
Abstract
A missing cornerstone in the development of tough micro/nano fibre systems is an understanding of the fibre failure mechanisms, which stems from the limitation in observing the fracture of objects with dimensions one hundredth of the width of a hair strand. Tensile testing in the electron microscope is herein adopted to reveal the fracture behaviour of a novel type of toughened electrospun poly(methyl methacrylate)/poly(ethylene oxide) fibre mats for biomedical applications. These fibres showed a toughness more than two orders of magnitude greater than that of pristine PMMA fibres. The in-situ microscopy revealed that the toughness were not only dependent on the initial molecular alignment after spinning, but also on the polymer formulation that could promote further molecular orientation during the formation of micro/nano-necking. The true fibre strength was greater than 150 MPa, which was considerably higher than that of the unmodified PMMA (17 MPa). This necking phenomenon was prohibited by high aspect ratio cellulose nanocrystal fillers in the ultra–tough fibres, leading to a decrease in toughness by more than one order of magnitude. The reported necking mechanism may have broad implications also within more traditional melt–spinning research.
Similar content being viewed by others

Introduction
Significant efforts are being made to introduce electrospun fibres on a commercial scale using large-scale electrospinning processing equipment1,2. Electrospun ultrathin fibres are currently used in filters and membranes and they are also being aimed towards emerging applications such as transparent composites, scaffold materials for tissue and templates for cell growth and reinforcement in biomedical silicone materials1,3,4,5.
A major challenge in reaching the targeted applications is the lack of fibre toughness, due to the fine fibre dimensions that make electrospun fibres sensitive to handle6,7. Electrospun fibres have, therefore, mostly been applied as non-woven mats where the fibres support one another, or collected on a support material as in filter applications1. Attempts to reinforce electrospun polymer fibres by embedding “rod–like” nanofillers which serve as load-carriers have been reported. Carbon nanotubes (CNTs) have received considerable attention due to their high strength and high elastic modulus combined with a high aspect ratio >100, but extracted cellulose nanofibrils that show more flexibility have also been considered2,8,9,10,11,12. Composite fibres with a higher modulus and strength were obtained in both cases, but the fibres were more brittle2,10,11. The absence of toughness was due to the problematic embedding of the often bent, coiled or spiral-shaped CNTs (occasionally protruding from the surface of the fibres) and to inadequate filler/matrix adhesion and poor dispersion of the nanofiller inside the polymer matrix2,10,11. Recently, Papkov et al.6 and Asran et al.13 demonstrated that tough nanofibres can be obtained by optimizing molecular orientation/alignment instead of preparing composite fibres. When the diameter of electrospun fibres is reduced (larger draw ratios), an increase in strength and strain-at-break is generally observed6,14,15,16,17,18,19,20,21. However, no direct observation of the fibre failure mechanism has so far been reported for toughness-improved fibres and their electrospun fibre mats.
In this paper, data for electrospun fibres based on blends of poly(methyl methacrylate) (PMMA) and poly(ethylene oxide) (PEO), with a toughness more than two orders of magnitude greater than that of pristine PMMA are presented. Tensile testing was carried out in-situ in the scanning electron microscope on samples consisting of ca. 100 000 parallel fibres. A template transfer method was used to ensure the non-destructive and reproducible transfer of the fibres to the micromechanical stage, without any premature stretching of the fibres22 after collection of the mats on a rotating cylinder operated at 2000 rpm. The testing revealed that the toughness improvement depend not only on spinning thin fibres but also on finding a polymer formulation that promoted extensive fibre necking prior to fracture, i.e. molecular alignment during fibre stretching. Flexible cellulose nanofibrils were evaluated as rod-like load-bearers inside the toughest PMMA/PEO blend to demonstrate their influence on the fracture and mechanical fibre performance. The materials presented are biocompatible23,24,25 and they offer the possibility to achieve toughened PMMA-based electrospun fibre systems in applications where non-harmful formulations are required.
Results and Discussion
Assessment of tensile data for electrospun PMMA/PEO fibre blends
Fig. 1a shows the stress–strain behaviour of aligned PMMA/PEO fibres with PEO contents ranging from 0 to 25 wt% (note the split logarithmic strain axis). In this concentration range the polymers are miscible26,27,28,29. All the fibres had a diameter of ca. 2 µm, which allowed an unbiased analysis of the fibre failure mechanisms of the different polymer blends. Fig. 1b and Table 1 show that the toughness of the fibres based on a 5 wt% PEO blend was twice that of the fibres based on pristine PMMA. A lower concentration of PEO (2 wt%) had essentially no effect on the tensile properties of the fibres (Table 1). However, the toughness increased strongly with the content of PEO: 26 and 134 times that of pristine PMMA for 12.5 and 25 wt%, respectively and the fracture energy increased from 0.13 MJ/m3 for PMMA to 17.1 MJ/m3 for the blend with 25 wt% PEO. The glass transition temperature (Tg) decreased from 124°C for the pristine PMMA to 75°C for the PMMA blend with 25 wt% PEO, confirming that a more ductile but glassy fibre material was formed. It should be noted that both the Young's modulus and the tensile strength of the fibres containing 5 and 12.5 wt% PEO were higher than those of the pristine PMMA fibres. The increase in Young's modulus was 18 and 11% and the tensile strength 11 and 77%, respectively. For comparison, the mechanical properties of solvent-cast films with 25 wt% PEO were evaluated under the same testing conditions to determine whether the improvement in toughness was entirely related to the polymer formulation. These solvent-cast films showed almost the same strength and modulus as the electrospun fibres (Table 1), but with 61% lower toughness than the fibres. It was assumed that the greater toughness of the fibres was solely due to the greater molecular alignment within the fibres, resulting from the rapid fibre stretching in the electrical field, as suggested by Papkov et al.6 However, the in-situ microscopy characterization revealed that the molecular alignment from the electrospinning was only part of the explanation of the observed increase in fibre toughness.

(a) Stress-strain curves of fibre mats of PMMA/PEO blends with 0, 5, 12.5 and 25 wt% of PEO (note the split strain-axis to reveal the strain at break for the fibre mats with the highest toughness. (b) Toughness as a function of PEO content and cellulose content ( : 4 wt% BC,
: 4 wt% BC,  : 10 wt% BC). (c) Fibre diameter distribution data of fibre mat samples. (d) Scanning electron micrographs of aligned fibre mats.
: 10 wt% BC). (c) Fibre diameter distribution data of fibre mat samples. (d) Scanning electron micrographs of aligned fibre mats.
Necking phenomena in toughness-improved PMMA fibres
Fig. 2a shows the tensile testing curve recorded during in-situ SEM analysis of the fibre mat consisting of PMMA fibres with 25 wt% PEO. The testing stage was stopped at different strain levels; 4, 8, 20, 40 and 100% – to investigate the failure behaviour of the fibre mat (Figs. 2 c–f). A progressive thinning and sequential failure of individual fibres explained the gradual decrease in stress with increasing strain, when individual fibres fractured. Higher magnification micrographs of individual fibres showed that all the fibres deformed via extensive necking and plastic deformation before failure. Necking was observed already at the early stage of the deformation (8% strain, Fig. 2d) and when the strain was increased the necked regions propagated uniformly along the fibres until complete fibre mat failure occurred between 130 and 150% strain. The formation of the necks resulted in an average reduction in fibre cross–sectional area from 3.8 µm2 to 0.5 µm2 (10 observed fibre necks). The true stress in the fibres necks could therefore be calculated to be ca. 150 MPa before failures started to appear at 40% strain and an engineering stress of 14.8 MPa.

(a) In-situ SEM tensile testing curve of electrospun PMMA with 25 wt% PEO fibres; (b) high resolution micrograph of an early developed fibre neck; (c) the fibre mat prior to tensile testing; (d) multiple parallel necking of individual fibres at 8% strain; (e) progressive thinning of fibre mat and; (f) breakage/collapse of aligned and stretched fibres.
Fig. 2b shows a close-up inspection of an isolated unloaded fibre at the early stage of necking, where a 4 µm neck had developed at 20% strain. The wrinkled surface in the neck was not observed in-situ and it is assumed that it originates from an elastic recovery when the fibre was unloaded for high-resolution microscopy. For comparison, Fig. 2d displays several 10–15 µm-long necked regions at 8% strain under load (in-situ). The smaller neck in the unloaded fibre exposed to an apparently larger strain may originate from an uneven loading of this particular fibre due to differences in the fibre alignment (Table 1) or stretching. This highlights the challenge of small-scale measurements. However, this necking length difference over the entire 5 mm sample corresponded to a variation of only ca. 0.2% over the entire mat deformation. In contrast, the necking was always absent in pristine PMMA fibres because fibre tolerated a strain of only 1.5%, which resulted in crisp and undeformed fracture surfaces (Fig. 3b) at a true stress close to that of the entire mat, i.e. ca. 17 MPa. The shape of the stress-strain curve in Fig. 2a (compared to that in Fig. 1a) shows stress relaxation, because the tensile stage was stopped for long periods (total ca. 30 min) to take the micrographs. This relaxation is common for polymers if the deformation is interrupted and was not a result of exposure to the electron beam since only a fraction of the entire mat was exposed. When the load was reinstated, a slightly higher stress was noted, probably due different relaxations among the fibres (some fibres were not optimally aligned), which in turn resulted initially in a more even load distribution over the fibres. The relaxation was not due to moisture since there was no moisture in the SEM chamber and a water uptake less than 0.1 wt% was measured when the samples were removed from the vacuum chamber and placed in an environment with a humidity of 50% RH.

(a) Stress-strain curves of aligned PMMA/PEO fibre mats containing different amounts of bacterial cellulose (BC) (3–5) compared to those of pristine PMMA and PMMA fibres with 25 wt% PEO; SEM fibre fracture cross sections of (a) pristine PMMA, (b) PMMA with 25 wt% PEO, (d) PMMA with 25 wt% PEO and 10 wt% BC; (e) TEM of 10 wt% BC in PMMA with 25 wt% PEO.
In order to see whether the observed necking phenomenon was a unique feature of the electrospun fibres, a blend based on the same PMMA with 25 wt% PEO was extruded into 2.7 mm thick continuous bars for comparison. A mini twin–screw extruder with a short sleeve die (2 mm) was used to induce minimal molecular orientation along the bars. The melt-extruded bars showed no signs of necking during tensile testing at the same strain rate as that used for the electrospun fibre mats. Only a small narrowing just prior to fracture was observed and the necking phenomenon was thus unique for the electrospun fibres. The maximal elongation of the bars was ca. 36%, with an insignificant toughness increase compared to that of the same formulation used in the electrospun fibres; see mechanical data in Table 1 and the photograph of the bar in supplementary information, figure S4.
Effect of rod–like cellulose nanofibrils as reinforcement in necking fibres
In order to investigate the effect of rod-like reinforcements in the electrospun fibres (PMMA with 25 wt% PEO), bacterial cellulose (BC) nanofibrils were embedded in the fibres at 4 and 10 wt%. The cellulose nanofibrils were extracted from bacterial cellulose (procedure found in supplementary information S1) and had a crystal content of 72%30. The reported modulus of such cellulose fibrils/crystals is ca. 100 GPa and they have a strength of the order of 7–8 GPa30,31,32,33,34,35. The average length of the cellulose fibrils was 1100 ± 700 nm, with an aspect ratio (L/h) of ca. 9430. The BC fibrils were dispersed and aligned parallel with the fibre axis in the electrospun fibres due to their favourable compatibility with the PMMA and their flexibility in the oscillating electrospinning jet. The darker regions in Fig. 3e show the aligned bacterial cellulose crystals. The composite fibres with 10 wt% BC showed an increase in the Young's modulus and tensile strength of 171% and 55%, respectively (Table 2 and Fig. 3a (curves 2 and 4)) compared to the same fibres without cellulose. The electrospun fibres containing 4 wt% BC showed no change in strength but a 25% increase in the Young's modulus (Table 2 and Fig. 3a (curves 2 and 3)). However, the increase in modulus and strength came at the expense of a significant loss in toughness, by more than one order of magnitude (from 17 to 0.6 MJ/m3), Fig. 1b (Table 2). Figure 3a and Table 2 also show the previously reported increase in strength and modulus of PMMA with 4 wt% nanofibrillated cellulose extracted from wood (WC) (curves 1 and 5)22. Electron micrographs of the fractured BC containing fibres revealed uneven fibre necking and surface porosity development around the fracture surfaces and also pull-outs of the cellulose nanofibrils (Fig. 3d compared to Fig. 3c). Accordingly, even if a relatively well-dispersed cellulose nanofibre phase was present (without protruding nanofibres), it appeared that the considerably stiffer crystals prohibited necking of the electrospun fibres. The cellulose nanofibrils from wood, with a smaller aspect ratio and a lower crystallinity did, however, lead to a higher toughness than that of the brittle pristine PMMA polymer.
Fibre characterization by X-ray diffraction and IR-spectroscopy
X-ray diffraction was performed to see whether PEO crystals could have contributed to the improved mechanical properties of the fibres (or affected the necking characteristics) and IR-dichroism measurements was used to assess chain orientation. It has previously been shown that PMMA/PEO blends with PEO contents greater than 25 wt% are biphasic26 and that PEO crystallizes into PEO-rich spherulites with PMMA molecules confined in the inter-lamellar regions26,36. Fig. 4a shows diffractograms for the different blends. A fully X-ray amorphous structure was apparent for concentrations up to 12.5 wt% PEO, whereas a low crystallinity of 1.2 vol% was detected in the fibres based on the 25 wt% PEO blend. The samples with higher amounts of PEO, especially 25 wt% shows a slight shift of the amorphous halo towards smaller lattice spacing's, which may be explained by the lack of bulky side groups in PEO (allowing for smaller lattice space). Another possibility for this would be the presence of sub 3 nm crystals appearing amorphous (albeit having similar lattice spacing as the crystalline phase), which is below the detection limit for X-ray diffraction37.

WAXD diffractograms of the electrospun PMMA/PEO fibres with and without bacterial cellulose (a), IR spectrum for the toughest fibres containing 25 wt% PEO (b) with inset IR-dichroism (polarized IR) for the CH2 skeletal vibration peak in PMMA at 749 cm−1.
Electrospun fibres based on the polymer blend with 25 wt% PEO and varying amounts of BC, showed similar levels of PEO crystallinity (ca. 1.2 vol%, see Fig. 4a), however it was difficult to accurately assess the PEO crystallinity in this case, due to the presence of overlapping peaks from the crystalline cellulose.
Fig. 4b shows polarized IR spectra of the aligned electrospun PMMA with 25 wt% PEO. The absorption peak at 749 cm−1 of the skeletal vibrational motion of the CH2 group in PMMA38 showed IR-dichroism; the angle between the transition moment vector and the chain axis (denoted α) has been reported to be 17° 33. The IR-dichroism; R = A||/A⊥ = 1.228 (A||, A⊥ refer to the absorbance values for light polarized parallel to and perpendicular the fibre axis)39. The Herman's orientation function (f) was calculated according to40,41,42:

where ζ is the angle between chain axis and the director (fibre axis). The chain orientation f was 0.081. PMMA/PEO (ca. 25 wt% PEO) blend films required a draw ratio of 3 in order to achieve a similar orientation factor39. Since this measurement was made on a fibre mat with a non-perfect alignment of fibres (Fig. 1d and Table 1), the orientation of the individual fibres must have been slightly higher. However, the extensive increase in toughness of the electrospun fibres compared to previous data for PMMA/PEO, the cast films or the extruded bars, could be explained neither by the moderate initial molecular alignment nor by the crystallinity of the fibres after spinning.
Mechanisms for toughness improvement
The decrease in cross sectional area by a factor of 7.6 (3.8 to 0.5 um2) can be explained by considering that only a portion of the fibres undergo necking, i.e. via the necking deformation zone shown in Fig. 2 (whereas an affine deformation gives a reduction factor of only 3). The size of this deformation zone at any given time during deformation was ca. 100–200 nm in the case of the electrospun fibres. This length can be compared to the size of the PEO ( of 600 kDa) and the PMMA (
of 600 kDa) and the PMMA ( of 410 kDa) molecules, which would be 5.0 and 1.0 µm long if they were completely straight (0.154 nm bond length)42,43. It is therefore clear that the molecules could easily bridge the necking zone in a continuous stretching event (alignment) as the deformation zone propagated along the fibres during deformation. However, a more realistic explanation would be to relate the end-to-end distance of a random coil of the PEO polymer which in its coiled state can be approximately 66 nm (r = 61/2rg2)43, based on a radius of gyration (rg) of ca. 27 nm to the length scale of the deformation44. This end-to-end distance is smaller than the deformation zone but, since the electrospinning induces orientation of the polymer chains, the true end-to-end distance in the direction of the fibre can be expected to be significantly larger and to correspond to the initial molecular orientation, which was equivalent to a tensile draw ratio of 3 (Fig. 4b)39. The end-to-end distance along the direction of the fibre is therefore expected to cover about the same or a slightly larger distance than the deformation zone during the necking. This would allow a continuous stretching and energy build-up in the deformation zone, which greatly contributed to the toughness of the fibres. The molecules thus adopted an unprecedented degree of orientation after the deformation zone and this may resemble strain hardening45 and allow the necks to bear a higher load per cross–sectional area than the undeformed parts of the fibres.
of 410 kDa) molecules, which would be 5.0 and 1.0 µm long if they were completely straight (0.154 nm bond length)42,43. It is therefore clear that the molecules could easily bridge the necking zone in a continuous stretching event (alignment) as the deformation zone propagated along the fibres during deformation. However, a more realistic explanation would be to relate the end-to-end distance of a random coil of the PEO polymer which in its coiled state can be approximately 66 nm (r = 61/2rg2)43, based on a radius of gyration (rg) of ca. 27 nm to the length scale of the deformation44. This end-to-end distance is smaller than the deformation zone but, since the electrospinning induces orientation of the polymer chains, the true end-to-end distance in the direction of the fibre can be expected to be significantly larger and to correspond to the initial molecular orientation, which was equivalent to a tensile draw ratio of 3 (Fig. 4b)39. The end-to-end distance along the direction of the fibre is therefore expected to cover about the same or a slightly larger distance than the deformation zone during the necking. This would allow a continuous stretching and energy build-up in the deformation zone, which greatly contributed to the toughness of the fibres. The molecules thus adopted an unprecedented degree of orientation after the deformation zone and this may resemble strain hardening45 and allow the necks to bear a higher load per cross–sectional area than the undeformed parts of the fibres.
A possible contribution to the necking behaviour is the presence of a thin ductile surface layer with a lower glass transition temperature (Tg), which prevented crack initiation by more facile reorientation of molecules in the neck deformation zone. This surface layer of the same molecular composition as the interior of the fibres emanates from an enhanced molecular mobility in the proximity of the fibre surface46,47. Evidence of a gradually decreasing Tg with closer distance to the surface of polystyrene was recently reported, wherein a suppression of ca. 50°C (or more) was demonstrated 10–50 nm from the polymer surface, depending on the measurement technique used46,47. An equivalent suppression of the Tg for the 75/25 PMMA/PEO blend would result in a glass transition below room temperature, i.e. a ‘rubbery’ material at the surface of the fibres. To explore this hypothesis, the toughest fibre formulation (75/25 PMMA/PEO) was electrospun at the slowest feed–rate possible to generate uniform fibres (10 μL/min) with approx. a doubled specific surface area. The obtained fibres (diameter: 1.2 ± 0.1 μm) showed a Young's modulus that was reduced by ca. 50% and with similar strength as the 2 μm fibres. The lower modulus and more ductile fibres obtained therefore served as an indication that a larger portion of rubbery surface layer existed, assuming the existence of a rubbery surface layer of the same thickness. However, the obtained fibres also showed a ca. 65% decreased strain to failure, which was related to an increased molecular alignment for thinner fibres (as suggested by Papkov et al.)6, which prohibited them from aligning further during the tensile test.
It is also clear that the probability of encountering defects at any given time during deformation becomes smaller as the volume associated with the deformation decreases and that the mechanical properties of the entire mat are related to neck formation distributed in thousands of fibres deforming simultaneously. An approximate calculation shows that the deformation zone constitutes less than 1 ppm of the entire fibre length. Hence, even when a neck propagates upon a defect, the surrounding thousands of fibres can continue to stretch under load and absorb energy in the absence of defects. Based on the above reasoning and the assumption that the non-necking portion of the fibre did not deform at all, the maximum volume of the neck was calculated to be 23% of the total fibre volume (based on the reduction in fibre diameter via necking and the 150% strain at break). This “necked” volume of the fibres corresponded to 71% of the final fibre length. Hence, if the prepared fibre formulation had allowed complete propagation of the necks over the entire fibre length, the toughness of the fibres would have increased even more, i.e. if the remaining 77 vol% of the fibres had been allowed to form necks in the absence of defects. An upper limit for the toughness of electrospun fibres of a PMMA/PEO blend with 25% PEO could then be calculated to be approximately 75 MJ/m3, which is more than 300% the observed toughness and comparable to that of carbon steel48.
Conclusions
This work presents for the first time electrospinning as a method of preparing ultra-toughened biocompatible fibre networks based on blends of the inexpensive engineering polymers poly(methyl methacrylate) (PMMA) and polyethylene oxide (PEO). The key to the toughening was to prepare a sufficiently ductile polymer blend, which could undergo an extensive and energy-absorbing fibre necking phenomenon prior to fibre failure due to a suppressed glass transition temperature at the surface of the fibres. A simultaneous increase in toughness (27 times), modulus (10.7%) and tensile strength (77%) was observed for fibres containing 12.5 wt% PEO, 25 wt% PEO, the brittle PMMA showed an increase in toughness of more than two orders of magnitude. The toughness improvement was accompanied by an increase in true fibre strength of ca. one order of magnitude from 16.8 MPa to ca. 150 MPa, as verified in in-situ electron micrographs during the tensile testing. Cast films and extruded rods based on the same 25 wt% PEO blend showed respectively 61% and 82% lower toughness than the electrospun counterpart. It is suggested that molecular orientation at the energy-absorbing neck requires an initial degree of molecular alignment (obtained via electrospinning) that is sufficiently high so that the size of the polymer molecules in the direction of the fibre is of the same order of magnitude as the deformation zone. Fibres consequently break when the deformation zone reach a point where the initial alignment is insufficient. This reasoning is consistent with our findings that cast films and extruded rods, with a larger size and with presumably little alignment, have toughness inferior to that of the electrospun counterpart. To further investigate the necking ability of the electrospun fibres, experiments were performed with the addition of highly crystalline cellulose fibres (as rod-like reinforcement) to the PMMA/PEO solution prior to electrospinning. This resulted in hybrid composite fibres that were stiffer and stronger, but also showed dramatically reduced toughness due to an inability of the rod-like cellulose to adapt to the extensive deformation inherent in neck formation.
The reported phenomena are likely to change the views on how to develop simultaneously strong and strain compliant polymer formulations for thin-fibre applications, since the reported phenomena become apparent only when dimensions approach scales close to one hundredth of the width of a hair strand.
Methods
Preparation of fibre solutions
PMMA with a  of 410 kDa (Alfa Aesar) and PEO (Acros Organics) with a
of 410 kDa (Alfa Aesar) and PEO (Acros Organics) with a  of 600 kDa were dissolved in DMF (BDH Prolabo 99.8%), or in DMF/BC containing suspensions (the details of the preparation of BC with 72% crystalline content30 are provided in the Supplementary Information S1). The polymer solutions/suspensions were heated to 70°C and kept at this temperature under constant stirring until homogeneous and completely transparent suspensions were obtained (2 h). The solutions were electrospun within 24 h.
of 600 kDa were dissolved in DMF (BDH Prolabo 99.8%), or in DMF/BC containing suspensions (the details of the preparation of BC with 72% crystalline content30 are provided in the Supplementary Information S1). The polymer solutions/suspensions were heated to 70°C and kept at this temperature under constant stirring until homogeneous and completely transparent suspensions were obtained (2 h). The solutions were electrospun within 24 h.
Electrospinning and collection of fibre mats
The fibre solutions were continuously fed from a 5 mL solvent-resistant polypropylene syringe at a rate of 40 µL/min (±0.1%), via a PTFE tube, to a flat tip 18-gauge needle with internal diameter of 0.84 mm. The needle tip was positioned 240 mm vertically above the rotating collector and the electric field from the needle to the collecting surface was maintained at ca. 40–50 kV/m, where the more viscous solutions required a higher electric field. The electrospun fibres were deposited on the rotating cylindrical collector/drum at 2000 rpm (collector diameter = 50 mm). Thermo-gravimetric measurements were carried out on all the samples to ensure that the fibre mats contained no residual low volatile DMF solvent prior to tensile testing. Infrared (IR) spectroscopy was also performed for all samples prior to further characterization, in order to ensure that there was no solvent remaining inside the fibres (Supplementary Information S3).
Tensile testing of fibres
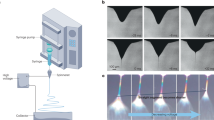
The tensile measurements were performed on a Deben Microtest stage, modified for direct A/D converter readout, which increased the resolution of the load cell (76 µN) and extensometer (167 nm). The measurements were carried out at a strain rate of 0.5 mm/min (10% of sample length per minute) and the tensile stress values were calculated by dividing the measured force by the cross sectional area of the fibre mat. The thickness of the fibre mat was derived from the area density (mass per unit area) of the electrospun fibre mat, as determined from the weight and area of an adjacent fibre mat on the same collector. The fibre tensile testing was performed according to the template transfer method (TTM)22. A layer of fluorinated ethylene propylene release film (thickness = 76 µm) was attached to the collector, followed by a pre-cut aluminium foil template (thickness = 30 µm) that was firmly attached to the surface of the collector directly over the release film with conductive copper tape (thickness = 66 µm). The template had a cut-out window (width: 10 mm, length: 5 mm) that acted as a gap where fibres could freely span. Fig. 5 shows an illustration of the TTM method.

(a) Template transfer configuration, illustrating the aligned fibres on the rotating collector and the aluminium foil cutout template. (b) Illustration of the fixed fibres on the template as mounted in the tensile stage during tensile testing.
The fibres were fixed on both sides of the window using alkoxy-ethyl-cyanoacrylate (Loctite 460, Henkel AG & Co. KGaA, Germany) before the aluminium template was moved to the tensile tester. The side panels of the pre-cut window were cut (without touching the fibres) immediately before testing when the template had been firmly mounted in the tensile stage22.
Preparation of cast films
The solution used for electrospinning was also used to cast films inside petri dishes by evaporating the solvent at a temperature of 50°C in vacuum for 2 days to ensure a complete evaporation of all the solvent. The films were cut and mounted for tensile testing in the same manner as the electrospun fibres.
Melt extrusion of bars
Powders of the same PMMA and PEO material were mixed in a DSM Xplore 5 ml micro twin-screw extruder at a temperature of 185°C. A short sleeve die was fitted with a 2 mm diameter that induced minimal molecular orientation to the final bars and this resulted in a final diameter of ca. 2.7 mm (due to die swelling). These much larger specimens than the cast films and electrospun fibres were tested in an Instron 5566 tensile testing machine with a specimen length of 40 mm, using the same strain rate as for all the other samples.
Microscopy
A Hitachi S-4800 cold–field–emission scanning electron microscope (SEM) was used. A ca. 2 nm coating of platinum–palladium was sputtered onto the surface of the samples (10 s at 80 mA) in a Cressington 208HR high-resolution sputter. The in-situ SEM tensile tests were performed in a Hitachi T-100 charge reduction SEM that allowed testing without sputtering in order not to alter the properties of the material.
X–ray diffraction (XRD)
XRD measurements were conducted on a PANalytical X'Pert Pro diffractometer using Cu–Kα radiation (45 kV, 35 mA) and a 1.00 arcmin step size. Crystallinity was calculated from the ratio of the integrated intensity of the crystalline peaks and the total intensity of the diffractograms (including the amorphous component).
Thermal analysis
Thermal characteristics of the electrospun mats were assessed by differential scanning calorimetry (DSC) using a Mettler Toledo DSC 1 with 40 µL aluminium cups and by thermal-gravimetric analysis (TGA) using a Mettler Toledo TGA/DSC 1 with 70 µL Al2O3 crucibles. The heating rate was 10°C/min and a nitrogen gas flow rate of 10 ml/min was used.
Infra red (IR) spectroscopy
IR spectroscopy was performed on a Perkin-Elmer Spectrum 2000 using a 1 cm−1 scan step equipped with a linear polarizer and a single reflection attenuated total reflectance stage (ATR) MKII Golden Gate unit (Specac Ltd., London, UK).
References
Luo, C. J., Stoyanov, S. D., Stride, E., Pelan, E. & Edirisinghe, M. Electrospinning versus fibre production methods: from specifics to technological convergence. Chem. Soc. Rev. 41, 4708–4735, 10.1039/c2cs35083a (2012).
Hou, H. Q. et al. Electrospun polyacrylonitrile nanofibers containing a high concentration of well-aligned multiwall carbon nanotubes. Chem. Mater. 17, 967–973, 10.1021/Cm0484955 (2005).
Jiang, S., Hou, H., Greiner, A. & Agarwal, S. Tough and transparent nylon-6 electrospun nanofiber reinforced melamine-formaldehyde composites. ACS Appl Mater Interfaces 4, 2597–2603, 10.1021/am300286m (2012).
Bergshoef, M. M. & Vancso, G. J. Transparent nanocomposites with ultrathin, electrospun nylon-4,6 fiber reinforcement. Adv. Mater. 11, 1362–1365, 10.1002/(Sici)1521-4095(199911)11:161362::Aid-Adma1362>3.0.Co;2-X (1999).
Swart, M., Olsson, R. T., Hedenqvist, M. S. & Mallon, P. E. Organic-Inorganic Hybrid Copolymer Fibers and Their Use in Silicone Laminate Composites. Polym. Eng. Sci. 50, 2143–2152, 10.1002/pen.21749 (2010).
Papkov, D. et al. Simultaneously Strong and Tough Ultrafine Continuous Nanofibers. ACS Nano 7, 3324–3331, 10.1021/nn400028p (2013).
Li, D., Wang, Y. L. & Xia, Y. N. Electrospinning of polymeric and ceramic nanofibers as uniaxially aligned arrays. Nano Lett. 3, 1167–1171, 10.1021/Nl0344256 (2003).
Zhou, C., Chu, R., Wu, R. & Wu, Q. Electrospun Polyethylene Oxide/Cellulose Nanocrystal Composite Nanofibrous Mats with Homogeneous and Heterogeneous Microstructures. Biomacromolecules 12, 2617–2625, 10.1021/bm200401p (2011).
Demczyk, B. G. et al. Direct mechanical measurement of the tensile strength and elastic modulus of multiwalled carbon nanotubes. Mat Sci Eng a-Struct 334, 173–178, 10.1016/S0921-5093(01)01807-X (2002).
Sung, J. H., Kim, H. S., Jin, H. J., Choi, H. J. & Chin, I. J. Nanofibrous membranes prepared by multiwalled carbon nanotube/poly(methyl methacrylate) composites. Macromolecules 37, 9899–9902, 10.1021/Ma048355g (2004).
Huang, C. B., Chen, S. L., Reneker, D. H., Lai, C. L. & Hou, H. Q. High-strength mats from electrospun poly(p-phenylene biphenyltetracarboximide) nanofibers. Adv. Mater. 18, 668, 10.1002/adma.200501806 (2006).
Olsson, R. T. et al. Extraction of Microfibrils from Bacterial Cellulose Networks for Electrospinning of Anisotropic Biohybrid Fiber Yarns. Macromolecules 43, 4201–4209, 10.1021/ma100217q (2010).
Asran, A. S., Seydewitz, V. & Michler, G. H. Micromechanical properties and ductile behavior of electrospun polystyrene nanofibers. J. Appl. Polym. Sci. 125, 1663–1673, 10.1002/App.34847 (2012).
Tan, E. P. S. & Lim, C. T. Physical properties of a single polymeric nanofiber. Appl. Phys. Lett. 84, 1603–1605, 10.1063/1.1651643 (2004).
Lim, C. T., Tan, E. P. S. & Ng, S. Y. Effects of crystalline morphology on the tensile properties of electrospun polymer nanofibers. Appl. Phys. Lett. 92, 10.1063/1.2857478 (2008).
Wong, S. C., Baji, A. & Leng, S. W. Effect of fiber diameter on tensile properties of electrospun poly(epsilon-caprolactone). Polymer 49, 4713–4722, 0.1016/j.polymer.2008.08.022 (2008).
Arinstein, A., Burman, M., Gendelman, O. & Zussman, E. Effect of supramolecular structure on polymer nanofibre elasticity. Nat Nanotechnol 2, 59–62, 10.1038/nnano.2006.172 (2007).
Pai, C. L., Boyce, M. C. & Rutledge, G. C. Mechanical properties of individual electrospun PA 6(3)T fibers and their variation with fiber diameter. Polymer 52, 2295–2301, 10.1016/j.polymer.2011.03.041 (2011).
Chew, S. Y., Hufnagel, T. C., Lim, C. T. & Leong, K. W. Mechanical properties of single electrospun drug-encapsulated nanofibres. Nanotechnology 17, 3880–3891, 10.1088/0957-4484/17/15/045 (2006).
Shin, M. K. et al. Size-dependent elastic modulus of single electroactive polymer nanofibers. Appl. Phys. Lett. 89, 10.1063/1.2402941 (2006).
Naraghi, M., Arshad, S. N. & Chasiotis, I. Molecular orientation and mechanical property size effects in electrospun polyacrylonitrile nanofibers. Polymer 52, 1612–1618, 10.1016/j.polymer.2011.02.013 (2011).
Andersson, R. L. et al. Micromechanical Tensile Testing of Cellulose-Reinforced Electrospun Fibers Using a Template Transfer Method (TTM). J. Polym. Environ. 20, 967–975, 10.1007/s10924-012-0486-6 (2012).
Allan, B. Closer to nature: new biomaterials and tissue engineering in ophthalmology. Brit J Ophthalmol 83, 1235–1240, 10.1136/bjo.83.11.1235 (1999).
Amon, M. & Menapace, R. Cellular invasion on hydrogel and poly(methyl methacrylate) implants: An in vivo study. J. Cataract Refract. Surg. 17, 774–779, 10.1016/S0886-3350(13)80410-5 (1991).
Dugan, J. M., Gough, J. E. & Eichhorn, S. J. Bacterial cellulose scaffolds and cellulose nanowhiskers for tissue engineering. Nanomedicine 8, 287–298 (2013).
Shi, W. & Han, C. C. Dynamic Competition between Crystallization and Phase Separation at the Growth Interface of a PMMA/PEO Blend. Macromolecules 45, 336–346, 10.1021/ma201940m (2012).
Li Xuan, W. Y. & Yang, Yang . Studies of the crystallization behavior in the crystalline/amorphous polymer blends: poly(ethylene oxide)/poly(methyl methacrylate) and poly(ethylene oxide)/poly(vinyl acetate). Polym. Commun. 280–288 (1985).
Lodge, T. P., Wood, E. R. & Haley, J. C. Two calorimetric glass transitions do not necessarily indicate immiscibility: The case of PEO/PMMA. J. Polym. Sci., Part B: Polym. Phys. 44, 756–763, 10.1002/polb.20735 (2006).
Schwahn, D., Pipich, V. & Richter, D. Composition and Long-Range Density Fluctuations in PEO/PMMA Polymer Blends: A Result of Asymmetric Component Mobility. Macromolecules 45, 2035–2049, 10.1021/ma2019123 (2012).
Sacui, I. et al. Comparison of the properties of cellulose nanocrystals and cellulose nanofibrils isolated from bacteria, tunicate and wood processed using acid, enzymatic, mechanical and oxidative methods. ACS Appl Mater Interfaces 6, 6127–6138, 10.1021/am500359f (2014).
Nishiyama, Y. Structure and properties of the cellulose microfibril. J. Wood Sci. 55, 241–249, 10.1007/s10086-009-1029-1 (2009).
Iwamoto, S., Kai, W., Isogai, A. & Iwata, T. Elastic Modulus of Single Cellulose Microfibrils from Tunicate Measured by Atomic Force Microscopy. Biomacromolecules 10, 2571–2576, 10.1021/bm900520n (2009).
Hepworth, D. G. & Bruce, D. M. A method of calculating the mechanical properties of nanoscopic plant cell wall components from tissue properties. J Mater Sci 35, 5861–5865, 10.1023/a,1026716710498 (2000).
Mark, R. E. Cell wall mechanics of tracheids. (Yale University Press, 1967).
Kroonbatenburg, L. M. J., Kroon, J. & Northolt, M. G. Chain Modulus and Intermolecular Hydrogen Bonding in Native and Regenerated Cellulose Fibres. Polym. Commun. 27, 290–292 (1986).
Martuscelli, E., Silvestre, C., Addonizio, M. L. & Amelino, L. Phase-Structure and Compatibility Studies in Poly(Ethylene Oxide) Poly(Methyl Methacrylate) Blends. Makromol Chem 187, 1557–1571 (1986).
Lezcano-Gonzalez, I. et al. Chemical deactivation of Cu-SSZ-13 ammonia selective catalytic reduction (NH3-SCR) systems. Applied Catalysis B: Environmental 154–155, 339–349, 10.1016/j.apcatb.2014.02.037 (2014).
Nagai, H. Infrared spectra of stereoregular polymethyl methacrylate. J. Appl. Polym. Sci. 7, 1697–1714, 10.1002/app.1963.070070512 (1963).
Zhao, Y., Jasse, B. & Monnerie, L. Orientation and relaxation in uniaxially stretched poly(methyl methacrylate) poly(ethylene oxide) blends. Polymer 30, 1643–1650, 10.1016/0032-3861(89)90324-8 (1989).
Hermans, P. H. & Platzek, P. Beiträge zur Kenntnis des Deformationsmechanismus und der Feinstruktur der Hydratzellulose. Kolloid-Z 88, 68–72 (1939).
Hermans, P. H. Contribution to the physics of cellulose fibres. (Elsevier Publishing Company Inc., 1946).
Gedde, U. W. Polymer Physics. (Springer, 1995).
Debye, P. The intrinsic viscosity of polymer solutions. J. Chem. Phys 14, 636–639 (1946).
Branca, C. et al. Study of Conformational Properties of Poly(ethylene oxide) by SANS and PCS Techniques. Phys. Scr. 67, 551 (2003).
Birley, A. W., Haworth, B. & Batchelor, J. Physics of Plastics: Processing, Properties and Materials Engineering. (Hanser Gardner Publications, 1992).
Ellison, C. J. & Torkelson, J. M. The distribution of glass-transition temperatures in nanoscopically confined glass formers. Nat Mater 2, 695–700 (2003).
Bäumchen, O., McGraw, J. D., Forrest, J. A. & Dalnoki-Veress, K. Reduced Glass Transition Temperatures in Thin Polymer Films: Surface Effect or Artifact? Phys. Rev. Lett. 109, 055701 (2012).
Callister, W. D. & Rethwisch, D. G. Fundamentals of Materials Science and Engineering: An Integrated Approach. (Wiley, 2012).
Acknowledgements
The Swedish International Development Cooperation Agency (SIDA) is acknowledged for financial support. The authors also thank M. Salajkova for her assistance in the high resolution SEM imaging of PMMA/PEO cross sections.
Author information
Authors and Affiliations
Contributions
The project was planned and overseen by M.H., U.G., P.M. and R.O. The experimental measurements were performed by R.A. as well as preparation of figures. Data analysis was performed by V.S. All authors contributed to the manuscript preparation.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Andersson, R., Ström, V., Gedde, U. et al. Micromechanics of ultra-toughened electrospun PMMA/PEO fibres as revealed by in-situ tensile testing in an electron microscope. Sci Rep 4, 6335 (2014). https://doi.org/10.1038/srep06335
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06335
This article is cited by
-
Synthesis and Boosting the Morphological and Optical Characteristics of SiC/SrTiO3 Nanomaterials Doped PMMA/PEO for Tailored Optoelectronics Fields
Silicon (2024)
-
Crosslinking of genipin and autoclaving in chitosan-based nanofibrous scaffolds: structural and physiochemical properties
Journal of Materials Science (2019)
-
Fabrication and characterization of low-cost, bead-free, durable and hydrophobic electrospun membrane for 3D cell culture
Biomedical Microdevices (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
