
Abstract
Growth through controlled adsorption of ferromagnetic elements such as Fe, Co and Ni on two-dimensional silicene provides an alternative route for silicon-based spintronics. Plane wave DFT calculations show that Fe, Co and Ni adatoms are strongly chemisorbed via strong sigma bonds, with adsorption energies (1.55 - 2.29 eV) that are two to six times greater compared to adsorption on graphene. All adatoms adsorb more strongly at the hole site than at the atom site, with Ni adsorbing strongest. Of the dimer configurations investigated, the hole – hole, b-atom – hole, vertically stacked at hole, vertically stacked at b-atom and bridge sites were found to be stable. The Co and Ni dimers are most stable when adsorbed in the hole-hole configuration while the Fe dimer is most stable when adsorbed in the atom-hole configuration. Metal-to-silicene and interconfigurational s-to-d electron transfer processes underpin the trends observed in adsorption energies and magnetic moments for both adatoms and dimers. Adsorption of these metals induces a small band gap at the Dirac Cone. In particular Co adatom adsorption at the hole site induces the largest spin-polarized band gaps of 0.70 eV (spin-up) and 0.28 eV (spin-down) making it a potential material candidate for spintronics applications.
Similar content being viewed by others
Introduction
With the discovery of graphene1, two dimensional materials have been investigated intensely for both fundamental2,3,4,5,6 and technological reasons7,8,9,10,11,12,13. Graphene has led this intense chase for uncovering the rich physics associated with novel material structures that includes nanoribbons7, nanosheets14 and nanomeshes15 and applying them in unique ways. The question of a silicon analogue of graphene was touted to add much value to various technological industries.
The discovery of silicon nanoribbons that are just one atom thick atop a Ag (110) substrate was first reported in 2010 by Lalmi et al.16. This discovery was regarded controversial for three reasons: (1) no other results associated with the material were reported, (2) the extremely short Si-Si bond length of 1.9 Å that was reported was too short compared to an expected value of between 2.2–2.4 Å and (3) the hexagonal structures observed using the STM could have well been an unreconstructed Ag (111) surface16,17. Since the work of Lalmi et al., many more reports of silicene grown on Ag (110)17,18,19, Ag (111)20, Ir (111)21, CaSi222, graphene23, diboride films24, grown as alternating stacked layers with MoS225 or embedded in h-BN26 and AlN27. The various experimental and theoretical reports of silicene adsorbed on, or interacting with metallic and insulating substrates conclude that there is minimal substrate effect on the properties of the silicene. We thus assume in this work that free-standing silicene is sufficient to mimic supported silicene without significant chemical or physical interference.
The electronic properties and band structure associated with two dimensional materials, specifically the Dirac fermions that are now near-synonymous with graphene, motivate fundamental understanding and technological application of silicene based materials. It has been shown that silicene would have a band structure that is similar to the band structure of graphene9,14,17,28,29,30,31,32,33,34. Much of the work that would be done on silicene would mimic or parallel that done with graphene: defects35, adsorption of hydrogen14,36 and metal adatoms37,38,39. An extensive review of the importance of investigating adsorbed metal clusters, in the particular context of how their chemical and physical properties change as a result of the interconfigurational electronic transitions that occur, is provided by Johll et al.40. These clusters are inadvertently affected by the substrate upon which they are adsorbed. Ferromagnetic metal clusters deposited on graphene have been shown to exhibit magnetic properties that lend support to applying these structures in magnetic materials and spintronic devices. Johll et al.40 have shown that Fe, Co and Ni have adsorption energies that range from 0.2 eV to 1.4 eV when adsorbed as atoms and between 0.1 eV to 0.5 eV per atom when adsorbed as dimers. Further, the band structures of the Fe and Co systems indicate spin polarization and band gaps that have potential to be used in driving a spin current. These results motivate investigation of the properties of silicene adsorbed adatoms and dimers of ferromagnetic elements. Sahin and Peeters37, using plane-wave density functional theory calculations parameterized by the local density approximation of Ceperly and Alder41 and a projected augmented wave pseudization42, found that various alkali, alkali earth and transition metal adatoms adsorb much more strongly on free-standing silicene than on graphene. Interestingly, there is little work that discusses the structure, stability, electronic and magnetic properties of these adatoms and/or dimers in the same context as graphene.
In this work, we carry out plane-wave density functional theory calculations to investigate the adsorption site configurations and stabilities, projected wavefunction density of states (i.e. electron population or occupancy), magnetic moments and band structures of Fe, Co and Ni adatoms and homonuclear dimers adsorbed on silicene.
Methodology
The plane-wave based density functional theory program PWSCF (Quantum Espresso Version 5.0.1)43 was used to carry out all calculations. The Rappe-Rabe-Kaxiras-Joannopoulos (RRKJ) ultrasoft pseudopotential was used for all species44. The Perdew-Burke-Ernzerhof (PBE)45 generalized gradient approximation (GGA) correction formalism was employed for the exchange-correlation functional. All pseudopotentials were obtained from the PWSCF pseudopotential online reference46. As reported in our previous work40, we re-iterate that the use of an RRKJ ultrasoft pseudopotential alleviates issues associated with using a generalized gradient approximation47 for the exchange-correlation functional particularly for the unadsorbed or ‘free’ transition metal atoms.
The supercell of dimensions 15.36 Å × 15.36 Å × 15.36 Å in each of the unit cell vectors consists of 32 Si atoms and either 1 metal adatom or dimer. The periodic silicene substrate used in all our calculations is akin to a bilayer Si(111) surface. This implies a mix of both sp2 and sp3 hybridization for the Si atoms. Marzari-Vanderbilt smearing48 was used to aid convergence with a small Gaussian spread of 0.001 Ry (0.013 eV). Local states and populations were calculated by projecting the plane-wave onto specific orbitals. All k-points generated in our calculations were based on the Monkhorst-Pack algorithm49. Convergence with respect to cutoff energies for both the wave function and the electron density and the number of k-points, was checked by calculating the adsorption energy of the a single Fe atom adsorbed at the hole site of the silicene sheet. A cutoff energy of 40 Ry for the planewave and 480 Ry for the energy density, with a Monkhorst-Pack grid of 4 × 4 × 1 was determined through calibration. In calibrating these energies, we utilized the Monkhorst-Pack grid without any Brillouin Zone shift and a force convergence threshold of 10−3 a.u.
The adsorption energies, Ead, for the adatom (i = 1) or dimer (i = 2) systems are calculated based on the magnitude of the enthalpy change of the reaction represented in Equation 1.

Results
Adatoms
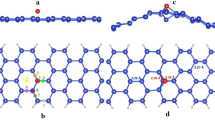
Relative to the adsorption plane, there are two Si atom types: an a-type atom that is closer to the adsorption plane and a b-type atom (see Figure 1). Three adatom adsorption sites were investigated in this work: the hole site, h (see Figure 1(i)), the above up-atom or a-site and the above down-atom or b-site (see Figure 1(ii)). Metal atoms initially adsorbed above the a-site geometrically transformed the system to be congruent with the case where the metal atom is adsorbed above the b-site. There are therefore just two adatom adsorption site configurations that are studied in this work, viz. the hole site and the b-site. We note that only adsorption energies and magnetic properties of converged or relaxed structures are reported.

Representations of silicene up and down atoms relative to adsorption plane of metal adatoms (represented generically in blue here).
There are just two adsorption sites that are stable: (i) metal adatom at the hole site and (ii) metal adatom adsorbed above the b-site. In (i), the metal adatom has a coordination number of 6 with two sets of bond lengths, ra and rb, respectively to a-type and b-type Si atoms. In (ii), the metal adatom has a coordination number of 4 (see Figure inset) with just one set of bond lengths. Fe, Co and Ni adatoms absorb more strongly at the hole site compared to the b-site.
Fe, Co and Ni adatoms adsorbed strongest at the hole site with adsorption energies of 0.2 eV (Fe and Co) and 0.3 eV (Ni) more than the corresponding down-atom (b) site adsorption (see Table 1). We account for this difference in adsorption energies by comparing the coordination numbers in each case. When adsorbed at the hole site, the metal adatom has a coordination number of 6 (see Figure 1(i)). This set of 6 bonds can be sub-categorized into two bond types: one with a-type Si atoms and one with b-type Si atoms. The adatom-Si bond lengths are 0.3A shorter in the former. When adsorbed at the atom site, the metal adatom has a coordination number of 4 (see Figure 1(ii)). All adatom-Si bond lengths in this configuration are equal to one another. Also, these bond lengths are equal to the shorter set of three bonds in the hole-site adsorption configuration.
Ni adatoms adsorbed strongest and Fe weakest, to silicene for all corresponding configurations. This trend is also found when these species are adsorbed on graphene. Similar to the case when these metal adatoms are adsorbed on graphene, there is a significant change in the localized electronic configurations of the adatoms compared to their free atomic or unbound state. We account for this increasing adsorption energy on going from Fe to Co to Ni by considering the interconfigurational energies associated with transferring one unit of charge from the s to the d orbitals: 0.87, 0.42 and −0.03 eV respectively50. Further to the normalized interconfigurational energies given here, we also find that Fe has the greatest s to d electron transfer of 0.9 electrons relative to the free atom (see Table 1), followed by Co (0.8 and 0.7 electrons when adsorbed at the hole site and b-site respectively) and Ni (0.4 electrons). Both the amount of charge and the interconfigurational energy change that accompanies the transfer of charge within the metal adatom explains strong Ni adsorption. These charge transfer processes also lowers the spin multiplicity on the metal adatoms and accounts for a decrease of 2 μB relative to the respective free atoms.
In comparison to adsorption on graphene40, the adsorption of Fe, Co and Ni adatoms to silicene is between two to six times stronger. We note that the comparison made here is justified on account that the computational parameters, in particular the plane wave cutoff energies, pseudopotential types and k-points used in the work done here and that presented by Johll et al. are the same. We propose two reasons for this. First, the interaction between the metal adatom and graphene is impeded by the π system of electrons. Second, there is stronger covalency and better orbital overlap when adsorbed on silicene compared to graphene. The electronegativities on the Pauling Scale of the metal adatoms (1.83, 1.88 and 1.91 for Fe, Co and Ni respectively51) and silicon (1.90)51 are comparable and is significantly different compared to carbon (2.55)51. The interaction between metal adatom and silicene is tantamount to a soft-soft orbital interaction; the interaction is soft-hard when adsorbed on graphene. The soft-soft orbital interactions between metal adatoms and the nearby Si atoms are indicative of better orbital overall compared to the case where these adatoms are adsorbed on graphene.
Adatom adsorption opens up a band gap at the K point (see Figure 2 and Table 2) of the irreducible Brillouin Zone of silicene. The opening of the band gap is accompanied with the lifting of the local s states of these metal atoms above the Fermi level, thus resulting in a de-population of the s state (see Table 1). The band gap that results from the adsorption of Fe and Ni on silicene is extremely small (see Table 2). In either case, the magnitude of the difference between the spin-up and spin-down gaps, assuming a direct-gap transition, is less than or equal to kT at room temperature. Fe and Ni adatom adsorption will not be useful in fabricating a device that has the potential of generating a spin current. On the other hand, the difference in band gap value for spin-up and spin-down states at the K point is 0.42 eV when Co is adsorbed at the hole site (see Table 2) will likely generate spin-polarized current even at elevated temperatures. Co therefore shows much promise in the context of developing two-dimensional materials with band structures that might be advantaged in the spintronics and electronics industries.

Spin polarized band structures of (i) Fe, (ii) Co, (iii) Ni adatoms when adsorbed at the hole site and (iv) band structure of clean silicene.
Both spin-up (red lines) and spin-down (blue lines) bands are visible for the Fe and Co systems. These bands overlap when Ni is adsorbed on silicene at the hole site.
Dimers
Seven dimer adsorption configurations were studied for each element in this work. The various configurations studied are:
-
1
hole site – hole site [h-h],
-
2
atom site – hole site [a-h],
-
3
atom site – hole site [b-h],
-
4
vertically stacked at the hole site [h-h*],
-
5
vertically stacked at the a-atom site [a-a*],
-
6
vertically stacked at the b-atom site [b-b*], and
-
7
adjacent and directly opposite atop bridge site within a hexagon [m-m].
Five of these seven dimer configurations were found to be stable for at least two of the three elements: the h-h, b-h, h-h*, b-b* and m-m dimers. The Ni h-h* configuration relaxed to a b-h configuration and the Fe b-b* configuration relaxed to the b-h configuration. All a-h and a-a* dimers, just like the corresponding adatom adsorption configuration, became b-h and b-b* dimers respectively. The structures, adsorption energies, magnetic moments and electronic configurations of the remaining 13 stable configurations are represented in Figure 3. The adsorption energies (per metal atom) and magnetic moments of the stable dimer configurations are summarized in Table 3.

Dimer adsorption configurations: hole site – hole site configuration [(i), (iii) and (v)], b-atom site – hole site configuration [(ii), (iv) and (vi)], vertically stacked at hole site [(vii) and (ix)], directly opposite bridge sites within the same hexagon [(viii), (x) and (xii)] and vertically stacked at b-atom site [(xi) and (xiii)].
All a-h and a-a* dimers, just like the corresponding adatom adsorption configuration, became b-h and b-b* dimers respectively. The adsorption energies, magnetic moments, charge, projected electronic configurations and geometric information of these dimers adsorbed on silicene are detailed within each sub-figure.
Co and Ni dimers adsorb strongest in the h-h configuration; the Fe dimer adsorbs strongest in the b-h configuration. In the h-h adsorption configuration, each Fe atom has the electron configuration 3d7.14s0.4 (see Figure 3(i)). However, when in the b-h configuration, Fe has the electron configuration 3d6.94s0.6 and 3d7.04s0.5 (see Figure 3(ii)). This lower average interconfigurational s-d transfer of 0.15 electrons in the Fe atoms when adsorbed in the b-h configuration is sufficient to offset the energy released from a greater coordination number with the underlying silicon atoms (4 cf. 6) when adsorbed in the h-h configuration. We note that the 3d-4s interconfigurational energy for Fe is larger than Co, which in turn is larger than Ni. As a result, there is substantial electron density between the Fe atoms when adsorbed as a dimer in the b-h configuration (see Figure 4(i)) while there is little electron density between the Co (see Figure 4(ii)) and Ni (see Figure 4(iii)) atoms when adsorbed as dimers in the h-h configuration. This indicates that the Co and Ni dimer adsorption is more akin to separate adatom adsorption. The m-m dimer configuration is weakest in all cases (see Figures 3(viii), 3(x) and 3(xii)). We propose here that this is due to increased metal atom – metal atom interaction and hence a lack of metal-Si coordination.

Electron density isosurfaces of the most stable (i-iii) and least stable (iv-vi) Fe, Co and Ni dimer adsorption configurations, respectively.
Co and Ni dimers adsorb strongest when there is little or negligible electron density between the two metal atoms (see Figures 4(ii) and 4(iii)). The Fe dimer adsorbs strongly even though there is substantial electron density (see Figure 4(i)) between the two Fe atoms.
We also calculated free dimer bond lengths for Fe and Co to be 1.96Å and that of Ni to be 2.10Å. At the h-h site, dimer bond lengths are about 1.5 Å more than the free dimer bond lengths (see Figure 3(i)), while the dimer bond lengths in the case of graphene are only about 0.3Å longer than the free dimer bond lengths. This supports the higher adsorption energy for dimers on silicene than on graphene at the h-h site, where it is about three times that of dimers on graphene40. This again can be attributed to the increase in σ interaction with the underlying Si atoms; in graphene, the π band impedes this interaction. The Eb of dimers at the h-h site is about 2 times that of the Ead of adatoms at the h site (see Tables 1 and 3), suggesting again these dimers more akin as separate adatoms.
The total magnetic moments of the most stable Co and Ni dimer systems, adsorbed in the h-h configuration, are quenched by 2 μB relative to the free dimer (see Table 3). The magnetic moment of the Co dimer system when adsorbed in the b-h configuration is also quenched by the same amount. When adsorbed in the configurations that have weaker adsorption energies (i.e. h-h*, b-b* and m-m), the Co dimer system has a magnetic moment that is equal or near equal (for the m-m case) to the free Co dimer. Similarly, the magnetic moments of all Ni dimer configurations except the b-b* configuration, is 0 μB. In the b-b* configuration, the Ni dimer is weakly adsorbed and has a magnetic moment of 2 μB. In the most stable Fe dimer system, adsorbed in the b-h configuration, the magnetic moment is equal to the free dimer magnetic moment; the magnetic moment of the adsorbed Fe dimer is only quenched, by 2 μB, when adsorbed in the h-h configuration. The magnetic moments of the dimer systems, which include a finite moment induced in the silicene substrate, is consistent with the argument that interconfigurational energies play a critical role in determining the adsorption energies of these species to silicene.
Similar to the adatoms, a band gap is observed at the K point (see Figure 5) when dimers are adsorbed on silicene. The difference between the spin-up and spin-down gaps for the Fe and Ni dimers adsorbed in the most stable configuration is less than or equal to kT at room temperature; only the Co dimer has an appreciable difference of 0.16 eV (see Table 4). The difference between the spin-polarized band gaps induced by the Co dimer is 0.26 eV lower than the gap difference induced when adsorbed as an adatom. In the context of developing a material suitable for spintronics-based applications, Co adatom adsorption is preferred over dimer formation from a growth perspective.

Band structures of the most stable dimer adsorption configurations for (i) Fe (adsorbed in the b-h configuration), (ii) Co (adsorbed in the h-h configuration) and (iii) Ni (adsorbed in the h-h configuration).
The general conic structure at the K point is preserved even with dimer adsorption. For these most stable dimer adsorption configurations, only Fe and Co dimers open a spin polarized band gap.
By considering adatom and dimer adsorption energies (see Tables 1 and 3), we find that Fe and Co adatoms tend to aggregate to form dimers, while Ni adatoms prefer to remain adsorbed as adatoms. The aggregation tendencies of Fe, Co and Ni adatoms, to form dimers, point to the average physical (and chemical) properties one might associate with the material system. We define the aggregation energies of adatoms to form dimers as the enthalpy change of the process shown in Equation 2.

In all cases, the hole site configuration was used for the pair of isolated single adatom ‘reactants’. The most stable dimer configuration was used for each of the species: b-h for Fe and h-h for both Co and Ni. Adatom aggregation to form dimers is thermodynamically favored for Fe and Co, but not for Ni, with aggregation enthalpies of −0.39 eV, −0.31 eV and +0.15 eV respectively (based on Equation 2). These results suggest that with sufficient growth control, Ni deposition would most likely result in isolated Ni adatoms that decorate the silicene. From a thermodynamic view, a low coverage of Co adsorbed on silicene is potentially most suitable for growing a material that has a significant difference between the spin-polarized band gaps.
Conclusion
We have carried out plane-wave density functional theory calculations to determine the adsorption energies, structures, electronic configurations, magnetizations and band structures of Fe, Co and Ni adatoms and dimers adsorbed in different configurations on silicene. Fe, Co and Ni adatoms adsorb strongest at the silicene hole site. They are less stable when adsorbed at the b-site. This stems from a difference in coordination number: 6 at the hole-site and just 4 at the b-site. Similar to the adsorption of these metal atoms on graphene, we find that Ni adsorbs strongest, followed by Co and then Fe. We attribute this trend to the decrease in 3d-4s interconfigurational energy on going from Fe to Co to Ni. Fe has the greatest s-to-d electron transfer when adsorbed on silicene. The net result is a lowering of the adsorption energy for Fe relative to Ni. The electron transfer processes also result in a reduction of 2 μB on each of the metal adatoms compared to their free atom state. The adatoms adsorb between two to six times more strongly to silicene than to graphene. The adatoms adsorb strongly via sigma bonds to silicene and just weakly via pi interactions with graphene. Small band gaps are induced via the adsorption of the metal adatoms on silicene. The band gaps induced by Fe and Ni are rather small; the gap induced by Co is large with values of 0.70 eV and 0.28 eV for spin-up and spin-down bands respectively. Therefore, at sufficiently low coverage, Co adsorbed on silicene has the potential to function has a spintronic device.
For Co and Ni dimers, the hole site – hole site configuration was found to be most stable. For the Fe dimer, the atop b-site – hole site dimer configuration was found to be most stable. Again, interconfigurational energies as well as coordination numbers were used to explain why these dimer configurations are most stable for the respective elements. In the case of Fe, preserving the integrity of the dimer leads to stronger adsorption to the substrate; in the case of Co and Ni, the lack of metal atom – metal atom interaction results in stronger adsorption. The band gap induced by Co dimer adsorption is 0.04 and 0.20 eV for spin-up and spin-down bands respectively. These gaps are less significant compared to when Co adatom configuration. Co adsorption on silicene is the best choice of material for spintronics application. Growth control for assembling Co as adatoms on silicene is critical for it to be applied for spintronics.
References
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
De Martino, A., Dell'Anna, L. & Egger, R. Magnetic confinement of massless dirac fermions in graphene. Phys. Rev. Lett. 98, 066802 (2007).
Stauber, T., Peres, N. M. R. & Geim, A. K. Optical conductivity of graphene in the visible region of the spectrum. Phys. Rev. B 78, 085432 (2008).
Zhou, Q., Coh, S., Cohen, M. L., Louie, S. G. & Zettl, A. Imprint of transition metal d orbitals on a graphene Dirac cone. Phys. Rev. B 88, 235431 (2013).
Ponomarenko, L. A. et al. Chaotic Dirac billiard in graphene quantum dots. Science 320, 356–358 (2008).
Novoselov, K. S. et al. Two-dimensional gas of massless Dirac fermions in graphene. Nature 438, 197–200 (2005).
Schwierz, F. Graphene transistors. Nat. Nanotechnol. 5, 487–496 (2010).
Stoller, M. D., Park, S., Yanwu, Z., An, J. & Ruoff, R. S. Graphene-Based ultracapacitors. Nano Lett. 8, 3498–3502 (2008).
Han, M. Y., Özyilmaz, B., Zhang, Y. & Kim, P. Energy band-gap engineering of graphene nanoribbons. Phys. Rev. Lett. 98, 206805 (2007).
Huang, X., Qi, X., Boey, F. & Zhang, H. Graphene-based composites. Chem. Soc. Rev. 41, 666–686 (2012).
Potts, J. R., Dreyer, D. R., Bielawski, C. W. & Ruoff, R. S. Graphene-based polymer nanocomposites. Polymer. 52, 5–25 (2011).
An, X. & Yu, J. C. Graphene-based photocatalytic composites. RSC Adv. 1, 1426–1434 (2011).
Kuila, T. et al. Recent advances in graphene-based biosensors. Biosens. Bioelectron. 26, 4637–4648 (2011).
Lew Yan Voon, L. C., Sandberg, E., Aga, R. S. & Farajian, A. A. Hydrogen compounds of group-IV nanosheets. Appl. Phys. Lett. 97, 163114 (2010).
Bai, J., Zhong, X., Jiang, S., Huang, Y. & Duan, X. Graphene nanomesh. Nat. Nanotechnol. 5, 190–194 (2010).
Lalmi, B. et al. Epitaxial growth of a silicene sheet. Appl. Phys. Lett. 97, 223109 (2010).
Kara, A. et al. A review on silicene - New candidate for electronics. Surf. Sci. Rep. 67, 1–18 (2012).
Aufray, B. et al. Graphene-like silicon nanoribbons on Ag(110): A possible formation of silicene. Appl. Phys. Lett. 96, 183102 (2010).
Enriquez, H., Vizzini, S., Kara, A., Lalmi, B. & Oughaddou, H. Silicene structures on silver surfaces. J. Phys. Condens. Matter 24, 314211 (2012).
Feng, B. et al. Evidence of silicene in honeycomb structures of silicon on Ag(111). Nano Lett. 12, 3507–3511 (2012).
Meng, L. et al. Buckled silicene formation on Ir(111). Nano Lett. 13, 685–690 (2013).
Ali, M. A. & Tchalala, M. R. Chemical Synthesis of Silicon Nanosheets from layered calcium disilicide. J. Phys. Conf. Ser. 491, 012009 (2014).
Cai, Y., Chuu, C.-P., Wei, C. M. & Chou, M. Y. Stability and electronic properties of two-dimensional silicene and germanene on graphene. Phys. Rev. B 88, 245408 (2013).
Fleurence, A. et al. Experimental evidence for epitaxial silicene on diboride thin films. Phys. Rev. Lett. 108, 245501 (2012).
Li, X., Wu, S., Zhou, S. & Zhu, Z. Structural and electronic properties of germanene/MoS2 monolayer and silicene/MoS2 monolayer superlattices. Nanoscale Res. Lett. 9, 110 (2014).
Liu, H., Gao, J. & Zhao, J. Silicene on substrates: A way to preserve or tune its electronic properties. J. Phys. Chem. C 117, 10353–10359 (2013).
Houssa, M., Pourtois, G., Afanas'Ev, V. V. & Stesmans, A. Can silicon behave like graphene? A first-principles study. Appl. Phys. Lett. 97, 112106 (2010).
Drummond, N. D., Zólyomi, V. & Fal'ko, V. I. Electrically tunable band gap in silicene. Phys. Rev. B - Condens. Matter Mater. Phys. 85, 075423 (2012).
De Padova, P. et al. Multilayer silicene nanoribbons. Nano Lett. 12, 5500–5503 (2012).
Ezawa, M. Valley-polarized metals and quantum anomalous hall effect in silicene. Phys. Rev. Lett. 109, 055502 (2012).
Ezawa, M. Hexagonally warped Dirac cones and topological phase transition in silicene superstructure. Eur. Phys. J. B 86, 139 (2013).
Gao, J., Zhang, J., Liu, H., Zhang, Q. & Zhao, J. Structures, mobilities, electronic and magnetic properties of point defects in silicene. Nanoscale 5, 9785–92 (2013).
Ni, Z. et al. Tunable bandgap in silicene and germanene. Nano Lett. 12, 113–118 (2012).
Lin, C. L. et al. Substrate-induced symmetry breaking in silicene. Phys. Rev. Lett. 110, 076801 (2013).
Gao, J., Zhang, J., Liu, H., Zhang, Q. & Zhao, J. Structures, mobilities, electronic and magnetic properties of point defects in silicene. Nanoscale 5, 9785–92 (2013).
Osborn, T. H., Farajian, A. A., Pupysheva, O. V., Aga, R. S. & Lew Yan Voon, L. C. Ab initio simulations of silicene hydrogenation. Chem. Phys. Lett. 511, 101–105 (2011).
Sahin, H. & Peeters, F. M. Adsorption of alkali, alkaline-earth and 3d transition metal atoms on silicene. Phys. Rev. B 87, 85423 (2013).
Kaloni, T. P., Gangopadhyay, S., Singh, N., Jones, B. & Schwingenschlögl, U. Electronic properties of Mn-decorated silicene on hexagonal boron nitride. Phys. Rev. B 88, 235418 (2013).
Sivek, J., Sahin, H., Partoens, B. & Peeters, F. M. Adsorption and absorption of boron, nitrogen, aluminum and phosphorus on silicene: Stability and electronic and phonon properties. Phys. Rev. B 87, 85444 (2013).
Johll, H., Kang, H. C. & Tok, E. S. Density functional theory study of Fe, Co and Ni adatoms and dimers adsorbed on graphene. Phys. Rev. B 79, 245416 (2009).
Ceperley, D. M. & Alder, B. J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 45, 566–569 (1980).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Rappe, A. M., Rabe, K. M., Kaxiras, E. & Joannopoulos, J. D. Optimized pseudopotentials. Phys. Rev. B 41, 1227–1230 (1990).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Baroni, S., Corso, A. D., de Gironcoli, S. & Giannozzi, P. Pseudopotentials - QUANTUMESPRESSO. Available at <http://www.quantum-espresso.org/pseudopotentials/> [Accessed January 2013].
Moroni, E. G., Kresse, G., Hafner, J. & Furthmüller, J. Ultrasoft pseudopotentials applied to magnetic Fe, Co and Ni: From atoms to solids. Phys. Rev. B 56, 15629–15646 (1997).
Marzari, N., Vanderbilt, D., De Vita, A. & Payne, M. C. Thermal Contraction and Disordering of the Al(110) Surface. Phys. Rev. Lett. 82, 3296–3299 (1999).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Yanagisawa, S., Tsuneda, T. & Hirao, K. An investigation of density functionals: The first-row transition metal dimer calculations. J. Chem. Phys. 112, 545 (2000).
Allred, A. Electronegativity values from thermochemical data. J. Inorg. Nucl. Chem. 17, 215–221 (1961).
Author information
Authors and Affiliations
Contributions
H.J., M.D.K.L. and S.P.N.N. performed the theoretical calculations. H.J., H.C.K. and E.S.T. analyzed the data and co-wrote the paper. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Johll, H., Lee, M., Ng, S. et al. Influence of Interconfigurational Electronic States on Fe, Co, Ni-Silicene Materials Selection for Spintronics. Sci Rep 4, 7594 (2014). https://doi.org/10.1038/srep07594
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07594
This article is cited by
-
A DFT investigation on the mechanical and structural properties of silicene nanosheets under doping of transition metals
Applied Physics A (2022)
-
First-principles study of medium-scale X-atoms-doped nickel clusters Nin−1X (X = C, Si, Ge, Sn, Pb; n = 19–23)
Theoretical Chemistry Accounts (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
