
Abstract
Nanobodies (Nbs) or single-domain antibodies are among the smallest and most stable binder scaffolds known. In vitro display is a powerful antibody discovery technique used worldwide. We describe the first adaptation of in vitro mRNA/cDNA display for the rapid, automatable discovery of Nbs against desired targets and use it to discover the first ever reported nanobody against the human full-length glucose transporter, GLUT-1. We envision our streamlined method as a bench-top platform technology, in combination with various molecular evolution techniques, for expedited Nb discovery.
Similar content being viewed by others
Introduction
Nearly all biomedical investigations require binders that specifically recognize desired targets. Although useful, common antibodies and their engineered variants (Fab – fragment antigen-binding; scFv – single-chain variable fragment) continue to have several limitations such as low stability, solubility and poor production feasibility. In contrast, the monomeric, hyper-variable, antigen-binding regions (VHH, termed Nanobodies® or Nbs) of homodimeric, heavy-chain-only antibodies (HCAbs) naturally found in camelids and sharks have superior physico-chemical properties (Figure 1)1.

Traditional antibodies and their fragments v/s nanobodies (Nbs).
Compared to regular antibodies (160 kDa) and their fragments (fragment antigen-binding, Fab – 65 kDa; single-chain variable fragment, scFv – 28 kDa), the molecular size of single-domain Nbs is the smallest (15 kDa). Nbs can be derived from the heavy-chain only antibodies (HCAbs) that occur naturally in camelids (VHH) and sharks (VNAR) (v-variable, c-constant, H-heavy chain, L- light chain).
Compared to regular antibodies (160 kDa) and their fragments (Fab – 65 kDa; scFv – 28 kDa), the molecular size of single-domain Nbs is the smallest (15 kDa) (Figure 1). Nbs are also extremely robust, highly resistant to denaturation/thermal degradation, have high aqueous solubility and in general, are highly and functionally expressed using standard microbial expression systems. Furthermore, Nbs also have superior body distribution, tissue penetration and blood clearance properties, making them highly attractive in the clinic2,3.
Although raising antibodies through animal immunization has been quite successful4, more so for soluble antigens than for membrane protein targets, the approach is limited by high costs, time, animal usage, bloodstream stability and antigenicity v/s toxicity of the target and overall practical feasibility of scale-up. Various in vitro/non-animal based approaches have been developed to address these issues. In these methods, each gene within a library of ~105–1015 variant sequences is linked to its encoded antibody. Depending on the technique used to create the genotype-phenotype linkage, several ‘display’ methods currently exist, including cell-based display using yeast or E. coli cells, or phage particles and in vitro display on ribosomes or mRNA/cDNA5.
Cell-based display methods are limited by the transfection/transformation efficiencies of the host organism, while ribosome display employs non-covalent, gene-ribosome-protein ternary complexes6, preventing the use of stringent selection conditions. In contrast, mRNA/cDNA display is an in vitro technique that employs a covalent bond to link an encoding nucleic acid fragment to its encoded antibody7. This covalent linkage allows the maintenance of stringent, tailored antigen-binding conditions that ensure the selection of highly specific, stable and robust binders8. The stringency is particularly useful in the case of challenging targets such as integral membrane proteins, which are solubilised using detergents and contain large, non-specific hydrophobic patches9,10.
The human GLUT-1 transporter is an integral membrane protein responsible for glucose uptake in erythrocytes, glucose supply across the blood-brain barrier and owing to these functions, is implicated in several diseases such as De Vivo or GLUT-1 deficiency syndrome and various cancers11.
We present the first adaptation of mRNA/cDNA display to the micro-scale, rapid discovery of nanobodies and use this novel method to discover a high-affinity nanobody for GLUT-1 as a model integral membrane protein target.
Methods
Preparation of Nb Template
All primers were obtained from IDT (San Diego, CA). E. coli-codon-optimized, N-terminal FLAG-tagged Nb-GFP12 (Table I) (Genscript, NJ) was PCR-amplified using platinum Taq DNA polymerase (Life Technologies, Carlsbad, CA) and primers Nb-GFP_Disp_Fw & Nb-GFP_Disp_Rv (Table II). HuSdL (Creative Biolabs, CD Inc., NY) was purchased and amplified using HuSdL_Disp_Fw and HuSdL_Disp_Rv (Table II), followed by two rounds of overlap extension PCR with primers HuSdL_Disp_Fw1 & HuSdL_Disp_Rv first, followed by Nb-Lib_Disp_Fw & HuSdL_Disp_Rv (Table II) to add the 5′ flanking sequences required for in vitro transcription and translation. Freshly amplified and column-purified PCR product was cloned into E. coli Top 10 cells using pCR2.1-TA® cloning (Life Technologies) to analyze sequence diversity and ensure the addition of the 5′ extension sequences. To synthesize a mutagenic library based on the Nb-GFP template, error-prone PCR was performed as previously published13, using the 5′-triphosphates of 6-(2-deoxy-b-D-ribofuranosyl)-3,4-dihydro-8H-pyrimido-[4,5-C][1,2]oxazin-7-one (dP) and of 8-oxo-2′deoxyguano-sine (8-oxo-dG). 0.4 mM each of dATP, dCTP, 2′-dPTP (Trilink Biotechnologies, San Diego, USA) instead of dTTP and dGTP and 0.8 mM of 8-oxo-2′-dGTP (Trilink Biotechnologies, San Diego, USA) were used in the total dNTP mix that was used to amplify Nb-GFP with Nb-Lib_Disp_Fw and Nb-GFP_Disp_Rv (Table II). A second round of amplification was performed with regular dNTPs to ensure the absence of mutagenic nucleotides that could bias subsequent downstream procedures.
Preparation of mRNA
The Nb template/library was in vitro transcribed and treated with Turbo DNase using the Megascript® T7 Transcription kit (Life Technologies), as per the manufacturer's instructions. mRNA was purified using the Megaclear mRNA Transcription Clean-up kit (Life Technologies), as per the manufacturer's instructions. 250 ng of the Nb template/library PCR product typically yielded up to 100 uL of purified mRNA at 1–2 µg/µL. Highly purified mRNA was analyzed for integrity and size on a 0.8% TBE-agarose gel by electrophoresis against the Century RNA ladder (Life Technologies).
mRNA-puromycin linker ligation
Purified mRNA was ligated to the puromycin linker, Nb_Disp_Puro using a DNA splint, Nb-GFP_Disp_Splint or HuSdL_Disp_Splint (Table II). For each 60 µL reaction, 5 µg mRNA was first annealed to 2.67 µM splint and 2.67 µM puromycin linker in the presence of 1× T4 ssRNA ligase buffer. Following annealing, 1 mM ATP, 1 µL murine RNase inhibitor (New England Biolabs, NEB), 3 µL T4 ssRNA Ligase were added and the reaction was incubated at room temperature overnight. Unligated linker and splint were cleaved using 5 µL lambda exonuclease (NEB) in a total volume of 240 µL, containing 24 µL of lambda exonuclease buffer and subsequent incubation at 37°C for 45 mins. After the addition of 60 μL of 1 M Tris-HCl, pH 7.5, the enzyme was inactivated by incubation at 65°C for 5 mins and the reaction was placed on ice. The mRNA-puromycin linker ligation product was purified using 70 µL magnetic OligodT25 beads (NEB), as per the manufacturer's instructions and eluted twice in 30 uL (15 µL × 2) of 10 mM Tris-HCl, pH 7.5. The ligation efficiency of this method was typically between ~40–60%.
mRNA-Nb fusion formation
The ligation product was precipitated using co-precipitant, pink (Bioline) and resuspended in 6.5 µL of 10 mM Tris-HCl, pH 7.5. In vitro translation was performed using Solutions A & B of the PURExpress® kit (NEB). Resuspended ligation product was mixed with 1 µL murine RNase inhibitor (NEB), 10 µL Solution A and 7.5 µL Solution B. The reaction was incubated at 37°C for 1.5 hr, followed by incubation on ice for 10 min. 2.5 uL of 1 M MgCl2 and 12.5 µL of 2 M KCl were added to the reaction, followed by incubation at room temperature for 10 min. The reaction was incubated at −20°C overnight to allow stable fusion formation. Following this step, the fusions were re-purified using the magnetic OligodT25 beads (NEB), as per the manufacturer's instructions and eluted in 30 µL of 10 mM Tris-HCl, pH 7.5. Fusion formation was assayed by western blot using the anti-FLAG-M2 primary antibody (Sigma) to detect the engineered N-terminal FLAG-tag on all the Nbs. The unbound fractions from the oligodT25 purification were used as non-displayed Nb controls (Figure 3d and Figure 5).

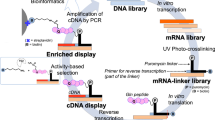
Schematic for mRNA/cDNA display of Nbs.
(a) Nb-encoding DNA is in vitro transcribed to (b) mRNA and (c) ligated to a 3′ puromycin (P) containing DNA linker. (d) Following in vitro translation and fusion formation, (e) reverse transcription is performed to synthesize cDNA. (f) mRNA-cDNA-Nb complexes are exposed to the antigen (Ag) of interest, which is immobolized on a solid-phase affinity matrix. Following washes and elution, PCR is performed to retrieve full-length Nb-encoding amplicons (N/C – N- & C-termini of displayed Nb).

mRNA/cDNA display of Nb-GFP.
(a) Nb-GFP-encoding DNA12 was in vitro transcribed to (b) mRNA and (c) ligated to a 3′ puromycin (P) containing DNA linker. The shift in molecular weight was tracked on an agarose gel. (d) In vitro translation resulted in displayed mRNA-Nb-GFP covalent fusions (F), detected on a western blot by an increase in molecular weight to ~170 kD, v/s non-displayed Nb-GFP (~15 kD). (e) Fusions (+ve) or untranslated ligations (−ve) were reverse transcribed and (f) binding-selection was performed on Ni-NTA-immobilized purified GFP protein. Binder Nbs were eluted and sequences were regenerated using PCR. The absence of PCR product from –ve control demonstrates successful antigenic selection. (M – molecular weight markers).

Specificity of antigenic selection during mRNA/cDNA display.
The mutant library that was constructed based on the Nb-GFP template was displayed using the mRNA/cDNA display method described in this manuscript and exposed to either GFP or mouse P-glycoprotein (mP-gp), which is an integral membrane transporter protein purified as previously published18. Following washes and elution, the binder sequences were regenerated using PCR. The agarose gel shows that for the same input material (library and RT reaction), Nb-specific binders were obtained for selection with GFP as the antigen, 40% sequences of which were confirmed to be Wt Nb-GFP, whereas Nb-specific PCR product was absent following exposure to mP-gp as the antigen, confirming the mRNA/cDNA display method was successful in removing non-specific binder Nbs.

mRNA/cDNA display of the Human Single-domain Library (HuSdL).
The HuSdL was displayed as described in the materials and methods section. OligodT25-purified and covalently linked mRNA-Nb fusion product was detected by a large increase in molecular weight (~170 kD) on a western blot using an anti-FLAG-mouse M2 primary antibody (Sigma). The non-displayed Nbs (~15 kD) were observed in the unbound fraction from the oligodT25-purification step. The lanes shown are from one and the same blot and are representative of 4 independent experiments.
Reverse Transcription
A 60 µL reaction was setup using the eluted fusion, 80 nM RT primer (Table II) and the remaining components of the superscript III first strand synthesis kit (Life Technologies), as per the manufacturer's instructions. The enzyme was inactivated at 70°C for 15 min.
Antigen-Nb binding and selection
1–3 µg His-tagged Pierce recombinant GFP (ThermoFisher) or purified GLUT-1 was bound to 50 µL of magnetic Ni-NTA agarose beads (Qiagen), pre-equilibrated in Bind buffer (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 0.05% Tween 20) at 4°C for 2 hr. 2 µg BSA (Amresco) was added to this reaction and further incubated at 4°C for 1 hr. Following a wash each with 5 column volumes of Bind and Wash/Interact buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% Tween 20, 20 mM imidazole, pH 8.0), the beads were resuspended in Wash/Interact with 2 µg BSA. The reverse transcription reaction was added to the beads and incubated on a room temperature tube rotator for 1 hr. The beads were washed 5 times with a total of 25 column volumes (5 × 5) of Wash/Interact, twice with a total of 10 column volumes (5 × 2) Tris-Wash buffer (20 mM Tris-HCl, pH 8.0, 0.05% Tween 20, 20 mM imidazole, pH 8.0) and eluted with 30 µL of Elution buffer (20 mM Tris-HCl, pH 8.0, 0.05% Tween 20, 300 mM imidazole, pH 8.0).
Sequence Retrieval and Analysis
Antigen-specific Nb sequences were retrieved through a PCR using 1 µL of the elution in a 100 µL reaction setup with platinum taq DNA polymerase and primers Nb-GFP_Disp_Fw and Nb-GFP_Disp_Rv or HuSdL_Disp_Fw2 and HuSdL_Disp_Rv (Table II). Freshly amplified and column-purified (Qiagen) PCR product was cloned into pCR2.1-TA® (Life Technologies) and transformed into E. coli Top 10 cells (Life Technologies) as per the manufacturer's instructions. Random colonies were sequenced using rolling circle amplification (RCA) and Sanger sequencing (Genewiz). Sequences were translated in silico using EXPASY-Translate and analyzed using protein BLAST®. Nb sequences of interest were amplified from the RCA product using Cloned Pfu polymerase (Agilent) and primers HuSdL_TOPO_Fw and HuSdL_TOPO_Rv (Table II). Trace plasmid in the reaction was digested using 1 µL Dpn1 (NEB) at 37°C for 1 hr. Freshly amplified and column-purified PCR product was cloned into pET100-D-TOPO® (Life Technologies) and transformed E. coli Top 10 cells. Plasmids from the transformant colonies were confirmed by sequencing to contain the Nb sequence for correct in-frame expression. Following sequence confirmation, plasmids were transformed into E. coli BL21 (DE3) Star (Life Technologies) for small-/large-scale Nb expression and purification.
Nb-GLUT-1 expression and purification
Small-scale expression tests for Nbs cloned into pET100-D-TOPO in E. coli BL21 (DE3) star cells were performed in 5 ml of Luria-Bertani (LB) medium containing 50 µg/ml carbenicillin (Omega Scientific). Cells were cultured at 37°C with shaking at 220 rpm for 16 hr. The lac promoter was induced by isopropyl-β-thiogalactoside (IPTG, Anatrace) for 1 hr. 500 µl of cells were harvested by centrifugation and resuspended in 100 µl of water and 100 µl of 2 X tris-glycine SDS sample buffer (Life Technologies). Samples were run on a 4–20% Mini PROTEAN TGX pre-cast gel (Bio-Rad Laboratories, Inc.). Expression was detected on western blot using mouse monoclonal His-probe antibody (Santa Cruz Biotechnology) as primary detection at 1:2000 dilution in TBST (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.005% Tween-20) and with goat-anti-mouse poly-HRP (Thermo Scientific) as secondary detection at 1:10,000 dilution in TBST. SuperSignal West Dura Chemiluminescent Substrate was used to develop the blot (Thermo Scientific). Highly expressed clones were selected for large-scale purifications.
For large-scale purifications Nbs were overexpressed in the BL21 (DE3) star cells in terrific broth (120 g of tryptone, 240 g of yeast extract and 40 ml of glycerol in 10 L) in a BioFlo 415 fermenter. The lac promoter was induced by IPTG (Anatrace) at OD of 1.5 for 2 hr. The cells were harvested and disrupted with a cell disruptor (Constant Systems) containing 20 mM Tris-HCl pH 8.0, 100 mM NaCl and 15% glycerol. Cells were disrupted at 20 kpsi. Cell debris was removed by centrifugation with JLA 16.25 rotor at 16,000 rpm for 30 minutes. The supernatant was collected and incubated with nickel affinity resin (Ni-NTA Agarose, Qiagen) at 4°C for 1.5 hours. The resin was rinsed with the buffers containing different concentrations of imidazole (30 mM, 60 mM and 80 mM) in 20 mM HEPES pH 7.5 and 150 mM NaCl. The protein was eluted from the resin with wash buffer plus 300 mM imidazole. The eluate was then diluted 10-fold again and incubated with Ni-NTA Agarose for 1.5 hours. The resin was rinsed again with the buffers containing different concentrations of imidazole (30 mM and 60 mM) in 20 mM HEPES pH 7.5 and 150 mM NaCl. The protein was eluted from the resin with wash buffer plus 300 mM imidazole. The protein was concentrated to about 5 mg/ml before applying to sizing exclusion chromatography column (Superdex 200 10/300; GE Healthcare) in the buffer containing 20 mM HEPES pH 7.5 and 150 mM NaCl). The largest peak fractions were collected and flash-frozen in liquid nitrogen for SPR and western blot experiments.
GLUT-1 cloning
Human cDNA was obtained from the transporter collection (Open Biosystems, Pittsburgh, PA). An epitope flag tag (5′-ATG GAC TAC AAG GAC GAT GAC GAT AAG-3′) was introduced at the 5′-end and TEV-10xHis tag (5′-GAG AAC TTG TAC TTC CAG GGT AGA GGT TCT CAC CAC CAT CAC CAC CAT CAC CAC CAT CAC-3′) was inserted at 3′-end of SLC2A1, then subcloned into the Pichia pastoris expression vector, pPICZ-A (Life Technologies, Carlsbad, CA), at EcoRI and SalI sites. 5 ug of plasmids were linearized with PmeI (NEB, Ipswich, MA) and introduced into P. pastoris KM71H electro-competent cells (Life Technologies, Carlsbad, CA). Total 48 colonies were chosen to small-scale expression with 0.5% methanol to test for expression of GLUT-1, which was detected with anti-flag antibody (Sigma, St. Louis, MO) or anti-his antibody (Clontech, Mountain View, CA) by western blot.
GLUT-1 purification
The full-length GLUT-1 with an N-terminal Flag tag and a C-terminal 10X His tag was over-expressed in Pichia pastoris in minimal media in a BioFlo 415 fermenter. Eighteen hours after induction with 5% methanol, cells were harvested and disrupted in 20 mM Tris-HCl pH 8.0, 100 mM NaCl and 15% glycerol with a cell disruptor (Constant Systems) at 40 kpsi. Cell debris was removed by centrifugation with JLA 8.1 rotor at 5000 rpm for 20 minutes and membrane fraction was isolated using centrifugation with JLA 16.25 rotor at 16,000 rpm for 3 hours. The protein was extracted from membrane fraction with 0.5% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace) in 20 mM Tris-HCl pH 8.0, 100 mM NaCl and 15% glycerol at 4°C for 2.5 hours. The detergent-soluble fraction was collected and incubated with nickel affinity resin (Ni-NTA Superflow, Qiagen) at 4°C for 1.5 hours. The resin was rinsed with the buffers containing different concentrations of imidazole (30 mM, 60 mM and 80 mM) in 20 mM HEPES pH 7.5, 150 mM NaCl, 0.001% (w/v) LMNG. The protein was eluted with the wash buffer plus 300 mM imidazole. The eluate was concentrated to about 1 mg/ml before applying to a desalting column (HiPrep 26/10; GE Healthcare) in the buffer containing 20 mM HEPES pH 6.7, 0.001% LMNG). The peak fractions were collected and flash frozen in liquid nitrogen for SPR.
Surface plasmon resonance (SPR)
SPR measurements were performed using the 404pi (Bioptix). For each experiment, about 3 µM of GLUT-1 in 20 mM HEPES, pH 6.7, 0.001% LMNG was immobilized on a CMD-200/150 sensorchip (Xantec/Bioptix) surface, using an amine coupling reaction with activation through 231 mg/mL EDC (1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide) and 33 mg/mL NHS (N-Hydroxysuccinimide) and post-immobilization deactivation using 1 M Tris-HCl, pH 7.5. GLUT-1 was immobilized at a flow-rate of 20 µL/min to 2,000 response units (RUs), while the control surface was treated with 20 mM HEPES, pH 6.7, 0.001% LMNG with identical activation and deactivation procedures. The instrument running buffer used during this step was 20 mM HEPES, pH 7.5, 20 mM NaCl, 50 µM EDTA, 0.001% LMNG, 0.005% Tween 20. The running buffer was then exchanged to 20 mM HEPES, pH 7.5, 150 mM NaCl, 50 µM EDTA, 0.001% LMNG, 0.005% Tween 20. Nb-GLUT-1 (or other Nbs) was injected at a flow-rate of 50 µL/min and at concentrations ranging from 1 nM up to 1 µM, with several intermediate buffer blank injections. Regenerations were performed with 5 µL of 0.1 M NaOH. Data were analyzed on Scrubber2 (BioLogic Software) using the double-subtraction method. On/association-rate (ka), off/dissociation-rate (kd) and dissociation constant (KD) values were computed from the fit function available on the software.
Western blot detection of GLUT-1 using Nb-GLUT-1
Full-length GLUT-1 and Nb-GLUT-1 were purified according to the protocol above and the concentrations were determined by comparing with the albumin standards (Thermo Scientific) on Coomassie-stained gels. Purified GLUT-1 and Nb-GLUT-1 samples were run on 12% or 4-20% gradient Mini PROTEAN TGX pre-cast SDS-PAGE gels (Bio-Rad Laboratories, Inc.). The samples on the gels were transferred on to nitrocellulose/PVDF membranes with the Trans-Blot Turbo Transfer System (Bio-Rad Laboratories, Inc.). Blocking was performed with either 5% skimmed milk in TBST buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.05% Tween-20; up to 0.1% Tween-20 was used at times to reduce background noise) for 30 min or PVA (polyvinyl alcohol) diluted 1:50 in TBST for 2 min. Washes and antibody treatments were conducted either traditionally or with the SNAPid system (EMD Millipore Corporation). For detecting GLUT-1, purified Nb-GLUT-1 at 2 mg/mL was used as the primary antibody at 1:1000 dilution in TBST and HRP-rProtein A (Life Technologies) as the secondary antibody at 1:5000 dilution in TBST. For detecting Nb-GLUT-1, 1:5000 HRP-rProtein A was used directly without requiring a secondary antibody. SuperSignal West Femto Chemiluminescent Substrate was used to develop the blots (Thermo Scientific).
Results
Since its invention, many variant versions of mRNA/cDNA display have been developed5,7,8,14,15,16. These methods usually require the use of higher microgram-milligram quantities of starting material (mRNA), or contain several gel-extraction or dialysis steps that pose hindrances to automation. To date, mRNA/cDNA display has never been reported for discovering Nbs. Here, we describe a highly integrated, streamlined, micro-scale mRNA/cDNA display method, which is amenable to high-throughput automation and demonstrate its adaptation for discovering high-affinity Nbs against full-length human integral membrane protein targets, which has been a serious challenge using conventional methods9,10,17.
In order to develop and optimize the mRNA/cDNA method for displaying Nbs, we chose a control Nb scaffold of known specificity to green fluorescent protein (GFP)12, termed Nb-GFP (Table I). mRNA/cDNA display makes use of the antibiotic, puromycin that mimics the aminoacyl moiety of tRNA, enters the ribosomes during translation and accepts the growing polypeptide chain via a peptide bond (Figure 2). Thus, when mRNA is modified at its 3′ end with puromycin, the C-terminus of the encoded/translated protein gets covalently bound to the 3′ end of the nucleic acid7 (Figure 2).
The detailed protocol for mRNA/cDNA display of Nbs is provided in the methods section and a schematic in Figure 2. Briefly, we in vitro transcribed the E. coli-codon-optimized Nb-GFP gene (Figure 3a, b) and used a DNA splint-assisted RNA-DNA ligation method to link a DNA oligo, containing a polyA sequence and a puromycin group at its 3′, to the 3′ end of the Nb mRNA (Figure 3c). In contrast to gel extraction that has been historically used to separate the ligated products from un-reacted molecules7,15,16, we used enzymatic cleavage coupled with oligodT-affinity chromatography to purify the ligated product (Figure 3c). In vitro Nb translation was performed using the PURExpress® In Vitro Protein Synthesis Kit (New England Biolabs) and mRNA-Nb covalent ‘fusions’ were created by the post-translational addition of Mg+2 and K+1 salts and incubation at −20°C overnight (Figure 3d). The fusions were purified using oligodT-affinity chromatography, reverse transcribed (Figure 3e) and exposed to the histidine-tagged target antigen, which was pre-immobilized on a Ni-NTA affinity solid phase. Following washes to remove unbound Nb, Fusion-target complexes were eluted from the solid phase and full-length Nb-encoding genes were re-generated using PCR. Sequences were analyzed by TA Cloning® (Life Technologies).
In order to validate our method, we constructed a mutant Nb-GFP library using error-prone PCR13, containing a calculated diversity of ~1 × 1010 unique sequences bearing 68-100% identity with the Wt Nb-GFP sequence. This library was displayed against GFP and, after only one round of binding and selection, 40% of the selected and sequenced binders were found to be Wt Nb-GFP (Figure 4). To validate functional Nb-Ag specificity, we replaced GFP with an integral membrane protein, mouse P-glycoprotein (mP-gp)18 as the antigen. Following elution, the selection with mP-gp resulted in failure to retrieve the full-length Nb-specific PCR product (Figure 4), leaving the off-target 200–300 bp amplicons to dominate the PCR. These non-specific amplicons were found to be a common result of mispriming-led templates from reverse transcription, typically increasing in proportion in the absence of Nb-specific templates.
Thus, our mRNA/cDNA display method was successful in retrieving an enriched parental sequence population among ~1010 mutated variants after the complete cycle. Our method was also successful in producing Nbs that maintain high antigenic specificity.
In demonstrating the widespread utility of our method, we performed mRNA/cDNA display using a commercially available library of Nbs. We chose the Human Single-domain antibody Library or HuSdL® (Creative Biolabs), which was constructed, based on the camelised human VH3 with elongated and randomized HCDR3 sequences and has a calculated diversity of 1.5 × 109 as per the manufacturer. We introduced the sequence elements necessary for in vitro expression using overlap-extension PCR and displayed the HuSdL using our optimized method (Figure 5). Successful covalent mRNA-Nb fusions were detectable by western blot, which showed a high molecular weight band (~170 kD) for displayed Nbs v/s non-displayed Nb (~15 kD) (Figure 5). As a test antigen, we used the full-length human integral membrane protein GLUT-1 (protein product of the gene SLC2A1), which is responsible for mediating glucose uptake in erythrocytes, glucose supply to the brain and is implicated in various diseases11.
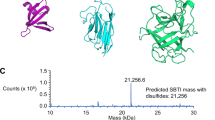
GLUT-1 was expressed at high levels in the yeast, Pichia pastoris and purified in detergent solution using affinity and size-exclusion chromatographies (Figure 6a). GLUT-1 is a 55 kD protein that runs at an apparent weight of ~43 kD on an SDS-PAGE gel (Figure 6a), probably due to the anomalous binding of SDS that is typical of integral membrane proteins19. Following three continuous rounds of mRNA/cDNA display-selection against purified GLUT-1, binder Nb sequences were retrieved. These Nbs were cloned, expressed in E. coli, purified using affinity and size-exclusion chromatographies (Figure 6a). The size-exclusion step is critical, as the Nbs from the human VH-derived HuSdL may dimerize20 (Figures 6a, 6c). The purified Nbs were ranked by binding affinity (KD) for detergent-purified GLUT-1 as measured by surface plasmon resonance (SPR). The best binder, named Nb-GLUT-1 (Table 1), has a mean KD of 22.3 ± 11.5 nM, with a mean association/on-rate (ka) of 1.2 ± 0.4 (× 105) M−1s−1 and a mean dissociation/off-rate (kd) of 1.6 ± 0.3 (× 10−3) s−1 (All data are Mean ± SEM, n = 4) (Figure 6b).

Nb-GLUT-1 binds and detects GLUT-1.
(a) Purified GLUT-1 and Nb-GLUT-1 were quantified by SDS-PAGE against bovine serum albumin (BSA) standards. (b) SPR was used to assess antigen-Nb binding. GLUT-1 or control detergent-buffer solution was immobilized on an amine-coupling sensor chip and Nb-GLUT-1 was flown as the analyte. Data shown are double-referenced and are from an experiment that was replicated 4 times with independent batches of purified proteins. (c) HRP-conjugated protein-A antibody directly detects (Left) Nb-GLUT-1, but not (Middle) GLUT-1 on a western blot. Right - Lower µg amounts of GLUT-1 were successfully detected on a western blot using Nb-GLUT-1 as the primary antibody, followed by HRP-conjugated protein-A as the secondary antibody. Data shown have been replicated 5 times with independent batches of purified proteins.
The ability to discover target-specific Nbs ‘on-the-fly’ can be particularly useful for the rapid detection of untagged targets by western blot. To this end, we show that Nb-GLUT-1 can be used as a primary antibody to detect lower µg amounts of GLUT-1 by western blot (Figure 6c). This result also demonstrates that Nb-GLUT-1 indeed binds to GLUT-1 and not to any background contaminants that could be present in the purified protein preparations.
Taken together, the display-selection cycles based on antigenic co-precipitation in the presence of BSA, the SPR data in the presence of detergent solution and the western blot results demonstrate that Nb-GLUT-1 binds to GLUT-1 with high affinity. This Nb is the first-ever reported for GLUT-1 and represents a first-generation Nb discovered without any deliberate affinity maturation procedures.
Discussion
In summary, we demonstrate the first use of mRNA/cDNA display for the discovery of Nbs against target antigens, including “hard” targets such as membrane proteins. A total of only <20 µg of mRNA (5 ug/round of display), <10 µg of purified target antigen (1-3 ug/round of display) and commercial readily-available materials are required for this method. We propose the use of our method for Nb discovery in cases where animal immunization4 may not be appropriate or when in vitro molecular evolution procedures are to be employed13,21,22.
Care is advised while handling RNA in vitro due to omnipresent RNases, although the inclusion of RNase inhibitors can significantly reduce losses. In our experience, the mRNA-puromycin linker ligation step should be optimized for each library, as this process appears to be dependent on the heterogeneity in size of mRNA fragments. Typically, ligation yields increase from the second round onwards due to the presence of more defined sequences of Nbs that bind to a particular antigen. Another area of care is the fusion formation step when mRNA covalently bonds with the Nb through puromycin. This step is critically dependent on the input ligation concentration, puromycin linker length and composition, in vitro translation system, incubation times and salt concentrations. Our recommended protocol is optimized for successful mRNA/cDNA display of Nbs.
The Nb discovery procedure described herein can be completed within 2–3 weeks per antigen and also multiplexed for several antigens in parallel. Furthermore, our method has the potential to become fully automated as the entire reaction is totally in vitro and requires mechanical procedures such as accurate aspirations, incubations, centrifugation, magnetic separations and temperature cycling. Nbs are extremely resistant to thermo-degradation allowing for a bench-top device to run the display cycles over a span of several days. Human interference may only be required during troubleshooting through quality control analytical gels and for sequence analysis, which could also perhaps be automated with the help of machine-learning algorithms.
The successful adoption of this method into modern synthetic biology may be bolstered through improvements such as novel defined libraries9, the use of non-natural amino acids23 and the addition of in vitro affinity maturation techniques to the display cycle13,21,22. We envision our method to form the basis for an automated bench-top antibody discovery platform that could receive antigenic inputs and following a few cycles of display, selection and affinity maturation, result in high affinity binder scaffold outputs.
References
Muyldermans, S. Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775 (2013).
De Meyer, T., Muyldermans, S. & Depicker, A. Nanobody-based products as research and diagnostic tools. Trends Biotechnol. 32, 263 (2014).
Williams, S. C. P. Small nanobody drugs win big backing from pharma. Nat. Methods 19, 1355 (2013).
Pardon, E. et al. A general protocol for the generation of nanobodies for structural biology. Nature protocols 9, 674 (2014).
Wada, A. Development of next-generation peptide binders using in vitro display technologies and their potential applications. Front. Immunol. 4, 10.3389/fimmu.2013.00224 (2013).
Hanes, J. & Plückthun, A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. USA 94, 4937 (1997).
Roberts, R. W. & Szostak, J. W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. USA 94, 12297 (1997).
Wang, H. & Liu, R. Advantages of mRNA display selections over other selection techniques for investigation of protein-protein interactions. Expert Rev. Proteomics 8, 335 (2011).
Seeger, M. A. et al. Design, construction and characterization of a second-generation DARPin library with reduced hydrophobicity. Protein Sci. 22, 1239 (2013).
Seddon, A. M., Curnow, P. & Booth, P. J. Membrane proteins, lipids and detergents: not just a soap opera. Biochim. Biophys. Acta Biomembranes 1666, 105 (2004).
Leen, W. G. et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain 133, 655 (2010).
Rothbauer, U. et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 3, 887 (2006).
Zaccolo, M., Williams, D. M., Brown, D. M. & Gherardi, E. An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol. 255, 589 (1996).
Seelig, B. mRNA display for the selection and evolution of enzymes from in vitro-translated protein libraries. Nat. Protocols 6, 540 (2011).
Olson, C. A. et al. Rapid mRNA-display selection of an IL-6 inhibitor using continuous-flow magnetic separation. Angew. Chem. Int. Ed. Engl. 50, 8295 (2011).
Keefe, A. D. in Current Protocols in Molecular Biology (John Wiley & Sons, Inc., 2001).
Hino, T., Iwata, S. & Murata, T. Generation of functional antibodies for mammalian membrane protein crystallography. Curr. Opin. Struct. Biol. 23, 563 (2013).
Ward, A. B. et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 110, 13386 (2013).
Rath, A., Glibowicka, M., Nadeau, V. G., Chen, G. & Deber, C. M. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proceedings of the National Academy of Sciences 106, 1760 (2009).
Baral, T. N. et al. Crystal structure of a human single domain antibody dimer formed through VH-VH non-covalent interactions. PloS one 7, e30149 (2012).
Zhao, H., Giver, L., Shao, Z., Affholter, J. A. & Arnold, F. H. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat. Biotechnol. 16, 258 (1998).
Stemmer, W. P. C. Rapid evolution of a protein in vitro by DNA shuffling. Nature 370, 389 (1994).
Murray, C. J. & Baliga, R. Cell-free translation of peptides and proteins:from high throughput screening to clinical production. Curr. Opin. Chem. Biol. 17, 420 (2013).
Acknowledgements
This work was supported by the NIH EUREKA grant GM94955.
Author information
Authors and Affiliations
Contributions
R.D. and G.C. designed, conducted and analyzed experiments and wrote the paper. C.R.T.V., B.R.C. and C.W.L. designed and conducted parts of the experiments. N.H. provided technical assistance under R.D.'s supervision.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Doshi, R., Chen, B., Vibat, C. et al. In vitro nanobody discovery for integral membrane protein targets. Sci Rep 4, 6760 (2014). https://doi.org/10.1038/srep06760
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06760
This article is cited by
-
Evaluation of the yeast surface display system for screening of functional nanobodies
AMB Express (2020)
-
Functional characterization and discovery of modulators of SbMATE, the agronomically important aluminium tolerance transporter from Sorghum bicolor
Scientific Reports (2017)
-
VHH antibodies: emerging reagents for the analysis of environmental chemicals
Analytical and Bioanalytical Chemistry (2016)
-
Characterization and applications of Nanobodies against human procalcitonin selected from a novel naïve Nanobody phage display library
Journal of Nanobiotechnology (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
