
Abstract
Edge-decorated graphene nanoribbons are investigated with the density functional theory; they reveal three stable geometric structures. The first type is a tubular structure formed by the covalent bonds of decorating boron or nitrogen atoms. The second one consists of curved nanoribbons created by the dipole-dipole interactions between two edges when decorated with Be, Mg, or Al atoms. The final structure is a flat nanoribbon produced due to the repulsive force between two edges; most decorated structures belong to this type. Various decorating atoms, different curvature angles and the zigzag edge structure are reflected in the electronic properties, magnetic properties and bonding configurations. Most of the resulting structures are conductors with relatively high free carrier densities, whereas a few are semiconductors due to the zigzag-edge-induced anti-ferromagnetism.
Similar content being viewed by others


Introduction
One-dimensional graphene nanoribbons have attracted considerable attention in the fields of chemistry, materials science and physics1,2,3,4,5,6,7,8 and various studies on electronic9,10,11, transport12,13,14,15,16 and magnetic17,18 properties have been conducted. The electronic properties of these nanoribbons are dominated by the edge structure, ribbon width19,20 and bulk defects21. Graphene nanoribbons possess two achiral structures: zigzag and armchair nanoribbons. The former belong to middle-gap semiconductors and the anti-ferromagnetic configuration is a more realizable structure22. On the other hand, the latter show non-magnetic configurations which own three types of width-dependent energy gaps20. Recently, curved and flat graphene nanoribbons have been successfully produced through several methods by unzipping carbon nanotubes3,4,5,23,24,25,26,27,28,29, e.g., a wet chemical method involving acid reactions3, a catalytic approach using metal nano-clusters as scalpels23 and a physicochemical method employing argon plasma treatment25. The curved structures and dangling bonds provide a suitable environment for modulating the geometric structure and electronic properties.
The edge decoration of flat nanoribbons drastically change the electronic properties. Metallic atoms30,31, non-metallic atoms32,33 and organic molecules34,35 have been used as decorating elements in theoretical calculations and experimental measurements. The low-energy electronic structures and spin configuration are severely altered by these decorating atoms. We theoretically study the edge-decorated graphene nanoribbons by investigating the curvature effect on the geometric and the electronic properties. Decorating atoms that have atomic number of lower than 30 and are able to bond with carbon atoms are used to systematically investigate the optimal geometric structures, which are affected by the atom-carbon distances, atom-atom distances, charge transfers and edge-atom interactions. Two special stable structures can form: a highly curved edge-decorated structure and a re-zipped tubular structure. The bending effects induced by the curvature enhance the total energy, while the edge-edge interactions can reduce or increase the total energy. It is the relationship, either competitive or cooperative, between the two effects that decides the resulting decorated structure.
The effects of the curvature and the decorating atoms can give rise to feature-rich electronic properties. A semiconducting zigzag nanoribbon, which corresponds to an unzipped armchair nanotube, has two separate partial flat bands above and below the chemical potential (μ = 0). However, an armchair nanotube is a 1D metal with gapless linear intersecting bands36. The curvature of curved nanoribbons is expected to cause their electronic properties to behave between the two extremes. This work shows that edge decorations can possibly destroy the intersecting bands or merge the split partial flat bands, or even create new energy bands crossing μ = 0. Edge decorations can determine whether the ribbon is a conductor or semiconductor—the former is associated with the decorating atom and its covalence bond, while the latter is related to the edge-structure-induced magnetic moments. Decorated systems that belong to conductors could have a free carrier density higher than or comparable to that of armchair carbon nanotubes and the electrons may be transported along the edge atoms. The selectivity of decorated structures is potentially important for a wide variety of applications in nanoelectronic devices.
Results
The introduction of decorating elements in the unzipping process can drastically affect the resulting geometric structures, as indicated in Figs. 1a and 1b. This selectivity can be used to form flat or curved nanoribbons, or even restore a cylindrical nanotube. These processes and the resulting geometric structures are investigated by the first-principles methods and three optimal geometric structures are obtained. Type I, a zipper-line decorated carbon nanotube (Fig. 1c), is formed by the covalent bonds between decorating atoms that form a zigzag chain on the zipping line. Type II is an edge-decorated curved nanoribbon (Fig. 1d), made up of a metal chain with interlaced dipole-dipole interactions between two edges that stabilize such a structure. Type III is a flat graphene nanoribbon with edge passivation (Fig. 1e), which is a stable planar structure due to the saturated edge atoms.

Geometric structures.
(a) A single wall carbon nanotube is used as starting material. (b) The unzipped nanotube is put in an environment of decorating atoms. (c)–(e) Three possible geometric structures are formed by three types of decorating atoms with different edge-edge interactions.
When an armchair carbon nanotube is being cut longitudinally, two kinds of nanoribbons result, that is, nanoribbons with an odd-ribbon-width or an even-ribbon-width (ribbon width is characterized by Ny). Both can form the three types of geometric structures mentioned above, but the odd-width ribbon is more stable and has unique electronic properties. This work therefore focuses on odd-width ribbons. We first discuss the formation of each geometric structures through qualitative analysis of its total energy. Secondly, different electronic configurations of decorating atoms bring about various types of edge-atom interactions. These are categorized into three types and may be identified based on the charge transfer, C-C bond length, etc. Finally, we study the atom distribution, which is very useful for further understanding the electronic properties.
A curved graphene nanoribbon can be regarded as a partial section of a cylindrical carbon nanotube. Its geometric structure is specified by the arc angle θ and the curvature radius R = W/θ × 180°/π, where W is the ribbon width. θ represents the ribbon curvature and conditions θ = 0° and θ ≠ 0° correspond to flat and curved ribbons, respectively. In order to determine the optimal geometric structures, we use various initial conditions of which the arc angles are from 0 to 360 degrees and calculate the energy difference (ΔEt) between the flat nanoribbon and each type of decorated structure. The critical angle θc is an angle with respect to the local maximum energy in Fig. 2a. Related to θc, the critical interaction distance dc is defined as the distance between the two ribbon edges. Depending on the critical angle, type I (II) is either a zipper line decorated nanotube (a highly curved edge-decorated nanoribbon) or a flat nanoribbon with edge decoration. When the initial angle is larger than the critical angle, zipper line decorated nanotube (type I) and highly curved ribbon (type II) are formed. Otherwise, the resulting structure is planar as the initial angle is smaller than the critical angle. Moreover, all type III structures are planar. The results indicate that the θ-dependent energy variation ΔEt is mainly attributed to the mechanical strain and the edge-edge atomic interactions, as shown in Fig. 2a. These two effects cooperate in deciding the resulting decorated structures for type III. However, this cooperation becomes a competition for type I and type II. Moreover, the θ-dependent energy for type II can be fitted as

where the first term is the strain energy which depends on the ribbon width Ny and the arc angle θ. The energy decreases monotonously as Ny increases. With a specific width, the increase of the energy is proportional to the square of θ, as shown by the black dash curve in Fig. 2a. This is obtained by fitting the data points from the previous study on curvature effect of nanoribbons37, On the other hand, the second term expresses the electric dipole moments from two ribbon edges based on a classical model of dipole-dipole interaction38. Further discussions will be provided later.

Formation energy.
(a) Arc-angle-dependent total energy per atom for various edge-decorated structures. The width-dependent energy barrier and the critical interaction distance of (b) a B-decorated nanotube and (c) a Be-decorated curved nanoribbon.
Different decorating atoms (B, Be, H and C) induce a dramatic change in the total energy and thus determine the optimal geometric structures. Based on the behavior of ΔEt, the θ-dependent energy can be separated into two zones (the Ny = 7 case in Fig. 2a). In zone I, the energy is almost the same as that of a flat graphene nanoribbon, irrespective of the type of edge-decoration. It shows a θ-square dependence in agreement with the first term of Eq. (1), as seen in carbon nanotubes37. The increase in the total energy is a consequence of the mechanical bending. Zone II refers to a region at sufficiently large θ's, where the energies with various edge decorations start to behave differently and diverge from one another. Edge-edge interactions must be taken into consideration when the distance between two edges is getting smaller. In general, various edge atoms result in three different types of edge interactions: covalent bonds, dipole-dipole interactions and repulsive forces. For type I, the boron case shows that its energy drastically decreases as the arc angle exceeds the critical value θc. The slopes in the cases of boron (blue circles) and carbon (black crosses) atoms are similar, so the resulting configuration is ascribed to the sp2 covalent bonds. The bonding energy can be fitted by the linear superposition of 1/d and 1/d2, where d is the distance between two edges. For type II, the quasi-stable structure in beryllium case (green triangles in Fig. 2a) is an unbonded curved nanoribbon with strong charge transfers between the Be and C atoms (discussed in detail later), which can be described by the dipole interaction model. In Eq. (1), the first nearest and second nearest dipole-dipole interactions match the energy curve well. In comparison with the boron and the carbon cases, the decrease in energy occurs at smaller angles, owing to the longer interactive distance of non-contact forces. The hydrogen-decorated nanoribbon belongs to type III, which is very different from types I and II. As the arc angle gradually increases, the repulsive interaction becomes stronger, leading to an increase in total energy (red diamonds in Fig. 2a). The edge atoms are in saturated states so that no bond forms between two edges and the charge transfer between H and C atoms is too weak to induce a dipole moment. Hence, quasi-stable structures do not exist, i.e., only flat nanoribbon is allowed.
The formation of the type II geometric structures deserves a more detailed discussion. The energy change in zone II can be regarded as an interaction of the electric dipole moments expressed in the second term of Eq. (1), where p is the dipole moment obtained through the Bader charge analysis39, α = 1 (2) represents the angle between a dipole and its first (second) nearest dipole,  and rα, respectively, denote the unit vector and the distance between the two dipoles, the factor 2 indicates a dipole interacting with one dipole on each side and
and rα, respectively, denote the unit vector and the distance between the two dipoles, the factor 2 indicates a dipole interacting with one dipole on each side and  is the vacuum permittivity. The reduction in the total energy due to an increase in the arc angle is attributed to two factors: (1) the shorter distance between two dipoles (rα) and (2) the larger values of
is the vacuum permittivity. The reduction in the total energy due to an increase in the arc angle is attributed to two factors: (1) the shorter distance between two dipoles (rα) and (2) the larger values of  and
and  in the second term of Eq. (1). The latter mainly results from the changing angle between two nearest dipoles caused by an increasing θ. Hence, the relative orientations of the dipoles are no longer parallel but approximately anti-parallel. At a specific angle, the energy local minimum happens (green triangle in Fig. 2a). The energy difference between black dash line (energy of a curved nanoribbon) and green triangle (energy of the Be-decorated structure) at this angle is obtained. This represents the energy difference induced by the dipole interaction. These energy differences are respectively 1.9 eV and 1.1 eV for beryllium and magnesium in the simulation. They turn out to be 2.1 eV and 1.2 eV by calculating the second term of Eq. 1. This means the dipole interaction model can express the comparable results obtained from the first principles. Consequently, type II decorating atoms exhibit similar behavior, hence producing highly curved nanoribbons, but various electric dipole moments result in different curvatures of the quasi-stable structures.
in the second term of Eq. (1). The latter mainly results from the changing angle between two nearest dipoles caused by an increasing θ. Hence, the relative orientations of the dipoles are no longer parallel but approximately anti-parallel. At a specific angle, the energy local minimum happens (green triangle in Fig. 2a). The energy difference between black dash line (energy of a curved nanoribbon) and green triangle (energy of the Be-decorated structure) at this angle is obtained. This represents the energy difference induced by the dipole interaction. These energy differences are respectively 1.9 eV and 1.1 eV for beryllium and magnesium in the simulation. They turn out to be 2.1 eV and 1.2 eV by calculating the second term of Eq. 1. This means the dipole interaction model can express the comparable results obtained from the first principles. Consequently, type II decorating atoms exhibit similar behavior, hence producing highly curved nanoribbons, but various electric dipole moments result in different curvatures of the quasi-stable structures.
The critical interaction distance (dc) and the energy barrier (Eb) of type I and type II for preventing the production of flat ribbon are obviously influenced by the ribbon width, or the number of the zigzag lines Ny, as shown in Figs. 2b and 2c. Because of the lower bending energy, wider ribbons only require relatively weak interactions from edge atoms in order to prevent flat ribbons from occurring. This accounts for the straightforward increasing relationship between Ny and dc. Eb, the total energy difference between the stable (quasi-stable) structure and E(θc), for boron (beryllium) will gradually rise with the increment of Ny. At Ny greater than 15, the tendency of Eb and dc levels off. This indicates the dominant bending energy approaches a lower value, leading to nearly constant Eb and dc. In short, such behavior of dc suggests that larger-width ribbons of type I and type II can form spontaneously when the interaction distance is less than 7 Angstroms.
The three types of edge-decorated systems exhibit very rich characteristics. The calculated charge transfers, atom-carbon distances, atom-atom distances and magnetic moments for various atoms are listed in Table 1. For boron and nitrogen, classified as type I, covalent bonds are formed between each decorating atom. To form type I structures, it is essential that the atom-carbon distances, atom-atom distances and electronegativity are all close to those of carbon. A boron atom has three valence electrons. It is assumed that two of them go into the two B-B single bonds that form a zigzag atom chain, while the third one makes the B-C single bond that zips the curved ribbon into a cylindrical nanotube. On the other hand, a nitrogen atom has two N-N single bonds and one N-C single bond; two unshared electrons are left for making up a full octet. Such a bonding configuration leads to a seriously deformed nanotube (see Fig. S1a, Supporting Information), in which the zigzag nitrogen atoms protruding from the cylindrical surface are associated with the antiferromagnetism of the edge carbon atoms. Similar magnetism is also found in deformed boron nitride nanotubes40. Except for boron and nitrogen, other non-metal elements, e.g., O, S, F, Cl, Si and P, can bond only with carbon atoms, but not form a tubular structure owing to their lack of the three above-mentioned essential conditions. These elements can thus only form edge-decorated flat nanoribbons.
Transition metals, well known for their catalytic roles, are very useful in fabricating carbon nanotubes41,42,43. Seven of the transition metals in the fourth period of the periodic table can form a tubular structure (type I), as listed in Table 1. Owing to the abundance of 3d orbitals, these elements provide empty hybrid orbitals and thus induce the single bond between a carbon atom and a transition metal atom. The atom-atom distances in transition metals are larger than those of boron and nitrogen, while they still remain stable in a somewhat deformed cylindrical structure. Magnetic moments do not exist in boron or carbon systems of type I, while the deformed nitrogen case being a special one. However, transition metals can give rise to magnetic moments on the edge carbon atoms. Among them, the non-ferrous metal copper has a magnetic moment of nearly zero. As for Zn and Sc, electrons fully occupy the 3d orbitals of the former and one 3d electron appears in the valence shell for the latter. These atoms can only bond with carbon atoms, but do not have the ability to bond with each other. Since their charge transfer is too low to form any dipole moment, Zn and Sc are categorized as type III.
When the system is decorated with metal elements, the high charge transfer between the metal and carbon allows the possible formation of dipoles on the edge, which are responsible for stabilizing the type II curved nanoribbon structures. The self-consistent calculations reveal that the atom-atom distances here are much greater than those in type I; as a consequence, such systems do not take the form of closed cylindrical structure because long interatomic distances make it impossible for the metal atoms to share electrons in single bonds. Both Be- and Mg-decorated structures belong to type II owing to the appropriate atom-carbon distance and the relatively high electron transfer, which are regarded as essential conditions. The distance between the decorating atom and the carbon atom is obviously greater than that in type I, so the electronic dipole effect becomes dominant. An Al-decorated structure constitutes a special case, where the low charge transfer still induces sufficient dipole moments to keep the ribbon in its curved form. Titanium, which has the highest electron transfer among the transition metals, shows similar results. In addition, titanium gives rise to ferromagnetic moments of edge carbon atoms, while magnetic moments generally do not exist in type II. However, alkali metal atoms have only one valence electron and thus cannot provide enough charge transfer for forming type II structure. The decoration by other metal elements such as Ca does not result in type II structure when the bond length between the decorating and carbon atoms is too long. This can be attributed to the lengthy bond that induces electric monopoles, which in turn exert a repulsive force between the edges. Li, Na, K and Ca, for instance, form stable structures of flat graphene nanoribbons with edge passivation.
The electronic properties of carbon-related systems are very sensitive to the curvature variation and the selected decorating element. For pure carbon systems, the cylindrical armchair nanotube possesses two linear bands intersecting at μ = 0 (type I in Fig. 3a), while the flat zigzag nanoribbon has a pair of partial flat bands separated by a band gap22. The presence of decorating atoms considerably changes the main features of the band structures of pure carbon systems. A cylindrical B-decorated system demonstrates two unique features (Fig. 3b): three energy bands crossing μ = 0 and a pair of partial flat bands located below μ = 0. The first linear energy band crossing μ = 0 near the wave vector k ~ 1/2 mainly comes from the B-B bond in the B-zigzag atom chain, with its contribution indicated by the circle radius in Fig. 3b. Such special features also remain the same in larger width cases (Ny = 11), as shown in Fig. S2a, Supporting Information. The other two nonlinear bands are closely associated with B-C bonds and most C-C bonds. This suggests that the electron transport along the edge B-B and B-C bonds is quite important. On the other side, a pair of partial flat bands attributed to edge C-C bonds becomes gapless at the zone boundary. This can be explained by the absence of AFM at both edges. It is also noticed that the partial flat bands and one linear band are absent in other nanotube-related systems where boron atoms are uniformly distributed, e.g., BC3 and BC3 nanotubes44. The concentrative distribution of boron atoms is responsible for the differences between them. Such differences clearly illustrate that the special geometric structure dominates the unique electronic properties.

Energy bands.
Energy bands of (a) an armchair nanotube, (b) B-, (c) Be- and (d) H-decorated systems. The circle radii represent the contributions of the decorating atoms.
The energy bands of a highly-curved nanoribbon behave somewhat in between the two extreme cases of an H-terminated flat nanoribbon (Fig. 3d) and a carbon nanotube (Fig. 3a). There exist two important features: a pair of partial flat bands mainly contributed by the edge atoms and the linear bands intersecting at μ = 0. For the Be-decorated nanoribbon, the partial flat bands are situated below μ = 0 (Fig. 3c); as for the H-terminated flat nanoribbon, they are symmetric about μ = 0. This means that the Be-decorated nanoribbon is a conductor, whereas the H-terminated one is not. Two linear intersecting bands at μ = 0 appear at a larger wave vector k ~ 4/5. One of them is contributed by all the carbon atoms and the other by the Be-C bonds, that is, the origin of the band intersection is quite different from the case of carbon nanotubes. This indicates that electrons can be transported either specifically through the decorating atoms or uniformly through the carbon atoms. These characteristics can also be found in larger width cases, where the linear intersecting bands still exist and one of them is mainly from the Be-C bonds, as shown in Fig. S2b. Other type II systems, such as those decorated with Mg and Al, also exhibit the similar energy band features. However, the linear bands for Al decoration cross above μ = 0 (see Fig. S1c, Supporting Information), a different behavior compared to the decoration by Be and Mg atoms. Although the origins of linear bands are different, the type II systems exhibit almost the same peculiar feature as the armchair carbon nanotubes: the metal-like electrical conductivity required for use as nanowires, which is a promising feature for application in future nanoelectronics.
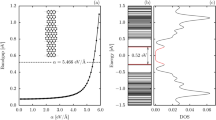
The density of states (DOS) directly reflects the primary characteristics of the band structures. The DOS in this study exhibits a plateau near μ = 0, two prominent peaks and a lot of square-root divergent asymmetric peaks, respectively, coming from the intersecting linearly bands, the partial flat bands and the parabolic bands, as in Figs. 4a–4d. The plateau DOS is presented by the armchair nanotube and the Be-decorated nanoribbon; its ω-dependence is negligible near μ = 0 and no peaks exist. However, the DOS of type I has no plateau near μ = 0 owing to the two nonlinear energy bands. It is worth noting that the DOS near μ = 0 determines the number of free carriers. Type I has the highest DOS; nevertheless, type I, type II and armchair nanotubes possess a metal-like electrical conductivity. As to the two prominent peaks (red triangles in Figs. 4b–4d), they are located separately above and below μ = 0 for the H-decorated system, while systems decorated by B and Be atoms show their peaks beneath μ = 0. These prominent peaks come from the the carbon atoms nearest to the decorating atoms, which are clearly revealed by the local density states (red curves in Fig. 4b and 4c). Moreover, B- and H-decorated systems each possess an additional peak near the prominent peaks, as marked by the blue circle in Figs. 4b and 4d. The former arises from the band edge states near k = 0 of the nonlinear band (Fig. 3b) and the latter originates from the extra band edge states at k ≠ 1 of the partial flat band (Fig. 3d). In addition, the local density states clear illustrate that this peak (blue circle in Fig. 4b) comes from the boron atoms (blue curve in Fig. 4b). The feature-rich DOS near μ = 0 could be verified by of scanning tunneling microscopy measurements36,45,46.

Density of states.
Densities of states and local densities of states of (a) an armchair nanotube, (b) B-, (c) Be- and (d) H-decorated systems. The red triangles mark the prominent peaks.
Whether a decorated system is a conductor or a semiconductor is determined by the DOS near μ = 0, as listed in Table 1. In general, most decorating atoms provide free carriers and thus increase the DOS, i.e., the decorated systems are conductors. Compared to the metallic armchair carbon nanotube, they possess a much higher DOS, except for Be- and Mg-decorated systems that only have a comparable DOS. When the anti-ferromagnetic configuration, induced by zigzag edge structure, is taken into consideration, the partial flat bands near μ = 0 start to split and result in vanishing DOS, i.e., the system becomes a semiconductor, such as in the cases of the H-, Li- and F-terminated nanoribbons. However, Si-decorated system is a conductor even though it owns an AFM configuration. This is because Si atoms have much more valence electrons and the energy bands associated with the valence electrons cross μ = 0. Systems decorated by transition-metal elements tend to retain the properties of the decorating atoms and hence are conductors. The only exception is a system when decorated by chromium atoms (see Fig. S1b, Supporting Information), which has a tiny band gap that may disappear in larger systems.
Discussion
The geometric and electronic properties of edge-decorated graphene nanoribbons are investigated by ab initio density functional theory calculations. Remarkably, three types of optimal geometric structures are obtained, originating from three different edge interactions: covalent bonds, dipole-dipole interactions and repulsive forces. Elements with an atomic number of lower than 30 are all studied and classified into the three types based on their charge transfer, atom-carbon distance, atom-atom distance and magnetic moment. These decorated structures each have their special electronic and magnetic properties that can be tuned by various decorating atoms. In type II systems, energy bands behave between the two extreme cases: the intersecting linear bands of the armchair nanotube and a pair of partial flat bands at the zone boundary of the H-terminated nanoribbon. Most of the decorated systems are classified as conductors with a DOS at μ = 0 much higher than or comparable to that of a metallic armchair nanotube, while a few are semiconductors with a band gap stemming from the anti-ferromagnetic configuration. Our results suggest a possible route to selectively fabricate specific types of edge-decorated structures and thus avoid the difficulties in separating the mixed products obtained in unzipping experiments. This selectivity of the geometric structures and the electronic properties might be potentially important for edge-decorated systems in the application of nanoelectronic devices.
Methods
Our first-principles calculations are based on the density functional theory (DFT) implemented by the Vienna ab initio simulation package (VASP)47. The generalized gradient approximation (GGA) in the Perdew-Burke-Ernzerhof (PBE) form are chosen for the DFT calculations and the projector augmented wave (PAW) is utilized to describe the electron-ion interactions. A vacuum space of 15 angstroms is inserted between periodic images to avoid interactions and the cutoff energies of the wave function expanded by plane waves were chosen to be 500 eV. For calculation of the electronic properties and the optimized geometric structures, the first Brillouin zones are sampled by 300 × 1 × 1 and 12 × 1 × 1 k-points by the Monkhorst-Pack scheme. The convergence of the Helmann-Feymann force is set to 0.01 eV Å−1.
References
Geim, A. K. Graphene: status and prospects. Science 324, 1530–1534 (2009).
Li, X. et al. Chemically derived, ultrasmooth graphene nanoribbon semiconductors. Science 319, 1229–1232 (2008).
Kosynkin, D. V. et al. Longitudinal unzipping of carbon nanotubes to form graphene nanoribbons. Nature 458, 872–876 (2009).
Cataldo, F. et al. Graphene nanoribbons produced by the oxidative unzipping of single-wall carbon nanotubes. Carbon 48, 2596–2602 (2010).
Shinde, D. B., Majumder, M. & Pillai, V. K. Counter-ion Dependent, Longitudinal Unzipping of Multi-Walled Carbon Nanotubes to Highly Conductive and Transparent Graphene Nanoribbons. Sci. Rep. 4, 4363 (2014).
Huang, H. et al. Spatially resolved electronic structures of atomically precise armchair graphene nanoribbons. Sci. Rep. 2, 983 (2012).
Ni, Y. et al. Spin Seebeck Effect and Thermal Colossal Magnetoresistance in Graphene Nanoribbon Heterojunction. Sci. Rep. 3, 1380 (2013).
Liu, L. et al. Nanosphere Lithography for the Fabrication of Ultranarrow Graphene Nanoribbons and On-Chip Bandgap Tuning of Graphene. Adv. Mater. 23, 1246–1251 (2011).
Nduwimana, A. & Wang, X. Q. Energy gaps in supramolecular functionalized graphene nanoribbons. ACS Nano 3, 1995–1999 (2009).
Lambin, P., Amara, H., Ducastelle, F. & Henrard, L. Long-range interactions between substitutional nitrogen dopants in graphene: electronic properties calculations. Phys. Rev. B 86, 045448 (2012).
Wong, J. H., Wu, B. R. & Lin, M. F. Strain effect on the electronic properties of single layer and bilayer graphene. J. Phys. Chem. C 116, 8271–8277 (2012).
Haskins, J. et al. Control of thermal and electronic transport in defect-engineered graphene nanoribbons. ACS Nano 5, 3779–3787 (2011).
Wakabayashi, K. & Sigrist, M. Zero-conductance resonances due to flux states in nanographite ribbon junctions. Phys. Rev. Lett. 84, 3390 (2000).
Tada, K. & Watanabe, K. Ab initio study of field emission from graphitic ribbons. Phys. Rev. Lett. 88, 27601 (2002).
Xia, F., Farmer, D. B., Lin, Y. M. & Avouris, P. Graphene field-effect transistors with high on/off current ratio and large transport band gap at room temperature. Nano Lett. 10, 715–718 (2010).
Wakabayashi, K., Takane, Y., Yamamoto, M. & Sigrist, M. Edge effect on electronic transport properties of graphene nanoribbons and presence of perfectly conducting channel. Carbon 47, 124–137 (2009).
Cantele, G., Lee, Y. S., Ninno, D. & Marzari, N. Spin channels in functionalized graphene nanoribbons. Nano Lett. 9, 3425–3429 (2009).
Lin, C. Y., Chen, S. C., Wu, J. Y. & Lin, M. F. Curvature Effects on Magnetoelectronic Properties of Nanographene Ribbons. J. Phys. Soc. Jpn. 81, 4719 (2012).
Ritter, K. A. & Lyding, J. W. The influence of edge structure on the electronic properties of graphene quantum dots and nanoribbons. Nat. Mater. 8, 235–242 (2009).
Son, Y. W., Cohen, M. L. & Louie, S. G. Energy gaps in graphene nanoribbons. Phys. Rev. Lett. 97, 216803 (2006).
Chen, J. J., Wu, H. C., Yu, D. & Liao, Z. M. Magnetic moments in graphene with vacancies. Nanoscale 6, 8814 (2014).
Lee, H. et al. Magnetic ordering at the edges of graphitic fragments: Magnetic tail interactions between the edge-localized states. Phys. Rev. B 72, 174431 (2005).
Elías, A. L. et al. Longitudinal cutting of pure and doped carbon nanotubes to form graphitic nanoribbons using metal clusters as nanoscalpels. Nano Lett. 10, 366–372 (2009).
Dhakate, S. R., Chauhan, N., Sharma, S. & Mathur, R. B. The production of multi-layer graphene nanoribbons from thermally reduced unzipped multi-walled carbon nanotubes. Carbon 49, 4170–4178 (2011).
Jiao, L. et al. Narrow graphene nanoribbons from carbon nanotubes. Nature 458, 877–880 (2009).
Higginbotham, A. L. et al. Lower-defect graphene oxide nanoribbons from multiwalled carbon nanotubes. ACS Nano 4, 2059–2069 (2010).
Jiao, L. et al. Facile synthesis of high-quality graphene nanoribbons. Nat. Nanotechnol. 5, 321–325 (2010).
Cano-Márquez, A. G. et al. Ex-MWNTs: Graphene sheets and ribbons produced by lithium intercalation and exfoliation of carbon nanotubes. Nano Lett. 9, 1527–1533 (2009).
Kosynkin, D. V. et al. Highly conductive graphene nanoribbons by longitudinal splitting of carbon nanotubes using potassium vapor. ACS Nano 5, 968–974 (2011).
Xiao, B., Ding, Y. H. & Sun, C. C. Beryllium and boron decoration forms planar tetracoordinate carbon strips at the edge of graphene nanoribbons. Phys. Chem. Chem. Phys. 13, 2732–2737 (2011).
Karamanis, P. & Pouchan, C. Second-Hyperpolarizability (γ) Enhancement in Metal-Decorated Zigzag Graphene Flakes and Ribbons: The Size Effect. J. Phys. Chem. C 117, 3134–3140 (2013).
Kudin, K. N. Zigzag graphene nanoribbons with saturated edges. ACS Nano 2, 516–522 (2008).
Simbeck, A. J. et al. Electronic structure of oxygen-functionalized armchair graphene nanoribbons. Phys. Rev. B 88, 035413 (2013).
Sinitskii, A. et al. Kinetics of diazonium functionalization of chemically converted graphene nanoribbons. ACS Nano 4, 1949–1954 (2010).
Wu, M., Pei, Y. & Zeng, X. C. Planar tetracoordinate carbon strips in edge decorated graphene nanoribbon. J. Am. Chem. Soc. 132, 5554–5555 (2010).
Ouyang, M., Huang, J. L., Cheung, C. L. & Lieber, C. M. Energy gaps in “metallic” single-walled carbon nanotubes. Science 292, 702–705 (2001).
Chang, S. L., Wu, B. R., Yang, P. H. & Lin, M. F. Curvature effects on electronic properties of armchair graphene nanoribbons without passivation. Phys. Chem. Chem. Phys., 14, 16409–16414 (2012).
Jackson, J. D. Classical Electrodynamics 3rd Ed.(Wiley, London, 1998).
Henkelman, G., Arnaldsson, A. & Jonsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 36, 354–360 (2006).
Zhang, Z. & Guo, W. Tunable ferromagnetic spin ordering in boron nitride nanotubes with topological fluorine adsorption. J. Am. Chem. Soc. 131, 6874–6879 (2009).
Iijima, S. & Ichihashi, T. Single-shell carbon nanotubes of 1-nm diameter. Nature 363, 603–605 (1993).
Thess, A. et al. Crystalline ropes of metallic carbon nanotubes. Science 273, 483–487 (1996).
Charlier, J. C., Amara, H. & Lambin, P. Catalytically assisted tip growth mechanism for single-wall carbon nanotubes. ACS Nano 1, 202–207 (2007).
Kim, Y. H., Sim, H. S. & Chang, K. J. Electronic structure of collapsed C, BN and BC3 nanotubes. Curr. Appl. Phys. 1, 39–44 (2001).
Wilder, J. W. et al. Electronic structure of atomically resolved carbon nanotubes. Nature 391, 59–62 (1998).
Odom, T. W., Huang, J. L., Kim, P. & Lieber, C. M. Atomic structure and electronic properties of single-walled carbon nanotubes. Nature 391, 62–64 (1998).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Acknowledgements
This work is supported by the NSC and NCTS (South) of Taiwan, under the grant Nos. NSC-102-2112-M-006-007-MY3 and NSC-101-2221-E-006-113.
Author information
Authors and Affiliations
Contributions
S.L.C. did numerical calculations under the guidance of M.F.L. S.Y.L., S.K.L. and C.H.L. contributed to the writing of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Figure S1, Figure S2
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Chang, SL., Lin, SY., Lin, SK. et al. Geometric and Electronic Properties of Edge-decorated Graphene Nanoribbons. Sci Rep 4, 6038 (2014). https://doi.org/10.1038/srep06038
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06038
This article is cited by
-
Alkali-created rich properties in grapheme nanoribbons: Chemical bondings
Scientific Reports (2017)
-
The Edge Stresses and Phase Transitions for Magnetic BN Zigzag Nanoribbons
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
