
Abstract
Lactobacillus brevis alcohol dehydrogenase (Lb-ADH) catalyzes reduction of prochiral carbonyl compounds to chiral alcohol and meanwhile consumes its cofactor NADH into NAD+, while the cofactor regeneration can be catalyzed by Candida boidinii formate dehydrogenase (Cb-FDH). This work presents three different Escherichia coli whole-cell biocatalyst systems expressing recombinant ADH/FDH, FDH-LIN1-ADH and FDH-LIN2-ADH, respectively, all of which display very high efficacies of prochiral carbonyl conversion with respect to conversion rates and enantiomeric excess values. ADH/FDH represents co-expression of Lb-ADH and Cb-FDH under different promoters in a single vector. Fusion of Lb-ADH and Cb-FDH by a linker peptide LIN1 (GGGGS)2 or LIN2 (EAAAK)2 generates the two bifunctional enzymes FDH-LIN1-ADH and FDH-LIN2-ADH, which enable efficient asymmetric reduction of prochiral ketones in whole-cell biotransformation.
Similar content being viewed by others
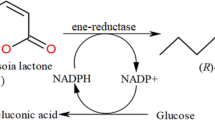
Introduction
Chiral alcohols are used as intermediates in a variety of multi-step chiral syntheses1. For instance, optically active phenylethanols and their derivatives are useful building blocks for synthesis of flavours, agrochemicals and pharmaceuticals2,3. Currently, chemical synthesis method is often used for industrial-scale production of chiral alcohols, but this method requires a rigorous condition and pollutes the environment and moreover the product has a low stereoselectivity. The use of oxidoreductases for asymmetric transformation of prochiral substrates has become an alternative to traditional chemical pathways to yield chiral intermediates at high purity. In particular, oxidoreductases are widely used in catalyzing asymmetric reduction of prochiral ketones to corresponding alcohols4.
Lactobacillus brevis alcohol dehydrogenase (Lb-ADH), an R-specific alcohol dehydrogenase belonging to the extended superfamily of short-chain dehydrogenases/reductases (SDRs), is a homotetrameric enzyme with 251 amino acid residues and 26,627 Da molecular weight for each monomer5. Lb-ADH is of industrial interest because it recognizes a broad variety of keto and diketo compounds, which contain a residue of bulky group such as phenyl, as its substrates. The preferred substrates of Lb-ADH are acetophenone and its derivates, which cannot be recognized as substrates by any of commercially available ADHs.
ADH is dependent on its cofactor NADH or NADPH for catalysis, but these cofactors are too expensive to be used in stoichiometric amounts6. Thus, the application of a second enzyme, e.g. formate dehydrogenase (FDH), for regeneration of cofactor NADH from NAD+ is of great importance in enzymatic research. The major advantage of using FDH for cofactor recycling is presence of almost irreversible catalytic reaction that provides close to 100% product yield at ambient conditions. In addition, formate and carbon dioxide themselves seldom influence activities of the partner enzymes that make use of generated NADH7. Candida boidinii FDH (Cb-FDH) has been employed as a workhorse for NADH-regeneration for decades8.
The most frequently applied strategy by far for producing chiral alcohols is whole-cell biotransformation via cells harboring two separated enzymes, using one enzyme for asymmetric transformation of prochiral substrates and the other one being an oxidoreductase in combination with a cheap co-substrate for cofactor regeneration9. In recent years, application of whole-cell biotransformation in commercial synthesis of chiral alcohols has undergone a revolution with the development of protein engineering10.
Bifunctional fusion proteins in which functional-coupled enzymes are connected through a linker have been constructed as an alternative to using two separated enzymes10. An adjacent active centre could reduce transfer distance of substrates and could decrease degradation of intermediate products11,12. A fusion protein of Mycobacterium vaccae FDH and Synechococcus KR [3-ketoacyl- (acyl-carrier-protein) reductase] displays an approximately two fold higher initial (1 h post-reaction) reaction rate in production of chiral alcohols than cells expressing the enzymes separately13.
In the present study, we constructed recombinant E. coli Rosetta (DE3) strains expressing bifunctional Lb-ADH-linker-Cb-FDH fusion enzymes to achieve high efficient whole-cell catalysis of prochiral carbonyl compounds into chiral alcohol (Figure 1).

Production of chiral alcohols with bifunctional fusion proteins.
Lb-ADH catalyzes reduction of prochiral ketones to (R)-alcohols, while Cb-FDH catalyzes regeneration of cofactor NADH.
Results
Over-expressed recombinant enzyme proteins
The two proteins Lb-ADH and Cb-FDH were co-expressed under different promoters in a single vector pETDuet-adh/fdh (Table 1). In addition, four different fusion proteins, namely ADH-LIN1-FDH, ADH-LIN2-FDH, FDH-LIN1-ADH and FDH-LIN2-ADH (each arranged from N- to C-terminus), were expressed with vectors pETDuet-adh-lin1-fdh, pETDuet-adh-lin2-fdh, pETDuet-fdh-lin1-adh, pETDuet-fdh-lin2-adh, respectively (Table 1). Expression of each recombinant protein in soluble form in E. coli Rosetta (DE3) cell supernatants was induced with isopropyl-β-D-thiogalactopyranoside (IPTG) and target proteins were separated through SDS-PAGE (Figure S1), digested with trypsin and analyzed by mass spectrum. The resulting peptide fragments were matched at coverage rates > 40% with target fusion proteins after theoretical trypsin digestion (Table 1 and Table S2).
Enzymatic activity in whole-cell extracts
The whole-cell extracts of induced E. coli Rosetta (DE3) cells expressing the above recombinant enzyme proteins were subjected for determination of ADH or FDH enzymatic activity (Table 1). Co-expressed ADH/FDH displayed an ADH activity of 103.90 ± 1.84 U/gCDW and a FDH activity of 32.47 ± 1.84 U/gCDW, while the enzymatic activities of the ADH or FDH moiety in the above four difunctional fusion proteins were ranged from 70.9 to 77.2% or from 30.8 to 384.6%, respectively (Table 1). Arrangement of ADH at the N- or C-terminus of the fusion proteins had little effect on its enzymatic activity; by contrast, the FDH moiety at N-terminus was much more active that at C-terminus.
Among these four bifunctional fusion enzymes, FDH-LIN2-ADH displayed the most active enzymatic activity and therefore was selected for analysis in detail using ADH/FDH as control. The residual ADH or FDH activity in whole-cell extract of Rosetta (DE3)-pETDuet-adh/fdh or Rosetta (DE3)-pETDuet-fdh-lin2-adh after incubation at 30 to 70°C for 10 min were determined; the ADH or FDH enzymatic stability for both ADH/FDH and FDH-LIN2-ADH decreased sharply with increasing of temperatures and notably FDH-LIN2-ADH displayed slightly higher stability than ADH/FDH at higher temperatures (40 to 70°C) (Figure 2). In addition, 0.2 M phosphate buffer at pH 7.0 was shown to be the optimized buffer condition for enzymatic reaction for both ADH/FDH and FDH-LIN2-ADH (Figure 3).

Effect of temperature on enzymatic activity.
Filled diamond or square indicates Rosetta (DE3)-pETDuet-fdh-adh or Rosetta (DE3)-pETDuet-fdh-lin2-adh, respectively. Experiments were done with three biological replicates (three independent bacterial cultures). Error bars indicate SD.

Effect of phosphate or pH on enzymatic activity.
Filled diamond or square indicates the ADH or FDH activity in Rosetta (DE3)-pETDuet-fdh-lin2-adh, respectively. Experiments were done with three biological replicates (three independent bacterial cultures). Error bars indicates SD.
Whole-cell biotransformation
Exemplary asymmetric reduction of acetophenone or 4-chloroacetophenone (4-Cl-AP) was conducted to evaluate whole-cell biotransformation efficacy in the six recombinant strains, Rosetta (DE3)-pETDuet-adh/fdh, Rosetta (DE3)-pETDuet-fdh-lin1-adh, Rosetta (DE3)-pETDuet-fdh-lin2-adh, Rosetta (DE3)-pETDuet-adh-lin1-fdh, Rosetta (DE3)-pETDuet-adh-lin2-fdh and Rosetta (DE3)-pETDuet (negative control), from 1 to 6 h post-reaction. Either acetophenone (Figure 4) or 4-Cl-AP (Figure 5) being as substrate, the former three strains displayed very high conversion rates (>85.0%) or e.e (enantiomeric excess) values [>95.0% (R)] at 6 h post-reaction, while the later two worked like the negative control. Notably, the initial (1 h post-reaction) rate of 4-Cl-AP conversation catalyzed by Rosetta (DE3)-pETDuet-fdh-lin2-adh was about 1.7 fold higher than that by Rosetta (DE3)-pETDuet-adh/fdh.

Efficiency of coupled acetophenone reduction.
□, Rosetta (DE3)-pETDuet-adh/fdh; *, Rosetta (DE3)-pETDuet-adh-lin1-fdh; ×, Rosetta (DE3)-pETDuet-adh-lin2-fdh; Δ, Rosetta (DE3)-pETDuet-fdh-lin1-adh; ◊, Rosetta (DE3)-pETDuet-fdh-lin2-adh; and ○, Rosetta (DE3)-pETDuet (negative control). Broken lines indicate conversion rates, while scattered diagrams denote e.e. values. Experiments were done with three biological replicates (three independent bacterial cultures). Error bars indicate SD.

Efficiency of coupled 4-Cl-AP reduction.
□, Rosetta (DE3)-pETDuet-adh/fdh; *, Rosetta (DE3)-pETDuet-adh-lin1-fdh; ×, Rosetta (DE3)-pETDuet-adh-lin2-fdh; Δ, Rosetta (DE3)-pETDuet-fdh-lin1-adh; ◊, Rosetta (DE3)-pETDuet-fdh-lin2-adh; ○, Rosetta (DE3)-pETDuet (negative control). Broken lines indicate conversion rates, while scattered diagrams denote e.e. values. Experiments were done with three biological replicates (three independent bacterial cultures). Error bars indicate SD.
Discussion
The Cb-FDH monomer consists of 15 α-helices and 13 β-strands that are arranged into two domains, termed NAD+ binding domain and catalytic domain, respectively7. Each domain contains three layers of polypeptide chain. Most of the 364 amino acid residues of Cb-FDH monomer are well-defined in electron density, but an exception is at the C-terminus, where 11 residues (354 to 364) are disordered7. The deletion of this 11-residue region in Cb-FDH leads to a >70% loss of its enzymatic activity (data not shown), highlighting the necessity of disordered C-terminus for Cb-FDH enzymatic activity. The catalytic activity of Lb-ADH depends strongly on the binding of Mg2+. The magnesium ion is structurally coupled to the putative C-terminal hinge of the substrate-binding loop forming the substrate binding region5.
When two oligomeric enzymes are fused together with an appreciate linker peptide, a multimeric protein network of the two parent enzymes is usually generated13. Choosing of linker peptide is vital for constructing a bifunctional fusion protein. The flexible peptide LIN1 (GGGGS)2 and the α-helical peptide LIN2 (EAAAK)2 have been successfully used for constructing various bifunctional fusion enzymes14,15.
The E. coli Rosetta (DE3) strain compensates for a number of rare codon genes and thus is frequently used for high-level expression of eukaryotic or prokaryotic proteins. This work presents three different recombinant E. coli Rosetta (DE3) whole-cell biocatalyst systems expressing ADH/FDH, FDH-LIN1-ADH and FDH-LIN2-ADH, respectively, all of which display very high efficacies of prochiral carbonyl conversion. ADH/FDH represents co-expression of Lb-ADH and Cb-FDH under different promoters in a single vector. Fusion of Lb-ADH and Cb-FDH with LIN1 or LIN2 generates bifunctional fusion enzymes FDH-LIN1-ADH and FDH-LIN2-ADH. FDH-LIN1-ADH and FDH-LIN2-ADH consists of two oxidoreductases, of which one catalyzes reduction of prochiral ketones and the other one mediates regeneration of cofactors. For both FDH-LIN1-ADH and FDH-LIN2-ADH, the spatial separation of Lb-ADH and Cb-FDH subunits by an appropriate linker peptide enable two individual moieties fold independently and correctly and thereby ensure activity of both native enzymes. By contrast, there are probably hindered folding of FDH and thus reduced FDH activity for the two additional fusion proteins ADH-LIN1-FDH and ADH-LIN2-FDH constructed in this study.
The substrate conversion rates are very similar among ADH/FDH, FDH-LIN1-ADH and FDH-LIN2-ADH at 6 h post-reaction during whole cell biotransformation. Nevertheless, FDH-LIN1-ADH and FDH-LIN2-ADH exhibit higher e.e values than ADH/FDH when 4-Cl-AP is used as substrate, indicating that these two fusion proteins have elevated enantioselectivity compared with ADH/FDH. FDH-LIN1-ADH and FDH-LIN2-ADH exhibit very similar performances at 1 h and 6 h post-reaction during whole cell biotransformation, although FDH-LIN1-ADH has an FDH activity about 65% that of FDH-LIN2-ADH; this might be due to very similar balanced activities of FDH and ADH [the ideal ratio of activity of the two enzymes is 1:n (n ≥ 1)] between FDH-LIN1-ADH and FDH-LIN2-ADH.
Methods
Bacterial strains and growth
For co-expression of L. brevis adh and C. boidiniifdh with a single expression vector, these two genes were cloned into the two different multiple cloning sites of pETDuet™-1 (Novagen) to generate pETDuet-adh/fdh. These two genes were placed under two separate T7 lac promoters and expression of one gene would not affect that of the other16. All the primers designed in this study were listed in Table S1.
The linker peptides LIN1 (GGGGS)2 and LIN2 (EAAAK)2 were used to construct four different chimeric genes, namely adh-lin1-fdh, adh-lin2-fdh, fdh-lin1-adh and fdh-lin2-adh, by splicing-by-overlap extension method17. The intact chimeric genes were cloned into pETDuet-1 to generate pETDuet-adh-lin1-fdh, pETDuet-adh-lin2-fdh, pETDuet-fdh-lin1-adh and pETDuet-fdh-lin2-adh.
Each recombinant plasmid was transformed into E. coli Rosetta (DE3) for inducible over-expression of target recombinant proteins. Overnight cultures were diluted into fresh Luria-Bertani broth at an optical density at 600 nm (OD600) of 0.1 and allowed for grow at 37°C with shacking at 200 rpm to an OD600 of 0.8. After addition of 1 mM IPTG, the cell cultures were incubated at 20°C for 12 h with shacking at 200 rpm for cell harvest. 100 μg/m ampicillin and 20 μg/ml chloromycetin were added when necessary.
SDS-PAGE
Harvested cells were resuspended in distilled water to an OD600 of 3 and disrupted by sonication at 40 Hz and 4°C for 3 min, followed by centrifugation (10,000 × g, 4°C and 30 min). The supernatants were mixed with 2× Laemmli buffer (300 mM Tris/HCl, pH6.8, 50% glycerol, 10% SDS, 5% 2-mercaptoethanol and 0.05% Bromophenol Blue) and equal volumes of 1× Laemmli buffer were used to resuspend the corresponding insoluble pellets. The resulting samples were heated to 100°C for 5 min and separated on the SDS-12% PAGE gels followed by Coomassie Blue staining.
Peptide mass fingerprinting (PMF)
The presumed bands of target recombinant proteins on stained SDS-PAGE gels were carefully excised, destained and digested with trypsin. PMF assays were performed using an Ultraflex III TOF/TOF mass spectrometer (Bruker) at National Centre of Biomedical Analysis, Beijing, China. Data were analyzed with the online tool PeptideMass (http://web.expasy.org/peptide_mass/).
Enzymatic activity assays
The enzymatic activity of ADH was determined photometrically at 340 nm in an 1 ml reaction mixture composed of 50 mM acetophenone (Sigma-Aldrich), 100 μM NADH (Roche), 1 mM MgCl2, 200 mM potassium phosphate buffer (pH 7.0) and 100 μl of crude whole-cell extract after an incubation at 30°C for 2.5 h18. The 1 ml reaction mixture for FDH activity assay contained 100 mM sodium formate, 500 μM NAD+ (Roche), 200 mM potassium phosphate buffer (pH 7.0) and 100 μl of crude whole-cell extract19. For test of temperature stability, crude whole-cell extracts were incubated at 30 to 60°C for 10 min and then on ice for 5 min for further determination of residual enzymatic activity as above. For test of effect of phosphate or pH, the reaction mixtures with various concentrations of potassium phosphate (10 to 400 mM), or at different pH values (6 to 8), were subjected for enzymatic activity determination. One unit (U) of enzymatic activity was defined as the amount of enzyme catalyzing conversion of 1 μmol substrate/min/mg total protein at 30°C. The amounts of total protein in crude whole-cell extracts were determined with Thremo BCA Protein Assay Kit.
Whole-cell biotransformation
After induction with IPTG, bacteria cells were harvested by centrifugation at 5,000 × g for 5 min and washed with 200 mM potassium phosphate buffer (pH 7.0). 0.08 g (w.t.) of cell pellets were resuspended in 500 μl of reaction solution (corresponding to 6 g/cdw) containing 200 mM sodium formate, 10 mM acetophenone or 2.5 mM 4-Cl-AP, 1 mM MgCl2 and 200 mM potassium phosphate buffer (pH 7.0). Biotransformation was conducted at 37°C for indicated times in a mute mixture to prevent cell sedimentation.
High-performance liquid chromatography (HPLC)
500 μl of biotransformation suspension were extracted with the same volume of n-hexane and 10 μl of the resulting supernatant was analyzed using a HPLC chromatograph (Waters) connected to a CHIRALPAK OD-H column (Daicel Chemical Industries, Ltd., Tokyo, Japan). The mobile phase n-hexane and 2-propanol (90:10, v/v) were used for quantitative analysis of the bioconversion products of acetophenone or 4-Cl-AP. The conversion rates were calculated with the formula ‘100% × volumes of product after conversion/volume of substrate before conversion’. The e.e values were calculated with the formula ‘100% × (R − S)/(R + S)’. R = R-phenylethanol or (R)-4-Chloro-1-phenylethanol. S = S-phenylethanol or (S)-4-Chloro-1-phenylethanol.
References
Zheng, G. W. & Xu, J. H. New opportunities for biocatalysis: driving the synthesis of chiral chemicals. Curr. Opin. Biotech. 22, 784–792 (2011).
Zilbeyaz, K., Taskin, M., Kurbanoglu, E. B., Kurbanoglu, N. I. & Kilic, H. Production of (R)-1-phenylethanols through bioreduction of acetophenones by a new fungus isolate Trichothecium roseum. Chirality 22, 543–547 (2010).
Zilbeyaz, K. & Kurbanoglu, E. B. Production of (R)-1-(4-Bromo-phenyl)-ethanol by locally isolated Aspergillus niger using ram horn peptone. Bioresour Technol. 99, 1549–1552 (2008).
Ni, Y. & Xu, J. H. Biocatalytic ketone reduction: a green and efficient access to enantiopure alcohols. Biotechnol. Adv. 30, 1279–1288 (2012).
Niefind, K., Müller, J., Riebel, B., Hummel, W. & Schomburg, D. The crystal structure of R-specific alcohol dehydrogenase from Lactobacillus brevis suggests the structural basis of its metal dependency. J. Mol. Biol. 327, 317–328 (2003).
Macleod, S. M. & Stickler, D. J. Species interactions in mixed-community crystalline biofilms on urinary catheters. J. Med. Microbiol. 56, 1549–1557 (2007).
Schirwitz, K., Schmidt, A. & Lamzin, V. S. High-resolution structures of formate dehydrogenase from Candida boidinii. Protein Sci. 16, 1146–1156 (2007).
Kratzer, R., Pukl, M., Egger, S. & Nidetzky, B. Whole-cell bioreduction of aromatic alpha-keto esters using Candida tenuis xylose reductase and Candida boidinii formate dehydrogenase co-expressed in Escherichia coli. Microb. Cell Fact. 7, 37; 10.1186/1475-2859-7-37 (2008).
Goldberg, K., Schroer, K., Lutz, S. & Liese, A. Biocatalytic ketone reduction--a powerful tool for the production of chiral alcohols-part II: whole-cell reductions. Appl. Microbiol. Biot. 76, 249–255 (2007).
Prachayasittikul, V., Ljung, S., Isarankura-Na-Ayudhya, C. & Bulow, L. NAD(H) recycling activity of an engineered bifunctional enzyme galactose dehydrogenase/lactate dehydrogenase. Int. J. Biol. Sci. 2, 10–16 (2006).
Correa, A. & Oppezzo, P. Tuning different expression parameters to achieve soluble recombinant proteins in E. coli: advantages of high-throughput screening. Biotech. J. 6, 715–730 (2011).
Torres Pazmino, D. E. et al. Self-sufficient Baeyer-Villiger monooxygenases: effective coenzyme regeneration for biooxygenation by fusion engineering. Angew. Chem. Int. Edit. 47, 2275–2278 (2008).
Holsch, K. & Weuster-Botz, D. Enantioselective reduction of prochiral ketones by engineered bifunctional fusion proteins. Biotechnol. Appl. Bioc. 56, 131–140 (2010).
Arai, R., Ueda, H., Kitayama, A., Kamiya, N. & Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 14, 529–532 (2001).
Lu, P. & Feng, M. G. Bifunctional enhancement of a beta-glucanase-xylanase fusion enzyme by optimization of peptide linkers. Appl. Microbiol. Biot. 79, 579–587 (2008).
Xiao, Z. et al. A novel whole-cell biocatalyst with NAD+ regeneration for production of chiral chemicals. PLoS One 5, e8860, 10.1371/journal.pone.0008860 (2010).
Xiong, A. S. et al. PCR-based accurate synthesis of long DNA sequences. Nat. Protoc. 1, 791–797 (2006).
Ernst, M., Kaup, B., Muller, M., Bringer-Meyer, S. & Sahm, H. Enantioselective reduction of carbonyl compounds by whole-cell biotransformation, combining a formate dehydrogenase and a (R)-specific alcohol dehydrogenase. Appl. Microbiol. Biot. 66, 629–634 (2005).
Madje, K., Schmolzer, K., Nidetzky, B. & Kratzer, R. Host cell and expression engineering for development of an E. coli ketoreductase catalyst: enhancement of formate dehydrogenase activity for regeneration of NADH. Microb. Cell Fact. 11, 7; 10.1186/1475-2859-11-7 (2012).
Acknowledgements
This work is supported by National Natural Science Foundation of China (81000763). The English writing of this manuscript was polished by Elsevier's Language Editing Service.
Author information
Authors and Affiliations
Contributions
Y.L., D.Z. and H.C. designed experiments. B.L., Y.X.L., D.B., X.Z., H.Y., J.W., G.L., J.Y., Y.L., D.Z. and H.C. performed experiments. B.L., Y.L. and D.Z. analyzed data. B.L., Y.X.L., Y.L., D.Z. and H.C. contributed reagents, materials and analysis tools. D.Z., B.L., Y.L. and H.C. wrote this manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Dataset 1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Li, B., Li, Y., Bai, D. et al. Whole-cell biotransformation systems for reduction of prochiral carbonyl compounds to chiral alcohol in Escherichia coli. Sci Rep 4, 6750 (2014). https://doi.org/10.1038/srep06750
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06750
This article is cited by
-
Production of Tyrian purple indigoid dye from tryptophan in Escherichia coli
Nature Chemical Biology (2021)
-
Characterization and Application of a Robust Glucose Dehydrogenase from Paenibacillus pini for Cofactor Regeneration in Biocatalysis
Indian Journal of Microbiology (2020)
-
A Rieske oxygenase/epoxide hydrolase-catalysed reaction cascade creates oxygen heterocycles in mupirocin biosynthesis
Nature Catalysis (2018)
-
Enzymatic characterization of a recombinant carbonyl reductase from Acetobacter sp. CCTCC M209061
Bioresources and Bioprocessing (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
