008).
Sonstrom, P. et al. Colloidal Nanoparticles Embedded in Ceramers: toward Structurally Designed Catalysts. J. Phys. Chem. C. 114, 14224–14232 (2010).
Adam, M., Kocanis, S., Fey, T., Wilhelm, M. & Grathwohl, G. Hierarchically Ordered Foams Derived from Polysiloxanes with Catalytically Active Coatings. J. Eur. Ceram. Soc. 34, 1715–725 (2014).
Iqbal, M. J. & Ashiq, M. N. Adsorption of Dyes from Aqueous Solutions on Activated Charcoal. J. Hazard. Mater. 139, 57–66 (2007).
Garg, V. K., Amita, M., Kumar, R. & Gupta, R. Basic Dye (Methylene Blue) Removal from Simulated Wastewater by Adsorption Using Indian Rosewood Sawdust: a Ttimber Industry Waste. Dyes. Pigments. 63, 243–250 (2004).
Haque, E., Jun, J. W. & Jhung, S. H. Adsorptive Removal of Methyl Orange and Methylene Blue from Aqueous Solution with a Metal-Organic Framework Material, Iron Terephthalate (MOF-235). J. Hazard. Mater. 185, 507–511 (2011).
Robinson, T., Chandran, B. & Nigam, P. Removal of Dyes from a Synthetic Textile Dye Effluent by Biosorption on Apple Pomace and Wheat Straw. Water Res. 36, 2824–2830 (2002).
El-Gohary, F. & Tawfik, A. Decolorization and COD Reduction of Disperse and Reactive Dyes Wastewater Using Chemical-Coagulation Followed by Sequential Batch Reactor (SBR) Process. Desalination 249, 1159–1164 (2009).
Lachheb, H. et al. Photocatalytic Degradation of Various Types of Dyes (Alizarin S, Crocein Orange G, Methyl Red, Congo Red, Methylene Blue) in Water by UV-irradiated Titania. Appl. Catal. B: Environ. 39, 75–90 (2002).
He, C. & Hu, X. Anionic Dye Adsorption on Chemically Modified Ordered Mesoporous Carbons. Ind. Eng. Chem. Res. 50, 14070–14083 (2011).
Hameed, B. H. A Novel Sorbent for the Removal of Basic Dye from Aqueous Solution. J. Hazard. Mater. 166, 233–238 (2009).
Ho, Y. S., Chiu, W. T. & Wang, C. C. Regression Analysis for the Sorption Isotherms of Basic dyes on Sugarcane Dust. Bioresour. Technol. 96, 1285–1291 (2005).
Iram, M., Guo, C., Guan, Y., Ishfaq, A. & Liu, H. Adsorption and Magnetic Removal of Neutral Red Dye from Aqueous Solution Using Fe3O4 Hollow Nanospheres. J. Hazard. Mater. 181, 1039–1050 (2010).
Mohammadi, N., Khani, H., Gupta, V. K., Amereh, E. & Agarwal, S. Adsorption Process of Methyl Orange Dye onto Mesoporous Carbon Material-Kinetic and Thermodynamic Studies. J. Colloid Interface Sci. 362, 457–462 (2011).
Xu, J., Wang, L. & Zhu, Y. Decontamination of Bisphenol A from Aqueous Solution by Graphene Adsorption. Langmuir 28, 8418–8425 (2012).
Ziegler, G. et al. Synthesis, Microstructure and Properties of SiCN Ceramics Prepared from Tailored Polymers. Mater. Chem. Phys. 61, 55–63 (1999).
SeifollahiBazarjani, M. et al. Nanoporous Silicon Oxycarbonitride Ceramics Derived from Polysilazanes in Situ Modified with Nickel Nanoparticles. Chem. Mater. 23, 4112–4123 (2011).
Hurwitz, F. I., Heimann, P., Farmer, S. C. & Hembree Jr, D. M. Characterization of the Pyrolytic Conversion of Polysilsesquioxanes to Siliconoxycarbides. J. Mater. Sci. 28, 6622–6630 (1993).
Mera, G., Riedel, R., Poli, F. & Müller, K. Carbon-Rich SiCN Ceramics Derived from Phenyl-Containing Poly(silylcarbodiimides). J. Eur. Ceram. Soc. 29, 2873–2883 (2009).
Colombo, P., Mera, G., Riedel, R. & Sorarù, G. D. Polymer-Derived Ceramics: 40 Years of Research and Innovation in Advanced Ceramics. J. Am. Ceram. Soc. 93, 1805–1837 (2010).
Qi, X. et al. Magnetic Properties and Large-Scale Synthesis of Novel Carbon Nanocomposites via Benzene Decomposition over Ni Nanoparticles. J. Phys. Chem. C. 113, 2267–2272 (2009).
Karlin, S. & Colomban, P. Raman Study of the Chemical and Thermal Degradation of As-Received and Sol-Gel Embedded Nicalon and Hi-Nicalon SiC Fibres Used in Ceramic Matrix Composites. J. Raman Spectrosc. 28, 219–228 (1997).
Ferrari, A. C. & Robertson, J. Resonant Raman Spectroscopy of Disordered, Amorphous and Diamondlike Carbon. Phys. Rev. B. 64, 075414 (2001).
Waldrop, J. R., Grant, R. W., Wang, Y. C. & Davis, R. F. Metal Schottky barrier contacts to alpha 6H-SiC. J. Appl. Phys. 72, 4757 (1992).
Swain, B. P. The Analysis of Carbon Bonding Environment in HWCVD Deposited a-SiC:H Films by XPS and Raman Spectroscopy. Surf. Coat. Tech. 201, 1589–1593 (2006).
Saha, S. K., Howell, R. S. & Hatalis, M. K. Silicidation Reactions with Co-Ni Bilayers for Low Thermal Budget Microelectronic Applications. Thin Solid Films. 347, 278–283 (1999).
Huntley, D. R. The Mechanism of the Desulfurization of Benzenethiol by Nickel (110). J. Chem. Phys. 96, 4550–4558 (1992).
Rufael, T. S., Huntley, D. R., Mullins, D. R. & Gland, J. L. Adsorption and Reaction of Dimethyl Disulfide on the Ni (111) Surface. J. Phys. Chem. C. 102, 3431–3440 (1998).
Ingo, G. M., Zacchetti, N., Della Sala, D. & Coluzza, C. XPS Investigation on Amorphous Silion Nitride (a-SiNx) Chemical Structure. Surf. Interface Anal. 12, 323–324 (1988).
Schmidt, H., Koch, D., Grathwohl, G. & Colombo, P. Micro-/Macroporous Ceramics from Preceramic Precursors. J. Am. Ceram. Soc. 84, 2252–2255 (2001).
Kroke, E. et al. Silazane Derived Ceramics and Related Materials. Mat. Sci. Eng. R. 26, 97–199 (2000).
Wan, J., Gasch, M. J. & Mukherjee, A. K. In Situ Densification Behavior in the Pyrolysis Consolidation of Amorphous Si-N-C Bulk Ceramics from Polymer Precursors. J. Am. Ceram. Soc. 84, 2165–2169 (2001).
Ghaedi, M., Hassanzadeh, A. & Kokhdan, S. N. Multiwalled Carbon Nanotubes as Adsorbents for the Kinetic and Equilibrium Study of the Removal of Alizarin Red S and Morin. J. Chem. Eng. Data 56, 2511–2520 (2011).
Ho, Y. S. & McKay, G. Sorption of Dye from Aqueous Solution by Peat. Chem. Eng. J. 70, 115–124 (1998).
Ayad, M. M. & El-Nasr, A. A. Adsorption of Cationic Dye (Methylene Blue) from Water Using Polyaniline Nanotubes Base. J. Phys. Chem. C. 114, 14377–14383 (2010).
Weber, W. J. & Morris, J. C. Kinetics of Adsorption on Carbon from Solution. J. Sanit. Eng. Div. Am. Soc. Civ. Eng. 89, 31–60 (1963).
Ai, L., Zhang, C. & Meng, L. Adsorption of Methyl Orange from Aqueous Solution on Hydrothermal Synthesized Mg-Al Layered Double Hydroxide. J. Chem. Eng. Data 56, 4217–225 (2011).
Ghorai, S. et al. Enhanced Removal of Methylene Blue and Methyl Violet Dyes from Aqueous Solution Using a Nanocomposite of Hydrolyzed Polyacrylamide Grafted Xanthan Gum and Incorporated Nanosilica. ACS Appl. Mater. Interfaces 6, 4766–4777 (2014).
Langmuir, I. The Adsopption of Gases on Plane Surfaces of Glass, Mica and Platinum. J. Am. Chem. Soc. 40, 1361–1403 (1918).
Fan, L. et al. Fabrication of Novel Magnetic Chitosan Grafted with Graphene Oxide to Enhance Adsorption Properties for Methyl Blue. J. Hazard. Mater. 215, 272–279 (2012).
Shao, Y. et al. Application of Mn/MCM-41 as an Adsorbent to Remove Methyl Blue from Aqueous Solution. J. Colloid Interface Sci. 429, 25–33 (2014).
Hao, X. et al. Magnetic Field Assisted Adsorption of Methyl Blue onto Organo-Bentonite. Appl. Clay Sci. 55, 177–180 (2012).
Zhang, F. et al. Selective and Effective Adsorption of Methyl Blue by Barium Phosphate Nano-Flake. J. Colloid Interface Sci. 386, 277–284 (2012).
Fan, L. et al. Synthesis and Characterization of Magnetic β-Cyclodextrin-Chitosan Nanoparticles as Nano-Adsorbents for Removal of Methyl Blue. Int. J. Biol. Macromol. 50, 444–450 (2012).
Zhou, Y., Hu, X., Zhang, M., Zhuo, X. & Niu, J. Preparation and Characterization of Modified Cellulose for Adsorption of Cd(II), Hg(II) and Acid Fuchsin from Aqueous Solutions. Ind. Eng. Chem. Res. 52, 876–884 (2013).
Dutta, M. & Basu, J. K. Fixed-Bed Column Study for the Adsorptive Removal of Acid Fuchsin Using Carbon–Alumina Composite Pellet. Int. J. Environ. Sci. Tech. 11, 87–96 (2014).
Sui, Z., Meng, Q., Zhang, X., Ma, R. & Cao, B. Green Synthesis of Carbon Nanotube-Graphene Hybrid Aerogels and Their use as Versatile Agents for Water Purification. J. Mater. Chem. 22, 8767–8771 (2012).
Freundlich, H. Colloid and Capillary Chemistry; Methuen: London, 1926.
Ma, J. et al. Enhanced Adsorptive Removal of Methyl Orange and Methylene Blue from Aqueous Solution by Alkali-Activated Multiwalled Carbon Nanotubes. ACS Appl. Mater. Interfaces 4, 5749–5760 (2012).
Liu, Y. et al. An ONIOM Study of H2O Interacting with the C-Terminated Surface of Silicon Carbide. Chem. Phys. Lett. 501, 87–92 (2010).
Gerardin, C., Taulelle, F. & Livage, J. Pyrolysis of a Polyvinylsilazane,Polymeric Precursor for Silicon Carbonitride: Structural Investigation by 1H, 13C, 29Si, 15N and 14N Nuclear Magnetic Resonance. J. Chim. Phys. 89, 461–467 (1992).
Seitz, J., Bill, J., Egger, N. & Aldinger, F. Structural Investigations of Si/C/N-Ceramics from Polysilazane Precursors by Nuclear Magnetic Resonance. J. Eur. Ceram. Soc. 16, 885–91 (1996).
Bai, F., Yang, X. & Huang, W. Synthesis of Narrow or Monodisperse Poly (divinylbenzene) Microspheres by Distillation-Precipitation Polymerization. Macromolecules 37, 9746–9752 (2004).
Acknowledgements
This research is supported by the Natural Science Foundation of China (21174112) and the Program for New Century Excellent Talents of the Education Ministry of China (NCET-11-0817). J.K. acknowledges the support from and Aoxiang Younger Scholar Plan of Northwestern Polytechnical University. The help of Dr. G. Motz (CME, Universität Bayreuth) for the discussion is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
J.K. designed the experiments, analysed the data and led the project. L.M. and X.Z. executed the experiments and analysed the data. Y.T. analysed the data. K.S. performed the semiemperical quantumchemical calculation. L.M. and J.K. wrote the paper. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supporting Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Meng, L., Zhang, X., Tang, Y. et al. Hierarchically porous silicon–carbon–nitrogen hybrid materials towards highly efficient and selective adsorption of organic dyes. Sci Rep 5, 7910 (2015). https://doi.org/10.1038/srep07910
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07910
This article is cited by
-
Adsorption thermodynamics of cationic dye on hydrolysis lignin-acrylic acid adsorbent
Biomass Conversion and Biorefinery (2023)
-
Fast and effective removal of textile dyes from the wastewater using reusable porous nano-carbons: a study on adsorptive parameters and isotherms
Environmental Science and Pollution Research (2022)
-
Enhanced photocatalytic degradation of Reactive Red 120 dye under solar light using BiPO4@g-C3N4 nanocomposite photocatalyst
Environmental Science and Pollution Research (2022)
-
Fabricating porous monolithic ceramic materials via phase separations in solutions of poly(Vinyl alcohol) and sol nanoparticles
Journal of Sol-Gel Science and Technology (2022)
-
Single-source-precursor synthesis of porous W-containing SiC-based nanocomposites as hydrogen evolution reaction electrocatalysts
Journal of Advanced Ceramics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
