
Abstract
Integrins are heterodimeric membrane proteins that regulate essential processes: cell migration, cell growth, extracellular matrix assembly and tumor metastasis. Each integrin α or β subunit contains a large extracellular domain, a single transmembrane (TM) domain and a short cytoplasmic tail. The integrin TM domains are important for heterodimeric association and dissociation during the conversion from inactive to active states. Moreover, integrin clustering occurs by homo-oligomeric interactions between the TM helices. Here, the transmembrane and cytoplasmic (TMC) domains of integrin β1a were overexpressed and the protein was purified in detergent micelles and/or reconstituted in liposomes. To investigate the TM domain conformational properties of integrin β1a, 26 consecutive single cysteine mutants were generated for site-directed spin labeling and continuous-wave electron paramagnetic resonance (CW-EPR) mobility and accessibility analyses. The mobility analysis identified two integrin β1a-TM regions with different motional properties in micelles and a non-continuous integrin β1a-TM helix with high immobility in liposomes. The accessibility analysis verified the TM range (Val737-Lys752) of the integrin β1a-TMC in micelles. Further mobility and accessibility comparisons of the integrin β1a-TMC domains in micelles or liposomes identified distinctively different oligomeric states of integrin β1a-TM, namely a monomer embedded in detergent micelles and leucine-zipper-like homo-oligomeric clusters in liposomes.
Similar content being viewed by others


Introduction
Integrins constitute a large family of heterodimeric adhesion receptors that regulate essential processes associated with cell-cell and cell-matrix interactions such as cell migration, cell growth, extracellular matrix assembly and tumor metastasis1,2. Each integrin consists of an α and a β subunit, both of which contain a relatively large extracellular domain, a single transmembrane domain (TM) and a short cytoplasmic tail3. In humans, 18 α and 8 β subunits combine to form different integrins4. The TM domains of the integrin α and β subunits play critical roles in bidirectional signal transduction across the plasma membrane5,6,7. A series of mutational studies showed that a specific TM helix-helix packing in the integrin αIIbβ3 dimer represents the inactive state, whereas disruption of the inter-helical interaction activates signal transduction5,6. Specifically, the inactive integrin αIIbβ3 state is stabilized by the hydrophobic heterodimerization packing of the TM helices and electrostatic interactions in the TM and adjacent cytoplasmic regions, whereas integrin activation ensues from the separation of the TM domains7,8,9,10. Recent studies reported the formation of an active receptor cluster with inter-helical interactions between TM domains of homo-oligomeric integrins after ligand binding (Fig. 1a)11. The active integrin clusters were detected in many cell types and shown to localize to cell- extracellular matrix (ECM) contacts12. The integrin cluster forms the basis for cell-ECM adhesion complexes that transfer force between the cell and the ECM and facilitate intracellular signaling, leading to protein phosphorylation and cytoskeletal attachment12,13.

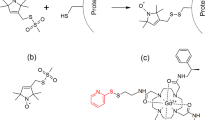
(a) Integrin architecture and potential mechanism for the activation and clustering of integrins. Specific contacts between the ectodomains, the TMH and cytoplasmic domains keep the integrin α (blue) and β (red) subunits proximal in the inactive state. Concomitant with activation, the transmembrane domains become separated and available for homomeric interactions. Homomeric association of the transmembrane domains leads to clustering on the cell surface11. (b) Primary sequence of the integrin β1a-TMC domain depicted with starting and ending residue numbers (V717 and K798). Residues selected for site-directed spin labeling one at a time (P731 to I756) are highlighted by framing in red. (c) SDS-PAGE analysis of integrin β1a-TMC in detergent micelles (left gel) and lipid liposomes (right gel). Lane 1 and lane 3 are molecular size markers. Lane 2 is integrin β1a variant L747C in detergent micelles while lane 4 is the variant L749C in lipid liposomes. L747C in detergent micelles migrated with a molecular weight of approximately 16 kDa as a monomer, while L749C in lipid liposomes migrated as multiple-bands of monomers, dimers and trimers. The gels have been run under the same experimental conditions and were cropped to improve the clarity and conciseness of the presentation. A vertical dividing line was drawn at the splice junction to show non-adjacent lanes in the SDS-PAGE for samples in lipid liposomes. Full-length gels are presented in Supplementary Figure S1.
Among the different β subunits, β1 integrin is the most abundantly expressed in adhesion-dependent cells14. The β1 integrin subunit can associate with at least 10 different α subunits to form distinct integrin heterodimers capable of interacting with various extracellular matrix molecules as well as some cell adhesion molecules14. A subgroup of collagen integrin receptors, namely α1/β1, α10/β1 and α11/β1, were found to mediate cell adhesion to the ECM15. Among them, integrin α1/β1 plays a role in fibrosis regulation16, cancer-related angiogenesis17, chronic inflammation18, the development of myopia19 and in the homing and differentiation of prostate cancer stem cells20. The transmembrane domain and cytoplasmic tails of most β subunits show significant sequence homology21,22. Several structural and functional studies have analyzed the transmembrane domain (or cytoplasmic tail) of several integrin proteins, such as integrin α1a, αIIb/β3, β1d and β3, using solution NMR methods23,24,25,26. However, no detailed reports focusing on the structural and biophysical characterization of integrin β1a have been published to date.
In the last decades, site-directed spin labeling (SDSL) electron paramagnetic resonance (EPR) spectroscopy has emerged as an effective method to study structural details, dynamics and conformational transitions of spin-labeled membrane proteins, especially in lipid bilayers27, or detergent micelles (a liposome mimic)28. In protein EPR studies, an unpaired electron is introduced by site-directed spin labeling of methanethiosulfonate (MTSL, R1) at a specific site through disulfide bond formation with a cysteine mutated from the native residue29. Acquired EPR signals of the introduced R1 groups can provide detailed information on side chain dynamics, polarity and topology profiles across the membrane lipid bilayer, as well as the distances between two spin labeled residues30. Unlike X-ray crystallography or solution NMR31,32, the high resolution three dimensional structure of membrane proteins is difficult to obtain by EPR. However, the combination of SDSL and EPR can provide dynamic and topological per residue information of membrane proteins in the presence of detergent micelles or liposomes. The higher gyromagnetic (γ) constant of electrons compared to that of nuclei (1H, 13C, 15N) guarantees an approximately 1000× sensitivity gain in EPR from that of NMR in protein dynamics studies, especially for eukaryotic membrane proteins, whose overexpression cannot be achieved in bacterial systems, or in isotope incorporation, which cannot be implemented in eukaryotic cell culture.
Here, the integrin β1a transmembrane domain and cytoplasmic tail (TMC) were overexpressed and purified in detergent micelles and reconstituted in liposomes. A series of cysteine mutations were introduced in the integrin β1a-transmembrane helix (TMH) domain for spin radical labeling (Fig. 1b). Then, SDSL-EPR was used to examine the structural and dynamic properties of the integrin β1a-TMH in detergent micelles or liposomes, using EPR line-shape analysis and accessibility estimation in the presence of paramagnetic regents (hydrophobic O2 or hydrophilic NiEDDA)33. The observed differences in dynamics or topology between the integrin β1a-TMC in detergent micelles and in liposomes suggested the presence of a monomeric integrin β1a-TMH in detergent micelles and homo-oligomers of the protein in liposomes, which could be attributed to leucine-zipper-like inter-helical interactions.
Results
The integrin β1a-TMC forms homo-oligomers in liposomes
The integrin β1a-TMC was overexpressed as inclusion bodies in E. coli. The protein was refolded and purified in buffers containing LDAO micelles after Ni2+-NTA affinity chromatography. Size exclusion chromatography was used for further protein purification. SDS-PAGE was used to further characterize protein purity and oligomeric states. A single band at approximately 16 kDa was observed in the sample of integrin β1a-TMC in LDAO micelles (Fig. 1c left). The observed molecular weight from SDS-PAGE was higher than the theoretical value of 10.3 kD (91 amino acids) for the integrin β1a-TMC. The slower migration rate in SDS-PAGE was possibly due to the strong interaction between the hydrophobic TMH and the detergents, resulting in a non-denatured state and a larger size than that of the pure protein34. To verify the lowest possible molecular weight of the band, the sample was boiled intensively before loading onto SDS-PAGE; however, no bands of smaller size (or faster migration rate) were observed (data not shown). These findings suggested that the protein existed as a monomer in micelles. More than three bands were observed for the sample containing the integrin β1a-TMC reconstituted in POPC/POPG liposomes (Fig. 1c right). These bands were 16, 26 and 37 kDa. The molecular weight differences derived from the SDS-PAGE bands were approximately 10 kDa and 11 kDa, which was consistent with the net protein size of the integrin β1a-TMC (10.3 kDa). This observation suggested that the 16 kDa band was the monomeric form of integrin β1a-TMC. At the same time, the detection of a series of protein bands by SDS-PAGE indicated the presence of homo-oligomeric integrin β1a-TMC and was evidence of strong inter-monomeric interactions (not disrupted in harsh detergent like SDS). The 16, 26 and 37 kDa bands possibly represented the monomer, dimer and trimer forms of the integrin β1a-TMC after reconstitution in liposomes. Several previous studies reported the presence of homo-oligomeric integrin protein clusters in the cell membrane, especially during cell metastasis35,36. However, the detailed mechanism driving the formation of integrin homo-oligomers remains to be elucidated.
Circular dichroism (CD) spectroscopy was used to estimate the secondary structure of the integrin β1a-TMC in detergent micelles (Supplementary. Fig. S2). No CD data of the protein in liposomes were obtained because of the large size of proteoliposomes and the consequent light scattering. Approximately 40% of the α-helical contents were derived from the CD data of the protein in LDAO micelles, which was consistent with the secondary structure prediction of the integrin β1a-TMC, namely a helical transmembrane domain and a random coil cytoplasmic tail. At the same time, high sequence homology was observed among integrins β1a, β1d and β3 (Supplementary. Fig. S3), especially the conserved residues in the transmembrane domains. The solution NMR structure of β1d and β3 demonstrated the helical secondary structure in the transmembrane domain23,26. Taken together, these findings suggested that the transmembrane domain of integrin β1a-TMC adopted the helical secondary structure.
CW-EPR spectra of the transmembrane residues of integrin β1a-TMC in LDAO micelles or POPC/POPG liposomes
After the mutation of cysteine residues in each site of the integrin β1a-TMH domain (from P731-I756), spin radical probe MTSL was performed in each cysteine site through disulfide bond formation of the purified integrin β1a-TMC in LDAO micelles and the protein was then reconstituted in POPC/POPG liposomes. CW-EPR spectra were acquired for a total of 52 samples (26 each) of MTSL-labeled integrin β1a-TMC in LDAO micelles (Fig. 2a) or POPC/POPG liposomes (Fig. 2b) at ambient temperature (298 K).

EPR spectra of spin-labeled integrin β1a-TMC scanning the transmembrane region P731–I756.
(a) EPR spectra of integrin β1a-TMC in LDAO micelles. (b) EPR spectra of integrin β1a-TMC reconstituted in POPC/POPG liposomes. Each spectrum was normalized by the height of the central peak. Asterisks indicate spectra that exhibit multiple components in lipid liposomes. “i” and “m” represent the “immobile” and “mobile” components, respectively, on the spectra of L749R1 and M755R1.
As shown in Figure 2a, all CW-EPR spectra of integrin β1a-TMC in LDAO micelles showed conventional three line spectra, with single values reflecting splitting of the hyperfine extrema 2A′zz (between mi = −1 and mi = +1), which indicated mono-dispersion of the conformational and motional properties of each residue in the integrin β1a-TMH in LDAO micelles. Overall broadening of spectral lines was observed in the CW-EPR spectra of integrin β1a-TMC in liposomes (Fig. 2b), indicating that the dynamic motion of the nitroxide spin-label was slower in liposomes than in micelles. Moreover, CW-EPR spectra with two populations of 2A′zz splitting were observed for some sites of integrin β1a-TMC in POPC/POPG liposomes at positions A746, L748, L749, I750, L753, L754 and M755 (* labeled in Figure 2b). This strongly indicated the presence of both immobilized (i) and mobilized (m) components at these sites. The two motional components detected in the EPR spectra suggested the presence of two different motional or conformational states of these residues of integrin β1a-TMC in liposomes.
Mobility analysis of the transmembrane residues of integrin β1a-TMC in LDAO micelles or POPC/POPG liposomes
The acquired EPR spectra permitted derivation of the rotational correlation time (τc) by empirical estimation37,38. The estimated τc values for several selected residues (G735R1, I740R1, G744R1, L749R1 and M755R1) of integrin β1a-TMC in detergent micelles and in liposomes are shown in Figure 3. For each residue, the rotational correlation time was longer in liposomes than in detergent micelles, consistent with the overall immobilization in liposomes, as revealed by spectral line broadening (Fig. 2). Furthermore, for residues of integrin β1a-TMC with two different dynamic components in liposomes (L749R1 and M755R1), the rotational correlation times corresponding to “mobile” and “immobile” components were quantified. The “immobile” component, with τc values of up to 20–40 nanoseconds, reflected a heavily restricted dynamic state of R1, which might have resulted from the interactions between the specific residue and other residues or lipid molecules.

Rotational correlation time (τc) estimation of integrin β1a-G735R1, -I740R1, -G744R1, -L749R1 and -M755R1 collected at 298K in LDAO micelles (a) and in POPC/POPG liposomes (b). Rotational correlation times are shown beside each spectrum. For integrin β1a-L749R1 and β1a-M755R1, two different motional components were observed. The “immobile” and “mobile” components were represented by “i” and “m”, respectively and the corresponding rotational correlation times (τc) are shown.
The inverse line width of the central mI = 0 resonance line (ΔH−1) and the inverse spectral second moment (<H2>−1) are normally used as comprehensive descriptors of nitroxide mobility and the corresponding structural features39, which may quantitatively reflect the motional modes of MTSL labeled sites in a semi-empirical way. Here, ΔH−1 and <H2>−1 were analyzed to further extract information on the mobility of the integrin β1a-TMC in detergent micelles and in liposomes (Fig. 4).

EPR Mobility analysis of integrin β1a-TMC in LDAO micelles and in POPC/POPG liposomes.
(a) Plot of the reciprocal of the central resonance line width ΔH−1 versus the residue number in micelles (black squares) and in liposomes (red dots). (b) Plot of the inverse spectral second moment of the EPR spectrum <H2>−1 versus the residue number in LDAO micelles (black squares) and in POPC/POPG liposomes (red dots). Sine waves were drawn to illustrate the observed periodicity of ΔH−1 and <H2>−1 indicative of a helical structure in (a) and (b). (c) Correlation diagram of <H2>−1 ~ ΔH−1 of integrin β1a-TMC in LDAO micelles. Two rectangular boxes were drawn to indicate the two different mobility groups. (d) Correlation diagram of <H2>−1 ~ ΔH−1 of integrin β1a-TMC in LDAO micelles (black dots) and in POPC/POPG liposomes (red triangles) were plotted in the same scale to show the difference between the two mediums. A blue box was drawn to include almost all residues while two red boxes were drawn as in (c).
For the samples of integrin β1a-TMC in LDAO micelles, the maximum and minimum motional τc values of the 26 MTSL labeled sites were 2.09 ns (L754R1) and 1.83 ns (G735R1), respectively. Theoretically, the periodic intensity oscillation of the ΔH−1 and <H2>−1 values in stretches of a protein segment reflect the secondary structure of the protein because of the heterogeneity of its local environment39. Here, no such obvious intensity oscillation periodicity was observed for the sample in LDAO micelles; however, the ΔH−1 (Fig. 4a, black squared data) decreased progressively from the N-terminus to the C-terminus residues of the transmembrane domain. The observed non-periodicity of the two mobility descriptors was consistent with a monomeric integrin β1a-TMC surrounded by detergent micelles, which provided only residue-detergent interactions and should be more homogeneous than the diverse inter-residue interactions in local tertiary cavities of soluble proteins40. Almost constant or slightly increasing <H2>−1 values were observed from P731R1 to G744R1, whereas decreasing <H2>−1 values after G744R1 indicated a higher degree of immobility for residues from L745R1 to I756R1 (Fig. 4b, black squared data). Splitting of the outer hyperfine extrema (2A′zz), which is also a measure mobility, showed similar pattern of change (Supplementary. Fig. S4, black squared data). The correlation diagram of ΔH−1 and <H2>−1 directly reflected the mobility distributions of residues in the integrin β1a-TMH domain. Two distinct regions were observed for samples in LDAO micelles (Fig. 4c): the residues from P735R1 to G744R1 were more mobile than the residues from L745R1 to I756R1, indicating the presence of two regions in the integrin β1a-TMH domain. The N-terminal segment was more mobile than the C-terminal segment, which could be attributed to the presence of three Gly and one Pro residues in the N-terminus compared to more Leu and Ile residues in the C-terminal region. Strong hydrophobic interactions between the Leu or Ile residues in the C-terminal region and the core of detergent micelles may have resulted in the relatively lower mobility.
For the sample of integrin β1a-TMC in POPC/POPG liposomes, large mobility variations were derived from the EPR spectra due to the presence of both mobile and immobile components of some residues in the TM domain (Fig. 2b). Both the ΔH−1 and <H2>−1 values derived from the EPR spectra showed high amplitude fluctuations (Fig. 4a and Fig. 4b, red dots data). The fit of the sine-wave, with a periodicity of 3.6 (α-helix), with the ΔH−1 and <H2>−1 values in two segments (P731R1-I743R1; L745R1-K752R1) indicated that the two segments adopted an α-helical secondary structure with the junction at G744. Periodic oscillation was also observed for 2A′zz values of integrin β1a-TMC in POPC/POPG liposomes (Supplementary. Fig. S4, red dots data). At the same time, the observed periodicity strongly implied the presence of inter-residue steric interactions in the sample of integrin β1a-TMC in POPC/POPG liposomes, which was consistent with the previously observed oligomeric bands in the SDS-PAGE analysis of integrin β1a-TMC in liposomes (Fig. 1c right). Under these conditions, the sites with lower mobility were mapped to V733R1, V737R1, I740R1, I743R1, A746R1, L749R1, L753R1 and L754R1. Among the residues with low ΔH−1 and <H2>−1 values, the L749R1, L753R1, L754R1 sites were found to have immobile components in the EPR spectra (Fig. 2b, marked with an asterisk). In the proposed homo-oligomers of integrin β1a-TMC in liposomes, these Leu sites could play major roles in inter-helical interactions, such as those found in the Leucine zipper domain41,42.
The ΔH−1 (Fig. 4a) and <H2>−1 (Fig. 4b) values and the ΔH−1 ~ <H2>−1 correlation diagram (Fig. 4d) were compared between samples of integrin β1a-TMC in LDAO micelles and in POPC/POPG liposomes. As described above, smaller values but much larger fluctuations of ΔH−1 and <H2>−1 were observed for the protein in liposomes than in detergent micelles, indicating decreased motion of the sites of the integrin β1a-TMC in liposomes. This was further verified by the correlation diagram in Figure 4d. The highly immobilized sites were likely due to the inter-reside interactions in the homo-oligomeric integrin β1a-TMC in liposomes compared to the residue-detergent interactions for monomeric integrin β1a-TMC in detergent micelles.
Accessibility analysis of the integrin β1a-TMH in LDAO micelles or POPC/POPG liposomes
For membrane protein topology analysis, the accessibility of protein side chains provides valuable information30. In EPR studies, the solvent accessibility of R1 in a membrane protein can be inferred from the collision frequency of the nitroxide radical probe with paramagnetic relaxing reagents in aqueous solution or in a hydrophobic environment. Normally, both molecular oxygen (O2) and 50 mM NiEDDA are applied for accessibility analysis of R133. The nonpolar molecule oxygen (O2) is more soluble in the hydrophobic interior of membranes or micelles than in aqueous solution. At the same time, the polar reagent NiEDDA shows high solubility in aqueous solution and low solubility in hydrophobic media. Therefore, the accessibility parameter (Π) is proportional to the collision frequency of nitroxide with either O2 or NiEDDA, which can be estimated using power saturation EPR techniques33. The conjugated accessibility analysis using ΠO2 and ΠNiEDDA can provide detailed information on protein-lipid interactions, especially in the junctional residues between the hydrophobic and hydrophilic regions. In the present study, both ΠO2 and ΠNiEDDA for R1 labeled in the transmembrane sites (P731–I756) of integrin β1a-TMC were collected for integrin β1a-TMH topology analysis using power saturation EPR methods. Figure 5 shows the recorded saturation curves for selected residues (V733R1, V741R1, A746R1 and M755R1) of the integrin β1a-TMC in detergent micelles and in liposomes after exposure to the paramagnetic relaxing reagent O2 or NiEDDA (N2 was used as a control). The ΠO2 and ΠNiEDDA values for each residue derived from fitting the saturation curves are shown in Figure 5. The derived ΠO2 and ΠNiEDDA values for all 26 residues of the integrin β1a-TMH in detergent micelles and in liposomes are shown in Figure 6.

EPR Power saturation curves for integrin β1a-V733R1, -V741R1, -A746R1 and -M755R1 in LDAO micelles (a) and in POPC/POPG liposomes (b). The power saturation curves were obtained under three conditions: molecular oxygen (O2), nitrogen (as a control) and the reagent NiEDDA (50 mM). ΠO2 and ΠNiEDDA values from fitting were shown.

EPR accessibility analysis of integrin β1a-TMC in LDAO micelles and in POPC/POPG liposomes using power saturation experiments.
Residue-specific environmental parameter profiles: NiEDDA accessibility parameter (ΠNiEDDA, panel a), oxygen accessibility parameter (ΠO2, panel b) and the corresponding depth parameter (Φ, panel c) in LDAO micelles (black squares) and in POPC/POPG liposomes (red dots) were plotted versus the residue number. Sine waves were drawn to illustrate the observed periodicity indicative of a helical structure in POPC/POPG liposomes.
For the sample of integrin β1a-TMC in LDAO micelles, the results of accessibility analysis of O2 and NiEDDA are shown in Figure 6a and 6b (black squared data), respectively. Correlational analysis of the conjugated ΠO2 and ΠNiEDDA values can provide topology information of the transmembrane sites labeled with spin radical R1. In Figure 6a, the ΠNiEDDA values decreased progressively from the V733R1 site to the A738R1 site, indicating a lower accessibility of these sites to the hydrophilic NiEDDA compound. Then, a series of low ΠNiEDDA values from A738R1 to W751R1, suggested that these sites were in a less hydrophilic region. The ΠNiEDDA values increased from the W751R1 site to the end point at I756R1. The ΠO2 values shown in Figure 6b also showed two levels of distribution, namely low values between P731R1 and V736R1 and between K752R1 and I756R1; and high values between A738R1 and W751R1. With the two conjugated properties of O2 and NiEDDA, the function Φ = ln[ΠO2/ΠNiEDDA] was normally applied to reduce the effect of steric factors and to provide a value proportional to the simple concentration ratio of the paramagnetic reagents in the immediate environment of R133. Thus, Φ could be at or near a local maximum for sites exposed to the hydrophobic interior, whereas low values were maintained at the interfacial regions proximal to the aqueous solvent. The calculated Φmicelle values for integrin β1a-TMC in LDAO micelles are shown in Figure 6c (black squared data) and the Φmicelle values showed a similar pattern to that of the ΠO2 values. Therefore, the Φmicelle values and the conjugated ΠO2 and ΠNiEDDA values of integrin β1a-TMC in LDAO micelles indicated a standard hydrophobic core and two hydrophilic ends of the spin radical labeled transmembrane sites. From the Φmicelle, ΠO2 and ΠNiEDDA values, the junction of the hydrophobic core and interfacial head group regions of LDAO micelles were mapped to around V737R1 and K752R1. However, in the report of glycosylation mapping studies of the integrin β1 subunit in microsomal membranes, the N-terminal membrane border resided approximately at P731, while the C-terminal membrane border resided approximately at I75643. In the present study, the correlational accessibility analysis using the ΠO2, ΠNiEDDA and Φmicelle values indicated that the hydrophobic core region was between V737R1 and K752R1, which was much shorter than the region defined in glycosylation mapping studies. Because of its small size, molecular oxygen (O2) can penetrate into the interior region of the detergent micelles; however, the glycosylation mapping assay can only access the sites in direct contact with the aqueous solvent and not even the interfacial regions defined by polar head groups of LDAO micelles. Therefore, the conjugated ΠO2 and ΠNiEDDA accessibility data provided a more accurate and clear topological analysis of the transmembrane sites of the integrin β1a-TMC in LDAO micelles.
For the sample of integrin β1a-TMC in POPC/POPG liposomes, the results of accessibility analysis of O2 and NiEDDA are shown in Figure 6a and 6b (red dots), respectively. Since detergent micelles are normally used to mimic the amphipathic liposomes (hydrophobic interior and hydrophilic exterior), we expected a similar ΠO2 and ΠNiEDDA value distribution of integrin β1a-TMC in POPC/POPG liposomes as that observed in detergent micelles. However, the ΠNiEDDA values were lower (approximately 0.025) (Fig. 6a, red dots), indicating low hydrophilic accessibility. The ΠO2 values were not in the high range, but showed large fluctuations from 0.1 to 0.5, with an average value of 0.3 (Fig. 6b, red dots). However, the derived Φliposome = ln[ΠO2/ ΠNiEDDA] was relatively high, although it showed large fluctuations. Therefore, the ΠNiEDDA, ΠO2 and Φliposome values indicated the relative hydrophobic regions along the primary amino acid sequence of the transmembrane domain of the integrin β1a-TMC in POPC/POPG liposomes. The fluctuations of the ΠO2 and Φliposome values were also fitted using the sine-wave with a period of 3.6, which strongly indicated an α-helical secondary structure (Fig. 6b, 6c). Considering the previous highly immobilized sites in the ΔH−1 − <H2>−1 diagram of the integrin β1a-TMC in POPC/POPG liposomes (Fig. 4d), it is likely that the transmembrane domain of the integrin β1a-TMC formed homo-oligomers through inter-helical interactions. Therefore, the observed low values of ΠNiEDDA may have been due to the low solubility of NiEDDA in the hydrophobic inter-helical contacts and the consequent homo-oligomeric clustering effects and not to the conventional hydrophobic interior environment of liposomes, which would result in a standard distribution as in detergent micelles. The proposed homo-oligomeric clustering of the integrin β1a-TMC was consistent with the observed multiple bands in the SDS-PAGE analysis (Fig. 1c right) of the protein in POPC/POPG liposomes.
Different oligomeric states of the integrin β1a-TMC in micelles or liposomes
As indicated above, the EPR mobility and accessibility analysis of the transmembrane domain of the integrin β1a-TMC in detergent micelles clearly demonstrated a transmembrane helix topology. Moreover, the lack of periodic fluctuation in mobility parameters (ΔH−1 and <H2>−1) and accessibility parameters (ΠNiEDDA, ΠO2 and the Πmicelle) indicated that there are only pure interactions between R1 in monomeric proteins and the hydrophobic core or interfacial regions of detergent micelles.
Detergent micelles are considered as good mimics of the native environment of lipid bilayers because detergent micelles can support the native stability, structure and functionality of membrane proteins44. Under this assumption, the mobility and accessibility data of the reconstituted integrin β1a-TMC in POPC/POPG liposomes might follow similar patterns as those obtained in LDAO micelles, provided the protein was also a monomer in liposomes. However, different mobility and accessibility patterns were observed for the integrin β1a-TMC in detergent micelles versus liposomes.
In POPC/POPG liposomes, a stronger overall immobilization of the integrin β1a-TMC was observed in liposomes than in LDAO micelles (Fig. 4d). Distinctive periodicity in the mobility parameters (ΔH−1 and <H2>−1) was also observed in liposomes (Fig. 4a, 4b) rather than in LDAO micelles, suggesting the existence of inter-residue tertiary interactions in the sample of integrin β1a-TMC in liposomes, in contrast to the interactions between detergent molecules and transmembrane residues of the monomeric integrin β1a-TMC in LDAO micelles.
The observed different accessibility patterns also suggested different oligomeric states of integrin β1a-TMC in liposomes than in detergent micelles. Overall, similar ascending and descending patterns were observed in detergent micelles and in liposomes, suggesting a similar topology or immersion depth of the transmembrane domain of the integrin β1a-TMC. However, the Φliposome showed a larger fluctuation amplitude than the Φmicelles, indicating the presence of hydrophobicity differences between the two samples. Through the accessibility data (including the ΠO2, ΠNiEDDA, Φmicelles), the center of the monomeric transmembrane domain was clearly identified for the integrin β1a-TMC in detergent micelles (black squares and line in Fig. 6). However, the center of the transmembrane domain was not clear for the clustered integrin β1a-TMC in liposomes (red dots and line in Fig. 6), which was due to the presence of protein-protein interactions (possibly of the leucine-zipper like hydrophobic interactions, Fig. 7b), besides the interactions between the side chains of transmembrane residues of integrin β1a-TMC and hydrophobic acyl-chain, hydrophilic glycerol skeleton, or polar head groups of liposomes. At the same time, very distinctive periodic patterns were observed for ΠNiEDDA, ΠO2 and Φliposome values in liposomes but not in detergent micelles (Fig. 6), suggesting the helical conformation (with periodicity of 3.6) and inter-helical interactions of the integrin β1a transmembrane domain in liposomes. The non-periodic patterns of the ΠNiEDDA, ΠO2 and Φmicelles values in LDAO were possibly due to the solely helix-detergent interactions. Furthermore, similarities in the phase of the period indicated that the O2 inaccessible residues, including I740R1, A746R1, L749R1 and L753R1 (Fig. 6b), were also the more immobilized residues (Fig. 2, 4a, 4b). Those O2 inaccessible and immobilized residues might interact with other residues, rather than directly with the hydrophobic tails of lipid molecules. If the inter-helical interactions occurred between two identical proteins, the Leu-Leu, Ala-Ala, Ile-Ile interaction pairs and the period numbers of 3 (749–746), 6(746–740) and 4 (753–749) demonstrated that those residues were lined on the same side of the inter-helical interaction (an ideal helix has a periodic pitch of 3.6 residue), forming a leucine-zipper pattern that is typical of the oligomerization behavior of the transmembrane domains of many membrane proteins41,45. A leucine-zipper-like motif was proposed to drive the strong self-association of the transmembrane domain of the discoidin family of receptor tyrosine kinases (DDR1 and DDR2)46 and the self-assembling erythropoietin receptor transmembrane segments47.

Schematic models of the integrin β1a-TMC in detergent micelles or in liposomes.
(a) The transmembrane domain of integrin β1a-TMC adopted a monomeric helix with a kink at residue G744. (b) Helical wheel diagram depicting the interface between the hydrophobic side chains of residues in the transmembrane domain (Ala746-Met755) of integrin β1a-TMC. Residues which might be involved in forming the hydrophobic interactions were colored in red. (c) Homo-oligomeric transmembrane domains of integrin β1a-TMC in liposomes. The clustering effect of transmembrane helices was represented by the formation of inter-helical dimers and trimers. The extracellular domain and cytoplasmic tail were eliminated for simplicity. Red areas represent the hydrophobic tail and blue areas are the hydrophilic head for the illustration of detergent and phospholipid molecules.
Discussion
The present EPR mobility and accessibility analysis indicated that the transmembrane domain of the integrin β1a-TMC forms a monomeric helix in LDAO micelles and clusters of homo-oligomeric helices of integrin β1a-TMC in POPC/POPG liposomes. These distinctive environmental influences imply that detergent micelles might not mimic lipid bilayers in high fidelity, especially because of their globular shape and spherical surface, which can only provide space for membrane proteins with a small number of helices. While liposomes provide space for the free lateral motion of membrane proteins, inter-helical interactions may drive the formation of integrin clusters. Tentative models of the integrin β1a-TMC in detergent micelles and liposomes are shown in Figure 7. Briefly, the integrin β1a-TMC adopted a single monomeric helical conformation with a non-helical secondary structure at site G744 (Fig. 7a), whereas the protein formed inter-helical clusters in liposomes (Fig. 7c) with undefined numbers of monomers, consistent with the multiple-bands observed in the SDS-PAGE analysis of samples from liposomes (Fig. 1c right). Residues showing low mobility and O2 accessibility, such as A746R1, L749R1, L753R1 and L754R1, might play an important role in the helix-helix interaction between the transmembrane domains of integrin β1a-TMC, in a way similar as Leucine zipper (Fig. 7b).
The kink G744 was derived from the observed mobility analysis data in detergent micelles. Although both the central line-width ΔH and the second moment <H2> were used to indicate mobility of the spin labeled site, the central line-width ΔH and the second moment <H2> are determined primarily by the degree of averaging of the anisotropic g tensors and the anisotropic hyperfine (A) tensor, respectively39. Moreover, for sites at which multiple populations of mobility states are observed, the central line-width ΔH is dominated by the most mobile component while the second moment <H2> is biased toward the immobilized component39. Therefore, with the drop of the second moment <H2> of residues after G744 in the transmembrane domain of integrin β1a-TMC, these residues were more immobilized, probably due to the strong stabilization interactions between hydrophobic side chains of the residues following G744 and the acyl-chain of detergents.
The lateral assembly of integrin clusters during cell adhesion, migration and signaling were found to be essential for cell survival and function in many cell types12. Homo- or hetero-oligomeric interactions between the transmembrane helices of integrin α or β subunits may play important roles in integrin activation and clustering48. In vitro studies have shown that the activation-induced transmembrane domain separation in integrins induces clustering, with the corresponding homo-oligomerization of the α or β transmembrane domains11,49,50. Homomeric interactions were observed for transmembrane helices of integrin αIIb in DPC micelles50, whereas no in vitro reports exist describing the homo-oligomeric integrin clusters in liposomes. In the present study, EPR mobility and accessibility analysis demonstrated that the transmembrane domain of the integrin β1a-TMC formed homo-oligomeric clusters in POPC/POPG liposomes resulting from hemophilic interactions between transmembrane helices, similar to the Leucine zipper conformation. Previous studies showed that the lipid bilayer could contribute to transmembrane helix oligomerization41,51. The present study demonstrated that phospholipid liposomes can induce and enhance the formation of integrin clusters, providing a more native-like environment for cluster formation in integrin proteins.
Methods
Construction of cysteine mutations in human integrin β1a-TMC
The wild-type oligonucleotides encoding the integrin β1a-TMC (V717-K798) were synthesized by Sangon (Shanghai). The cysteine at site 723 was first mutated to serine by site-directed mutagenesis using the PCR-based overlap extension method. The consequent cys-less sequence was then used as template for further mutagenesis. A total of 26 residues (from P731 to I756) were substituted with cysteine (Fig. 1b). The 26 single-cys mutants were introduced into the plasmid expression vector pET21b (Novagen), which carries a 6 × His-tag at the C-terminus. All mutant constructs were verified by DNA sequencing.
Protein expression and purification
Expression and purification of the integrin β1a-TMC were performed as described for integrin α1a-TMC in a previous report24. Briefly, integrin β1a-TMC expression was induced by addition of isopropyl-β-D-thio-galactoside (IPTG) to a final concentration of 0.8 mM in LB medium at a cell density of OD600 = 0.8. The inclusion body proteins were firstly solubilized in 0.5% SDS, then processed by detergent exchange to 0.2% LDAO on a Ni2+-NTA column. The proteins were further purified through size exclusion chromatography on a Superdex 200 10/300 GL column (GE healthcare) in a buffer consisting of 20 mM Tris, 100 mM NaCl, 0.2% LDAO, 2 mM DTT, pH 8.0. For proteins that were subsequently reconstituted into POPC/POPG liposomes, 0.2% LDAO was substituted by 0.2% (w/v) DM (n-decyl-β-D-maltopyranoside, Anatrace) during size exclusion chromatography. The concentration of the purified protein was determined by OD280 and the purity was analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). All mutants were at least 95% pure according to SDS-PAGE. All samples were boiled in SDS-PAGE loading buffer for 10 minutes to totally denature the protein before loading onto gels.
Site specific spin radical MTSL labeling
Prior to spin labeling, DTT was removed using a PD-10 gravity flow desalting column (GE Biosciences), eluting with binding buffer (20 mM Tris, 100 mM NaCl, pH 8.0) in the presence of 0.2% (w/v) LDAO. Single cysteine mutants were immediately reacted with a 10-fold molar excess of spin radical MTSL (R1: 1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrr-oline-3-methyl methanethiosulfonate, Toronto Research Chemicals, Ontario, Canada) at room temperature for 30 min and then at 4°C overnight. Excessive spin reagents were removed through gel filtration chromatography in binding buffer with 0.2% LDAO (0.2% DM for samples which were subsequently reconstituted into POPC/POPG liposome). For samples in LDAO micelles, Amicon Ultra-15 centrifugal filter units (Millipore) were used to concentrate the sample to a final concentration of approximately 200 uM. The spin-labeled samples in LDAO micelles were subsequently used for CW-EPR and power saturation EPR experiments. Labeling efficiency was determined by measuring the protein concentration and the spin concentration. As determined by double integration of the EPR spectra, the labeling efficiency for each mutant was beyond 90%. Spin labeled integrin β1a-TMC was designated by giving the single letter code for the original residue, the sequence number and the letter code “R1” for the introduced spin label (e.g. L742R1 means the R1 introduced at Leu742 site).
Reconstitution into POPC/POPG Liposomes
Powders of POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt), Avanti, Alabaster, AL) were mixed in chloroform (3:1 POPC:POPG molar ratio). The solution was gently dried under a nitrogen flow and then placed under a high vacuum overnight to further evaporate any residual solvent. The lipid film was rehydrated with binding buffer to yield a final concentration of approximately 4 mg/ml and dispersed through vigorous stirring followed by at least 10 rounds of freeze−thaw−sonication until a clear solution was obtained. The resulting solution was filtered 20 times through a 100 nm polycarbonate membrane filter (Whatman, Newton, MA) mounted on a mini-extruder. The spin-labeled integrin β1a-TMC (in binding buffer with 0.2% DM) and the extruded phospholipid liposomes were mixed to obtain a lipid/protein molar ratio of 200:1. Two cycles of freeze−thaw−sonication were performed to improve the even distribution of integrin β1a-TMC in liposomes. Membrane protein reconstitution was implemented by dialysis against 200-fold excessive binding buffer for a total of 48 h, with buffer changes every 12 h. After dialysis, the proteoliposome samples were pelleted down by ultracentrifugation at 300,000 g for 30 min. The pellet was then thoroughly re-suspended in binding buffer and extruded through the 100 nm filter again to obtain homogeneous protein-incorporated vesicles in the size range of 50–100 nm. The reconstituted spin-labeled samples in POPC/POPG liposomes were subsequently used for CW-EPR measurements and power saturation EPR experiments.
Circular dichroism (CD) spectroscopy analysis
Integrin β1a-L749R1 in LDAO micelles was diluted to 0.10 mM in 50 mM NaH2PO4–Na2HPO4 buffer, pH 8.0. CD measurements were performed on a Jasco-810 spectropolarimeter at 298 K. CD spectra were recorded over a wavelength range of 190–280 nm using a cuvette of 1-mm path length at a scanning speed of 20 nm/min and subjected to 10 scans. Acquired data were normalized by subtracting the baseline recorded for the buffer only and are shown in the supplementary data (Supplementary. Fig. S2). The acquired CD spectra of integrin β1a-L749R1 in LDAO micelles were used for secondary structure estimation using a curve-fitting method52. CD spectroscopy was not performed for samples in liposomes because of the strong perturbations in the CD signal due to depolarization caused by light scattering53,54.
Continuous wave EPR spectroscopy
CW-EPR spectroscopy was performed on a Bruker A300 spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany) at X-band (9.5 GHz) equipped with a high-sensitivity cavity (ER 4119HS, Bruker Biospin GmbH, Rheinstetten, Germany) at room temperature (298 K). Spectra were recorded at a microwave power of 2 mW over a scan width of 200 Gauss with a field modulation of 1 Gauss at a frequency of 100 kHz. Samples were placed in a glass capillary tube in a volume of approximately 20 μL. Data acquisition was performed 20–40 times to achieve a reasonable signal/noise ratio.
Rotational correlation time (τc) and mobility parameter calculation
The rotational correlation time (τc) of spin labeled integrin β1a-TMC in detergent micelles and in liposomes was estimated as described previously37,38. Briefly, continuous wave EPR spectra of the spin labeled protein in detergent micelles and in liposomes were acquired under two thermal conditions: at ambient temperature (298 K) and at its rigid limit (frozen solution at 150 K, data not shown). The rotational correlation time τc was evaluated from the expression,

where

ARzz is identical to the magnetic tensor Azz and A′zz is the generic room-temperature genera magnetic tensor37,38. The values of a = 8.52 × 10−10 sec and b = −1.16 × 10−10 sec are determined by evaluating the peak-to-peak derivative Lorentzian linewidth (δ) derived from the rigid limit EPR spectrum.
The inverse line width of the central mI = 0 resonance line (ΔH−1) and the inverse spectral second moment (<H2>−1) were analyzed to further extract information on the mobility of the integrin β1a-TMC in detergent micelles and in liposomes. The second moment of spectra was calculated to represent the spectral breadth:

which is defined based on the first moment <H> (the mean or geometrical center of the spectrum):

where B is the magnetic field and S(B) is the absorption spectrum of the spin-labeled protein39.
EPR power saturation studies and accessibility analysis
Power saturation experiments were performed on the same spectrometer coupled with an ER4123D CW resonator (Bruker BioSpin). Samples were loaded into gas permeable TPX capillary tubes with a total volume of 3–4 μL at a concentration of 50–100 μM. EPR data were collected using a modulation amplitude of 1 Gauss and a scan range of 15 Gauss. The range of the incident microwave power was 0.7 to 180 mW for power saturation experiments. Nitrogen was used as a control to purge oxygen and other paramagnetic relaxing reagents in the sample. The water-soluble paramagnetic reagent, Nickel(II)-EDDA complex (NiEDDA) was synthesized as previously described33. The power saturation curves were obtained for integrin β1a-TMC in LDAO micelles and POPC/POPG liposomes under three conditions: (1) equilibrated with the hydrophobic paramagnetic reagent 21% O2 (air), (2) equilibrated with nitrogen as a control and (3) equilibrated with nitrogen in the presence of the hydrophilic paramagnetic reagent NiEDDA (50 mM).
Power saturation curves were measured and the accessibility parameters ΠO2 and ΠNiEDDA were derived from fitting the saturation curves33. Briefly, the saturation curves were measured as the vertical peak-to-peak amplitude (A) of the first derivative Mi = 0 line as a function of incident microwave power (P)33. The data points were then fit using an R software script according to equation (5):

where I is a scaling factor, P1/2 is the power where the first derivative amplitude is reduced to half of its unsaturated value and ε is a measure of the homogeneity of saturation of the resonance line. The change in P1/2, ΔP1/2, is calculated as the difference in P1/2 values in the presence and absence of relaxing agent. The parameter ΔP1/2/ΔHpp is normalized to the same parameter for a reference sample to account for instrumental variations, where ΔHpp is the peak-to-peak line-width of the first derivative spectrum. The corresponding accessibility parameter, Π, is calculated using the following equation:

where P1/2 (DPPH) and ΔHpp (DPPH) are the P1/2 and line-width values determined for a standard sample of crystalline 2,2-diphenyl-1-picrylhydrazyl (DPPH) in KCl33.
References
Hynes, R. O. Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 (2002).
Ginsberg, M. H., Partridge, A. & Shattil, S. J. Integrin regulation. Curr Opin Cell Biol 17, 509–516 (2005).
Vinogradova, O. et al. A structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell 110, 587–597 (2002).
Hynes, R. O. Targeted mutations in cell adhesion genes: what have we learned from them? Dev Biol 180, 402–412 (1996).
Luo, B. H., Springer, T. A. & Takagi, J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol 2, e153 (2004).
Partridge, A. W., Liu, S., Kim, S., Bowie, J. U. & Ginsberg, M. H. Transmembrane domain helix packing stabilizes integrin alphaIIbbeta3 in the low affinity state. J Biol Chem 280, 7294–7300 (2005).
Lau, T. L., Dua, V. & Ulmer, T. S. Structure of the integrin alphaIIb transmembrane segment. J Biol Chem 283, 16162–16168 (2008).
Hughes, P. E. et al. Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J Biol Chem 271, 6571–6574 (1996).
Li, W. et al. A push-pull mechanism for regulating integrin function. Proc Natl Acad Sci U S A 102, 1424–1429 (2005).
Luo, B. H., Carman, C. V., Takagi, J. & Springer, T. A. Disrupting integrin transmembrane domain heterodimerization increases ligand binding affinity, not valency or clustering. Proc Natl Acad Sci U S A 102, 3679–3684 (2005).
Li, R. et al. Activation of integrin alphaIIbbeta3 by modulation of transmembrane helix associations. Science 300, 795–798 (2003).
Welf, E. S., Naik, U. P. & Ogunnaike, B. A. A spatial model for integrin clustering as a result of feedback between integrin activation and integrin binding. Biophys J 103, 1379–1389 (2012).
Miyamoto, S. et al. Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol 131, 791–805 (1995).
Hynes, R. O. Integrins: versatility, modulation and signaling in cell adhesion. Cell 69, 11–25 (1992).
Anthis, N. J. & Campbell, I. D. The tail of integrin activation. Trends Biochem Sci 36, 191–198 (2011).
Gardner, H. A. Integrin signaling in fibrosis and scleroderma. Curr Rheumatol Rep 1, 28–33 (1999).
Pozzi, A. et al. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci U S A 97, 2202–2207 (2000).
Ekholm, E. et al. Diminished callus size and cartilage synthesis in alpha 1 beta 1 integrin-deficient mice during bone fracture healing. Am J Pathol 160, 1779–1785 (2002).
McBrien, N. A., Metlapally, R., Jobling, A. I. & Gentle, A. Expression of collagen-binding integrin receptors in the mammalian sclera and their regulation during the development of myopia. Invest Ophthalmol Vis Sci 47, 4674–4682 (2006).
Rentala, S., Yalavarthy, P. D. & Mangamoori, L. N. Alpha1 and beta1 integrins enhance the homing and differentiation of cultured prostate cancer stem cells. Asian J Androl 12, 548–555 (2010).
Tamkun, J. W. et al. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell 46, 271–282 (1986).
Fitzgerald, L. A., Steiner, B., Rall, S. C., Jr, Lo, S. S. & Phillips, D. R. Protein sequence of endothelial glycoprotein IIIa derived from a cDNA clone. Identity with platelet glycoprotein IIIa and similarity to “integrin”. J Biol Chem 262, 3936–3939 (1987).
Anthis, N. J. et al. The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J 28, 3623–3632 (2009).
Lai, C., Liu, X., Tian, C. & Wu, F. Integrin alpha1 has a long helix, extending from the transmembrane region to the cytoplasmic tail in detergent micelles. PLoS One 8, e62954 (2013).
Lau, T. L., Kim, C., Ginsberg, M. H. & Ulmer, T. S. The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J 28, 1351–1361 (2009).
Lau, T. L., Partridge, A. W., Ginsberg, M. H. & Ulmer, T. S. Structure of the integrin beta3 transmembrane segment in phospholipid bicelles and detergent micelles. Biochemistry 47, 4008–4016 (2008).
Bordignon, E. & Polyhach, Y. H. EPR techniques to probe insertion and conformation of spin-labeled proteins in lipid bilayers. Methods Mol Biol 974, 329–355 (2013).
Smirnova, I. et al. Sugar binding induces an outward facing conformation of LacY. Proc Natl Acad Sci U S A 104, 16504–16509 (2007).
Yu, L. et al. Distance measurement between two flexible sites in proteins in high viscosity medium at physiological temperature using continuous wave EPR. Protein Cell 5, 334–337 (2014).
Bordignon, E. Site-directed spin labeling of membrane proteins. Top Curr Chem 321, 121-157 (2012).
Howell, S. C., Mesleh, M. F. & Opella, S. J. NMR structure determination of a membrane protein with two transmembrane helices in micelles: MerF of the bacterial mercury detoxification system. Biochemistry 44, 5196–5206 (2005).
Long, S. B., Tao, X., Campbell, E. B. & MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382 (2007).
Altenbach, C., Greenhalgh, D. A., Khorana, H. G. & Hubbell, W. L. A collision gradient method to determine the immersion depth of nitroxides in lipid bilayers: application to spin-labeled mutants of bacteriorhodopsin. Proc Natl Acad Sci U S A 91, 1667–1671 (1994).
Rath, A., Glibowicka, M., Nadeau, V. G., Chen, G. & Deber, C. M. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc Natl Acad Sci U S A 106, 1760–1765 (2009).
Bendas, G. & Borsig, L. Cancer cell adhesion and metastasis: selectins, integrins and the inhibitory potential of heparins. Int J Cell Biol 2012, 676731 (2012).
Huttenlocher, A. & Horwitz, A. R. Integrins in cell migration. Cold Spring Harb Perspect Biol 3, a005074 (2011).
Oppenheim, S. F., Buettner, G. R. & Rodgers, V. G. J. Relationship of rotational correlation time from EPR spectroscopy and protein-membrane interaction. J Memb Sci 118, 133–139 (1996).
Goldman, S. A., Bruno, G. V. & Freed, J. H. Estimating slow-motional rotational correlation times for nitroxides by electron spin resonance. J Phys Chem 76, 1858–1860 (1972).
McHaourab, H. S., Lietzow, M. A., Hideg, K. & Hubbell, W. L. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry 35, 7692–7704 (1996).
Zhou, Z. et al. Solution structure of the cytoplasmic domain of erythrocyte membrane band 3 determined by site-directed spin labeling. Biochemistry 44, 15115–15128 (2005).
Li, E., Wimley, W. C. & Hristova, K. Transmembrane helix dimerization: beyond the search for sequence motifs. Biochim Biophys Acta 1818, 183–193 (2012).
Gurezka, R. & Langosch, D. In vitro selection of membrane-spanning leucine zipper protein-protein interaction motifs using POSSYCCAT. J Biol Chem 276, 45580–45587 (2001).
Armulik, A., Velling, T. & Johansson, S. The integrin beta1 subunit transmembrane domain regulates phosphatidylinositol 3-kinase-dependent tyrosine phosphorylation of Crk-associated substrate. Mol Biol Cell 15, 2558–2567 (2004).
Coey, A. T. et al. Reconstitution of KCNE1 into lipid bilayers: comparing the structural, dynamic and activity differences in micelle and vesicle environments. Biochemistry 50, 10851–10859 (2011).
Oates, J., King, G. & Dixon, A. M. Strong oligomerization behavior of PDGFbeta receptor transmembrane domain and its regulation by the juxtamembrane regions. Biochim Biophys Acta 1798, 605–615 (2010).
Noordeen, N. A., Carafoli, F., Hohenester, E., Horton, M. A. & Leitinger, B. A transmembrane leucine zipper is required for activation of the dimeric receptor tyrosine kinase DDR1. J Biol Chem 281, 22744–22751 (2006).
Ruan, W., Becker, V., Klingmuller, U. & Langosch, D. The interface between self-assembling erythropoietin receptor transmembrane segments corresponds to a membrane-spanning leucine zipper. J Biol Chem 279, 3273–3279 (2004).
Li, R. et al. Dimerization of the transmembrane domain of Integrin alphaIIb subunit in cell membranes. J Biol Chem 279, 26666–26673 (2004).
Cluzel, C. et al. The mechanisms and dynamics of (alpha)v(beta)3 integrin clustering in living cells. J Cell Biol 171, 383–392 (2005).
Li, R. et al. Oligomerization of the integrin alphaIIbbeta3: roles of the transmembrane and cytoplasmic domains. Proc Natl Acad Sci U S A 98, 12462–12467 (2001).
Mokrab, Y., Stevens, T. J. & Mizuguchi, K. Lipophobicity and the residue environments of the transmembrane alpha-helical bundle. Proteins 74, 32–49 (2009).
Chang, C. T., Wu, C. S. & Yang, J. T. Circular dichroic analysis of protein conformation: inclusion of the beta-turns. Anal Biochem 91, 13–31 (1978).
Mao, D. & Wallace, B. A. Differential light scattering and absorption flattening optical effects are minimal in the circular dichroism spectra of small unilamellar vesicles. Biochemistry 23, 2667–2673 (1984).
Persson, D., Thoren, P. E. & Norden, B. Penetratin-induced aggregation and subsequent dissociation of negatively charged phospholipid vesicles. FEBS Lett 505, 307–312 (2001).
Acknowledgements
This work was supported by funds from the Ministry of Science and Technology of China (Grant numbers 2011CB911104 and 2013CB910202) and Projects U1332138 and 31100847 of the National Natural Science Foundation of China.
Author information
Authors and Affiliations
Contributions
L.Y., W.W., Yi.X. and C.T. designed the experiments, Ya.X. and Sh.L. made the mutations, Sa.L., C.L. and L.X. expressed the proteins, L.Y. and W.W. made the radical labeling and EPR measurements, L.Y., L.Z. and C.T. analyzed the data and wrote the paper. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Data
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yu, L., Wang, W., Ling, S. et al. CW-EPR studies revealed different motional properties and oligomeric states of the integrin β1a transmembrane domain in detergent micelles or liposomes. Sci Rep 5, 7848 (2015). https://doi.org/10.1038/srep07848
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07848
This article is cited by
-
Structural and dynamic origins of ESR lineshapes in spin-labeled GB1 domain: the insights from spin dynamics simulations based on long MD trajectories
Scientific Reports (2020)
-
Structure of an E. coli integral membrane sulfurtransferase and its structural transition upon SCN− binding defined by EPR-based hybrid method
Scientific Reports (2016)
-
Combined approaches of EPR and NMR illustrate only one transmembrane helix in the human IFITM3
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
