
Abstract
Thermal stability of composite cathodes for solid oxide fuel cells, the mixtures of (La0.8Sr0.2)0.95MnO3−δ (LSM) and (Y2O3)x(ZrO2)1−x (xYSZ, x = 3, 6, 8 and 10), is determined using in-situ neutron diffraction. Thanks to the most advanced high flux neutron source, our work highlights the visualization of the phase evolutions in heterogeneous material systems at high temperatures, along with the analysis of the diffusion activities of transition metal ions that reveal the reaction mechanism and kinetics. It is found that the tetragonal-to-cubic phase transition in YSZ at T > 900°C leads to a heterogeneous redistribution of Mn ions. The subsequent reaction of LSM and YSZ occurring at T > 1100°C is revealed as a three-stage kinetic process, yielding La2Zr2O7, SrZrO3 and MnO. The diffusion activities of Y, Mn and La ions in the heterogeneous systems at elevated temperatures are derived by the structural analysis and the three-stage reaction of YSZ and LSM is found strongly correlated to ions' behaviors as functions of temperature.
Similar content being viewed by others

Introduction
Heterogeneous materials, which consist of two or more phases, share many benefits in the energy-converting devices including fuel cells and batteries1,2,3,4,5. Taking the solid oxide fuel cell (SOFC) – an electrochemical conversion device with high efficiency, fuel flexibility and low emission, as an example, the electrode of SOFC performing as a reaction chamber requires a combination of both good ionic and electronic conductivities, high catalysis and sometimes good mechanical supporting6,7. Thanks to the concept of heterogeneous materials, those complex functions can be achieved at the same time. The properties are adjusted by controlling the architecture of the materials systems, including dimension, fraction and distribution of the phases8,9,10. However, the heterogeneity of the material system may lead to the loss of chemical stability under high temperature application. The SOFC typically operates at 500 ~ 1000°C and the materials themselves may also experience higher-temperature thermal treatment during the synthesis and cell assembling. The elevated temperature can possibly trigger the phase evolution and spur the atom's diffusion in the heterogeneous environment, leading to deterioration of the functional materials. As more novel materials for SOFC are proposed and studied11,12, it is important to understand the stability and compatibility of the materials system at the situation of high temperatures, to resolve the degradation mechanism of SOFCs and develop reliable and durable SOFCs. In this paper, we start with a widely used cathode material system consisting of lanthanum-strontium-manganese oxides (LSM) and yttria-stabilized zirconia (YSZ), utilizing the advantages of neuron diffraction to draw a detailed picture of the structural evolution and reveal the ions' activities in this heterogeneous system at high temperatures.
LSM and YSZ are the materials for cathodes and electrolytes of SOFCs, respectively13,14. The composite cathode such as LSM/YSZ will provide higher ionic conductivity and larger area of the triple phase boundaries (TPBs), to expand the reaction sites across the depth of the cathode and thus to improve the performance of the cathode15. However, near the joint of LSM and YSZ, the formation of secondary phases, such as lanthanum zirconate (La2Zr2O7, abbr. LZO) and strontium zirconate (SrZrO3, abbr. SZO), worsens the cathodes' performance because of their low ionic conductivity and the unfavorable mismatch of coefficient of thermal expansion with YSZ electrolyte16.
The formation of the secondary phases depends on the temperature, stoichiometry of LSM, content of yttrium in YSZ, mixing ratio of LSM and YSZ, atmosphere and other factors. Major formation of a La2Zr2O7 phase is expected during the sintering process of the electrode at higher temperature, although a lower growth rate at a typical operation temperature of 1000°C was reported17. In the reaction of lanthanum manganite and YSZ, La-deficient manganite shows less reactivity with YSZ than the stoichiometric one18,19, while excess Mn effectively hinders the formation of zirconate20,21. Mitterdorfer and Gauckler22 studied the kinetics of La2Zr2O7 formation at the interface of LSM/YSZ. They proposed a reaction mechanism in which the manganese concentration and excess lanthanum oxide affect the nucleation and the growth rate of the La2Zr2O7 phase. In addition to the influence by LSM, the increase of yttrium content in xYSZ (x = 8 ~ 12) leads to a longer induction period before La2Zr2O7 is formed23; that is, the yttrium dopant slows down the reaction. Chen et al.24 studied the long-term stability of LSM/YSZ system in different atmospheres. After being annealed at 1000°C in air for as long as 13 weeks, monoclinic ZrO2 forms and the amount increases with the initial LSM/YSZ ratio. On annealing in N2, both La2Zr2O7 and SrZrO3 form under YSZ-rich condition (25 wt%LSM–75 wt%YSZ). The La2Zr2O7 formation can be prevented with increased ratio of LSM/YSZ, while SrZrO3 formation cannot be prevented. Those findings above are based on ex-situ characterization methods, including X-ray diffraction (XRD), scanning electron microscope (SEM), energy dispersive spectroscopy (EDS), transmission electron microscope (TEM) and so forth. The reaction hypothesis was made on grounds of the post-reaction observations and the process of the reaction and/or the phase transition of the LSM/YSZ cathode composite at elevated temperature has not been directly observed.
In-situ phase investigation is a key to unravel the structural evolution during sintering. Neutron diffraction is superior for this purpose because the large cross-section of all components makes it possible to differentiate multiple phases. The high flux and deep penetration of neutrons facilitate the in-situ study of bulk materials at elevated temperatures, enabling us to derive the structural evolution during heating, annealing and cooling25,26. With the neutron capability, we carried out the in-situ measurement of (La0.8Sr0.2)0.95MnO3−δ (LSM) and x mol% Y2O3 stabilized ZrO2 (xYSZ, x = 3 ~ 10) heterogeneous materials system (xYSZ-LSM) at elevated temperatures up to 1500°C with a vacuum environment, in order to reveal the structural evolutions and the effects of Y doping. By extracting the real-time information, such as phase fractions, crystalline symmetries and lattice parameters, the diffusion activities of Y, Mn and La ions are drawn to unmask their impacts on the reaction process in the heterogeneous system.
Results
(1) Phase characterization
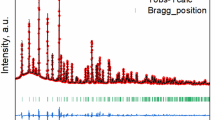
Through Rietveld refinement on the neutron diffractions of xYSZ-LSM (x = 3, 6, 8 and 10), we identified all phases that participated in the reaction. Figure 1 shows the example of 6YSZ-LSM before and after the thermal treatment. The starting compounds were verified as LSM and YSZ without detectable impurities. As expected, a mixture of cubic YSZ (YSZ-c) and tetragonal YSZ (YSZ-t) were found in 3YSZ and 6YSZ while only YSZ-c was observed in 8YSZ and 10YSZ. The reaction phases were solid solution phases of pyrochlore La2Zr2O7 (LZO), perovskite SrZrO3 (SZO), YSZ-c and rock salt MnO. The phase identification is consistent with the reactions in low O2 pressure atmosphere reported by Wiik27. The oxygen-deficient environment led to the formation of MnO28,29 rather than Mn2O3, which is often formed in O2-rich environments28. The final phases of LZO, SZO, YSZ and MnO were confirmed from the backscattering images of SEM and EDS mapping (see Supplementary Fig. S1 online). Instead of pure compounds, the formation of solid solutions was suggested, such as LZO with Y and Sr, SZO with La and Y, YSZ with La and Sr. Mn was found only in MnO grains, where no traces of La, Y, Sr or Zr were detected. As used here, the abbreviations of LZO, SZO and xYSZ stand for those kinds of solid solutions but not the exact nominal compositions.

Neutron diffraction patterns of 6YSZ-LSM before (a) and after (b) thermal treatment.
The tick markers indicate Bragg reflections of each phase, whose space group is given in the bracket.
Quantitatively, the phase fractions in each specimen after the reaction were deduced from Rietveld refinement (see Supplementary Table S1 online). LSM was finally exhausted in 6YSZ-LSM, 8YSZ-LSM and 10YSZ-LSM, but the fractions of the final phases show little dependence on the initial concentration of yttria represented by x. In contrast, there remains about 10.2 wt% LSM residue in 3YSZ-LSM. As another residual phase of YSZ, only cubic phase YSZ-c instead of tetragonal phase YSZ-t was found after the reaction, although 3YSZ-LSM and 6YSZ-LSM started with certain amounts of YSZ-t phase. The fractions of residual YSZ in 6YSZ-LSM, 8YSZ-LSM and 10YSZ-LSM are about 7 ~ 9 wt% while it is as high as 19.1 wt% in 3YSZ-LSM.
(2) Real-time observation of phase evolutions at elevated temperatures
The real-time phase evolution is directly presented by means of the intensity contour plots of neutron diffraction patterns during the reaction. Figure 2 shows an example of 3YSZ-LSM. It clearly reveals the phase formation, coexistence and disappearance during the process. With the Rietveld refinement applied to each diffraction pattern along the temperature/time axis, the quantitative phase evolutions are then shown as phase fractions versus temperature/time curves (Figure 3 and 4). Also, the real-time evolutions of lattice parameters are derived (Figure 5 and 6), which reflect the diffusion in the solid solution accompanying the reaction. The different numbers of experiment points for different samples or random point drops are due to the availability of neutron beam during these in-situ continuous heating experiments.

Contour plot of neutron diffraction showing the phase transformation and reaction in 3YSZ-LSM during heating from 800 ~ 1500°C and then 5 hours dwell.
Each run represents a time bin of 5 minutes.

Real-time plots of the phases' fractions in 3YSZ-LSM (a) and 6YSZ-LSM (b) by Rietveld refinement.
The dash-dotted lines indicate the borders for the three-stage reaction, as marked by the Roman numbers. The fitting error bars smaller than the symbols are invisible.

Real-time plots of the phases' fractions in 8YSZ-LSM (a) and 10YSZ-LSM (b) by Rietveld refinement.
The dash-dotted lines indicate the borders for the three-stage reaction, as marked by the Roman numbers. The fitting error bars smaller than the symbols are invisible.

Lattice evolutions of YSZ-c in xYSZ-LSM series.
The inset figure is the normalized values in 6YSZ-LSM, 8YSZ-LSM and 10YSZ-LSM by their lattice parameters at 200°C.  – tetragonal-to-cubic phase transition in YSZ;
– tetragonal-to-cubic phase transition in YSZ;  – pristine YSZ-c with few Mn dopants participating in the reaction with LSM;
– pristine YSZ-c with few Mn dopants participating in the reaction with LSM;  – Mn-rich zirconia reacting with LSM.
– Mn-rich zirconia reacting with LSM.

Lattice evolutions of LZO in xYSZ-LSM series.
In 3YSZ-LSM and 6YSZ-LSM, the phase evolution started with a tetragonal-to-cubic phase transition of YSZ-t, as shown in Figure 3(a) and 3(b). In 3YSZ-LSM, it started at 1000°C and continued till at about 1300°C accompanied by the reaction of YSZ and LSM. Of slight difference in 6YSZ-LSM, the transition took place at 900°C and finished before 1100°C, without YSZ-t participating in the reaction of YSZ and LSM. At the temperature of phase transition, a kink in the YSZ-c lattice parameter plots (Figure 5, marked as  ) indicates the ion diffusion accompanying the reaction, leading to lattice shrinkage. For a better show of the lattice change of YSZ-c in 6YSZ-LSM, Figure 5 (inset) plots the relative values normalized to the lattice parameter at 200°C. The data of 8YSZ-LSM and 10YSZ-LSM were normalized in the same way for comparisons. In contrast, no such transition was observed below 1100°C in 8YSZ-LSM and 10YSZ-LSM without YSZ-t.
) indicates the ion diffusion accompanying the reaction, leading to lattice shrinkage. For a better show of the lattice change of YSZ-c in 6YSZ-LSM, Figure 5 (inset) plots the relative values normalized to the lattice parameter at 200°C. The data of 8YSZ-LSM and 10YSZ-LSM were normalized in the same way for comparisons. In contrast, no such transition was observed below 1100°C in 8YSZ-LSM and 10YSZ-LSM without YSZ-t.
At T > 1100°C, the reaction of LSM and YSZ started in all samples. The new phases LZO, SZO and MnO gradually appeared while consuming YSZ and LSM. Varied reaction rates were noted during temperature ramping from 1100°C to 1500°C. Based on the reaction rate, the reaction is split into several segments. Taking 6YSZ-LSM as an example, we observed a typical three-stage reaction process in Figure 3(b). Stage I (1100 ~ 1350°C) showed constant reaction rate, as indicated by the linear changes of LSM, YSZ and LZO phase fractions. Stage II (1350 ~ 1450°C) started when the reaction proceeds under a very low rate. The coexistence of LSM, YSZ, LZO, SZO and MnO nearly reached equilibrium within this temperature range and a higher temperature was needed to retrigger the further reaction. Stage III (1450 ~ 1500°C) showed rapid reaction at higher temperature. When the threshold temperature of 1450°C passed, the reaction was reactivated and came to a rapid process so that LSM instantly exhausted in the third stage, with corresponding quick changes of other phases' fractions. The three-stage reaction was also observed in 8YSZ-LSM and 10YSZ-LSM, as noted in Figure 4(a) and 4(b), respectively, though the transitions between stages were not as sharp as in 6YSZ-LSM.
In 3YSZ-LSM, where the initial YSZ was mostly in the tetragonal form, the three-stage reaction was not observed; rather, the reaction rate showed discontinuity at 1300°C, indicated by the phase fraction plots of LSM and LZO in Figure 3(a). The reaction was accompanied by the structural transformation of YSZ. The YSZ-t vanished eventually at 1380°C while LSM did not disappear and the residual reagents and products coexisted until the end of the thermal treatment.
The reaction did not fully cease after the LSM vanished in 6YSZ-LSM, 8YSZ-LSM and 10YSZ-LSM. It was noticed that the LZO phase continued to gain mass of about 0.8 wt% at the isothermal dwell while the YSZ-c and MnO showed corresponding mass loss. Once the cooling step started, the mass exchange was reversed at 1500 ~ 800°C: the fraction of MnO was significantly increased while that of LZO decreased. The YSZ-c slightly lost mass during cooling as well.
Discussion
Based on the comprehensive phase analysis, we propose the sequence of phase transition and reaction in xYSZ-LSM systems, as shown in Figure 7. In detail, the cause and effect of the three-stage reaction is to be discussed below, by resolving the diffusion activities of Y, Mn and La ions associated with the YSZ-t phase transition in the heterogeneous system.

Scheme of the phase evolutions in xYSZ-LSM system.
(P.T. – tetragonal-to-cubic phase transition).
The yttria-doping effects in the series of xYSZ-LSM are directly reflected by the systematical differential in the lattice parameters of the related phases, e.g. YSZ-c and LZO. At the isothermal dwell, which is a relatively stable state, the cubic YSZ-c exhibits an increase of the lattice parameters with the increase of x in the xYSZ-LSM system (Figure 5). This increase is due to the larger ionic radius of Y3+ in comparison to Zr4+ and the trend agrees well with the Y concentration. Inversely, the product LZO shows decreased lattice parameters with more yttria doping (larger x) in the xYSZ-LSM series (Figure 6). It was assumed that Y3+ ions would stay at Zr-site in LZO30 because Y3+ ions substitute Zr4+ ions in YSZ. However, the Y3+ ions contributing to the shrinkage of the LZO lattice reveals that the smaller Y3+ ions replace the larger La3+ ions at the La-site of LZO, which agrees with the prediction based on EDX analysis in Ref. 31. Both lattices of YSZ-c and LZO exhibit significant correlation to the initial yttria content, represented by x. The influence of Y3+ in SZO is difficult to extract because of the small phase fraction as well as low crystal symmetry. Here it is hypothesized that the situation is similar to that in LZO. We conclude that the activity of Y3+ mostly follows the Zr4+ during the reaction – either staying in the residual YSZ-c or joining into LZO and SZO, with nearly the same Y/Zr mass ratio as in the initial xYSZ.
In contrast, the yttria content does not exhibit direct influence on the reaction occurrence. Thermodynamically, those yttria dopants do not prevent LZO formation as the previous consideration23,32. The reaction starts at about 1100°C for all samples independent of the number of yttria dopants in the range of 3 ~ 10 mol%. Kinetically, the reaction stages of xYSZ-LSM differ from each other but do not exhibit a simple dependence on the yttria content (Figure 3 and 4). In fact, the indirect yttria-doping effect on the YSZ-LSM reaction arises from the diffusion of Mn and La in zirconia: excess Mn hindered the reaction20,29 while the La diffusion promoted it22,31,33. Although the concentrations of Mn and La are not the variables in the xYSZ-LSM series, the activities of Mn and La ions are affected by the YSZ-t phase transition and heterogeneous diffusion at high temperatures, which provides an indirect link to the variable x, the initial yttria content.
The tetragonal-to-cubic phase transition of YSZ-t drives the heterogeneous Mn diffusion in YSZ at elevated temperature. In 3YSZ-LSM and 6YSZ-LSM with lower yttria content, the YSZ-t, which itself has low solubility (<3 mol%) of Mn34, gained Mn solute during the phase transition into YSZ-c. The lattice parameter of YSZ-c provided a clear indication of the Mn diffusion accompanying the phase transition of YSZ. As shown in Figure 5, an abrupt kink at 1000°C (noted with  ) is observed in the plots for 3YSZ-LSM, indicating the smaller lattice of the new product cubic YSZ. Such a kink of 6YSZ-LSM is also seen in Figure 5(inset). Because Mn has a smaller ionic radius than Y and Zr, the new transformed cubic YSZ with Mn stabilizer has a reduced lattice parameter in comparison with the pristine YSZ-c with little Mn doping. For comparison, the YSZ-c in 8YSZ-LSM and 10YSZ-LSM exhibit a nearly quadratic thermal expansion without interrupt at T < 1100°C since there is no YSZ-t or tetragonal-to-cubic phase transition. The lattice thermal expansion behavior is consistent with the bulk thermal expansion of 8YSZ measured by dilatometry in Ref. 35. Therefore, the phase transition of YSZ-t promotes Mn migrating from LSM to YSZ and we consider it is stabilization of YSZ-t by Mn. Some other ions rather than Mn are probably active around as well, such as La3+ and Sr2+, but they have too large radii to shrink the YSZ-c lattice by diffusion. It is thus justified by neutron diffraction that the Mn ions from LSM stabilized the tetragonal zirconia at T > 900°C.
) is observed in the plots for 3YSZ-LSM, indicating the smaller lattice of the new product cubic YSZ. Such a kink of 6YSZ-LSM is also seen in Figure 5(inset). Because Mn has a smaller ionic radius than Y and Zr, the new transformed cubic YSZ with Mn stabilizer has a reduced lattice parameter in comparison with the pristine YSZ-c with little Mn doping. For comparison, the YSZ-c in 8YSZ-LSM and 10YSZ-LSM exhibit a nearly quadratic thermal expansion without interrupt at T < 1100°C since there is no YSZ-t or tetragonal-to-cubic phase transition. The lattice thermal expansion behavior is consistent with the bulk thermal expansion of 8YSZ measured by dilatometry in Ref. 35. Therefore, the phase transition of YSZ-t promotes Mn migrating from LSM to YSZ and we consider it is stabilization of YSZ-t by Mn. Some other ions rather than Mn are probably active around as well, such as La3+ and Sr2+, but they have too large radii to shrink the YSZ-c lattice by diffusion. It is thus justified by neutron diffraction that the Mn ions from LSM stabilized the tetragonal zirconia at T > 900°C.
The Mn diffusion directly into YSZ-c during the temperature ramping is also allowed, since Mn has certain solubility ranging from 5.1% at 1000°C to 11.4% at 1300°C29. However, this process is slow and the quantity of mass transfer is not comparable to that via the stabilization process. The slight Mn-doped influence on lattice parameter of YSZ-c is barely visible in 8YSZ-LSM and 10YSZ-LSM at 800 ~ 1100°C.
As a result, the YSZ-t encounters a rapid process to take in Mn via the tetragonal-to-cubic phase transition and then it becomes a Mn-rich zirconia phase. The less Y doped, the more Mn diffuses into the zirconia. The Mn-rich zirconia yielded from YSZ-t, though cubic, is more retarded in the reaction with LSM due to the excess Mn. The initial YSZ-c takes in Mn slowly. Those YSZ-c grains with low Mn content, referred to pristine YSZ-c, are thus more active and more prior in the reaction with LSM.
Due to the uneven Mn distribution in YSZ, the reaction becomes heterogeneous corresponding to different YSZ sources. In Stage I, it is the pristine YSZ-c grains with low Mn content that dominate the YSZ source of the reaction in 6YSZ-LSM, 8YSZ-LSM and 10YSZ-LSM. At this stage, the Mn-rich zirconia grains are relatively inactive since the excess Mn hinders the reaction. This mechanism is reflected by the reduced lattice thermal expansion of YSZ-c, as noted with  in Figure 5 (inset). Because the pristine YSZ-c with larger lattice was consumed while the Mn-rich zirconia with smaller lattice survived in Stage I, the average lattice parameter of YSZ-c was decreased. Also, the LZO lattice provides another support. Mn doping in LZO would bring a smaller lattice in LZO due to the small ionic radius of Mn ions. In this case, the 6YSZ-LSM sample, whose YSZ gained more Mn dopants during the phase transition, would yield LZO with smaller lattice parameter than 8YSZ-LSM and 10YSZ-LSM. This deduction obviously conflicts with the results shown in Figure 6, in which LZO in 10YSZ-LSM has the smallest lattice. Thus little Mn migrates into LZO at this stage. It is YSZ-c but not Mn-rich zirconia that is first to react with LSM in Stage I and yttria is the major dopant in the product LZO at high temperature.
in Figure 5 (inset). Because the pristine YSZ-c with larger lattice was consumed while the Mn-rich zirconia with smaller lattice survived in Stage I, the average lattice parameter of YSZ-c was decreased. Also, the LZO lattice provides another support. Mn doping in LZO would bring a smaller lattice in LZO due to the small ionic radius of Mn ions. In this case, the 6YSZ-LSM sample, whose YSZ gained more Mn dopants during the phase transition, would yield LZO with smaller lattice parameter than 8YSZ-LSM and 10YSZ-LSM. This deduction obviously conflicts with the results shown in Figure 6, in which LZO in 10YSZ-LSM has the smallest lattice. Thus little Mn migrates into LZO at this stage. It is YSZ-c but not Mn-rich zirconia that is first to react with LSM in Stage I and yttria is the major dopant in the product LZO at high temperature.
Those pristine YSZ-c grains with low Mn doping disappear at the end of Stage I. It is read from Figure 3 and 4 that the YSZ fractions at this time are approximately 31 wt%, 28 wt% and 26 wt% in 6YSZ-LSM, 8YSZ-LSM and 10YSZ-LSM, respectively. The data indicate that the reaction proceeded further in the sample with more yttria dopant, e.g. in 10YSZ-LSM, because there is less Mn in YSZ that hinders the reaction. After Stage I, the rest of the YSZ-c is Mn-doped zirconia originally from either the stabilization of YSZ-t by Mn or the YSZ-c with extensive diffusion of Mn. Those Mn-rich zirconia grains remain inactive until a higher temperature is reached, followed by the rapid reaction in Stage III.
Correspondingly, the Mn-rich zirconia grains dominate the YSZ source in Stage III and the activity of Mn in this stage is clearly revealed by the YSZ-c and LZO lattice evolutions. The YSZ-c lattices experience a rapid expansion in 6YSZ-LSM, 8YSZ-LSM and 10YSZ-LSM, which are noted with  in the Figure 5 (inset). Meanwhile the LZO lattice parameters show a drop at the temperature of Stage III (Figure 6). Those instant changes reveal the smaller Mn ions leaving YSZ-c and coming into LZO. The suggested reaction mechanism is that the Mn-rich zirconia takes part in the reaction in Stage III to transport Mn from zirconia to LZO; moreover, the Mn solutes in zirconia spontaneously migrate to LZO, resulting in the expansion of the zirconia lattice. It is believed that the Mn ions are in the form of Mn2+ occupying the La-site in LZO and contributing to the reduced LZO lattice because the radius of Mn2+ is smaller than that of La3+. In Stage III, the LZO lattice parameter in 6YSZ-LSM shows a larger drop than those in 8YSZ-LSM and 10YSZ-LSM. It is consistent with our previous conclusion that the stabilization of YSZ-t in 6YSZ-LSM introduced more Mn in YSZ. Thereby, the lattice evolutions confirm that Stage III is the rapid reaction of LSM with Mn-rich zirconia.
in the Figure 5 (inset). Meanwhile the LZO lattice parameters show a drop at the temperature of Stage III (Figure 6). Those instant changes reveal the smaller Mn ions leaving YSZ-c and coming into LZO. The suggested reaction mechanism is that the Mn-rich zirconia takes part in the reaction in Stage III to transport Mn from zirconia to LZO; moreover, the Mn solutes in zirconia spontaneously migrate to LZO, resulting in the expansion of the zirconia lattice. It is believed that the Mn ions are in the form of Mn2+ occupying the La-site in LZO and contributing to the reduced LZO lattice because the radius of Mn2+ is smaller than that of La3+. In Stage III, the LZO lattice parameter in 6YSZ-LSM shows a larger drop than those in 8YSZ-LSM and 10YSZ-LSM. It is consistent with our previous conclusion that the stabilization of YSZ-t in 6YSZ-LSM introduced more Mn in YSZ. Thereby, the lattice evolutions confirm that Stage III is the rapid reaction of LSM with Mn-rich zirconia.
Beyond the mechanisms of Stage I and Stage III, a question is raised concerning the factor that activates the Mn-rich zirconia at higher temperature (Stage III). The key is to understand the diffusion behavior of Mn at high temperatures, reflected by the temperature-dependent solubility of MnO in LZO. As discussed before, Mn ions seldom participate in the reaction in Stage I; it is not the dominant dopant in LZO. In contrast, LZO has a large solubility of Mn ions above the critical temperature, probably when Stage III starts. The mass transfer from MnO to LZO is observed in 6YSZ-LSM, 8YSZ-LSM and 10YSZ-LSM at 1500°C after LSM vanishes. During the hold at 1500°C, the weight fraction of LZO keeps increasing (Figure 3 and 4) with the lattice parameter decreasing (Figure 6); at the same time, the MnO phase undergoes its mass loss (Figure 3 and 4) and the cubic YSZ shows a slight lattice expansion (Figure 5). Since LZO is able to accommodate MnO with Mn2+ freely and has shrinking lattice parameters, it can be hypothesized that it is the Mn2+ ions in the LZO and that Mn2+ occupies the La-site but not Zr-site – Mn2+ is smaller than La3+ but larger than Zr4+. Interestingly, the Mn ions, which are preferred in LZO at 1500°C, prevented the formation of LZO in Stage I and II at lower temperatures. The solubility of MnO in LZO rapidly decreases when it cools down from 1500°C to 800°C. The MnO becomes oversaturated and precipitates from LZO, as shown in Figure 3 and 4. Similarly in YSZ, the dual slopes of YSZ lattice during cooling also verified the loss of small Mn ions (Figure 5). The MnO precipitation is observed via SEM (see Supplementary Fig. S1 online) as a scatter of fine MnO grains locating at the grain boundaries of LZO and YSZ. Additionally, in the final phases, Mn atoms are found only in MnO but not in other phases.
Therefore, the activity of Mn in the reaction is established. The Mn ion with high valance and small radius, e.g. Mn3+ from LSM, is most likely situated in the cubic YSZ lattice and this Mn-rich cubic zirconia is well stabilized. In the formation of LZO, the Mn3+ ion does not prefer the La-site due to the large radius mismatch with La3+. It would not stay at the Zr-site of La2Zr2O7 instead of reverting to LaMnO3, either. That is the reason for retarding the reaction of LSM and Mn-rich zirconia. However, at the higher temperature in vacuum, it is possible that Mn3+ is reduced to Mn2+. The larger Mn2+ ion is acceptable in the La-site of LZO and at the same time, the Mn-stabilized zirconia just loses its stability due to the large mismatch of radius and valance between Mn2+ and Zr4+. Then, the Mn2+ starts to diffuse into La-site of LZO and meanwhile, the zirconia becomes active to boost the reaction in Stage III.
The yttria leads to heterogeneous distribution of Mn ions and thus the stages of reaction in xYSZ-LSM systems. The 3YSZ-LSM is another extreme case with little pristine YSZ-c but a lot of Mn-rich zirconia transformed from YSZ-t. Though Mn attempts to hinder the reaction, the large amount La2O3 around as solid solution, which forms for equal quantity of Mn ions leaving LSM36, stimulates the formation of LZO at high temperatures30. Thus, the reaction is slow at first and then speeds up at 1300 ~ 1450°C (Figure 3(a)), corresponding to the increase of Mn solubility in LZO. The Mn ions participate in the reaction from the very beginning and thus the lattice parameter of LZO is even smaller than that in 6YSZ-LSM at 1100 ~ 1200°C (Figure 6). Also, there is no kink of the LZO lattice plot during heating and no lattice shrinkage at dwelling, because Mn is almost saturated in LZO during the whole process. Eventually without exhaustion of LSM, the reagents and the products reach the equilibrium at 1500°C.
The yttria dopants also impact the La ions' diffusion and indirectly influence the reaction kinetic, though La ions are not as active as Mn ions in the xYSZ-LSM systems. The effect is observed in the samples with higher concentration of yttria dopants, e.g. 8YSZ-LSM and 10YSZ-LSM without pristine YSZ-t, in which the heterogeneous diffusion of Mn is less significant. It is noticed that less yttria content (8YSZ-LSM) leads to a relatively more rapid reaction, which is similar to the observation in the reaction of xYSZ and La0.9MnO323. That is probably caused by the diffusion of La. La3+ (r = 130 pm) has a much larger ionic radius than Y3+ (r = 115.9 pm) and Zr4+ (r = 98 pm) at 8-coordinate and the solid solubility of La2O3 in YSZ is limited20,29 in comparison with MnO. The 10YSZ has heavier Y3+ doping along with more oxygen vacancies than 8YSZ does and then 10YSZ is not able to accommodate as many La3+. The lack of La2O3 diffusion is a possible reason for slowing down the reaction in 10YSZ-LSM. Thus, 10YSZ-LSM experiences Stage II at 1400 ~ 1500°C and the LSM vanished after 1 h dwell at 1500°C, later than in 8YSZ-LSM. During the isothermal dwell, the LZO is still being produced with YSZ-c consumed after LSM vanishing (Figure 3 and 4). It implies that the La2O3 solutes in YSZ gradually segregate in the form of LZO in this period. This LZO forming without LSM verifies the important step in the reaction of YSZ-LSM that the La2O3 leaves LSM and diffuses into YSZ to produce LZO. The diffusion and solid solubility of La2O3 in YSZ are important factors of the reaction processing, but the solubility depends on the quantity of yttria dopant.
In summary, the thermal stability (RT ~ 1500°C in vacuum) of xYSZ-LSM heterogeneous systems for the SOFC cathode was revealed by in-situ neutron diffraction. The intensity contour mapping with time/temperature synchronization visualized phase evolutions in real time. It is found that the varying yttria contents led to the heterogeneous distribution of Mn ions in YSZ, either by tetragonal-to-cubic phase transition of YSZ or by diffusion at high temperature. The diffusion activity of Mn and La ions played very important roles in the three-stage reaction of LSM and YSZ. The reaction mechanisms were suggested based on quantitative analysis of the neutron diffraction data, especially on temperature dependences of the phase fractions and lattice constants.
Methods
(1) In-situ thermal treatment with neutron diffraction
In-situ heating neutron diffraction experiments were carried out at the Engineering Materials Diffractometer, VULCAN37,38,39, at the Spallation Neutron Source, Oak Ridge National Laboratory (SNS, ORNL). The commercial (La0.8Sr0.2)0.95MnO3−δ (LSM) powders with nominal particle size of 0.7 ~ 1 μm (NexTech Materials, USA) and x mol% yttria stabilized zirconia (xYSZ, x = 3, 6, 8 and 10) powders with nominal particle size less than 0.3 μm (Tosoh Corporation, Japan) were the starting materials. 50 wt% LSM and 50 wt% xYSZ were mixed with binders and then pressed to cylinders with nominal dimension of 12 mm in diameter and 25 mm in length. The cylindrical samples were densified by annealing at 800°C in air for 16 hours. During in-situ neutron diffraction, samples were mounted in a vacuum furnace and heated at 2°C/min from room temperature to 1500°C, held for 5 hours at 1500°C and then cooled down at 5°C/min. Neutron diffraction data were collected continuously during the heating/cooling process then were separated by a time bin of 5 minutes and synchronized with temperature profiles by the VDRIVE software (An, K. VDRIVE – Data reduction and interactive visualization software for event mode neutron diffraction. ORNL Report, Oak Ridge National Laboratory. ORNL-TM-2012-621, 2012). The data were analyzed using Rietveld refinement with the GSAS software and EXPGUI interface40 (Larson, A. C. & von Dreele, R. B. General structure analysis system (GSAS). Los Alamos National Laboratory Report LLAUR. 86–784, 2000).
(2) Characterization of microstructures
In order to study the microstructure and element distribution in the grains after the reaction of LSM and YSZ, backscattered electron mode scanning electron microscopy and energy-dispersive X-ray spectroscopy element mapping were done on polished surfaces with a Hitachi S-3400N SEM equipped with an EDS. The backscattering images and EDS maps were taken at 200× magnification at 15 kV.
References
Reifsnider, K. L. et al. Multiphysics Design and Development of Heterogeneous Functional Materials for Renewable Energy Devices: The HeteroFoaM Story. J. Electrochem. Soc. 160, F470–F481 (2013).
Li, T. et al. Carbon-Coated Fe−Mn−O Composites as Promising Anode Materials for Lithium-Ion Batteries. ACS Appl. Mater. Inter. 5, 9470–9477 (2013).
An, K., Reifsnider, K. L. & Gao, C. Y. Durability of (Pr0.7Sr0.3)MnO3±δ/8YSZ composite cathodes for solid oxide fuel cells. J. Power Sources. 158, 254–262 (2006).
Reifsnider, K., Huang, X., Ju, G., Feshler, M. & An, K. Mechanics of Composite Materials in Fuel Cell Systems. Mech. Compos. Mater. 41, 1–8 (2005).
Chen, Y. et al. Layered YSZ/SCSZ/YSZ electrolytes for intermediate temperature SOFC Part I: design and manufacturing. Fuel Cells. 12, 722–731 (2012).
Minh, N. Q. Solid oxide fuel cell technology-features and applications. Solid State Ionics. 174, 271–77 (2004).
O'Hayre, R. P., Cha, S., Colella, W. G. & Prinz, F. B. Fuel cell fundamentals, second edition, John Wiley & Sons, New York (2009).
Antunes, R., Jewulski, J. & Golec, T. Full Parametric Characterization of LSM/LSM-YSZ Cathodes by Electrochemical Impedance Spectroscopy. J. Fuel Cell Sci. Tech. 11, 011007 (2014).
Chen, Y., Orlovskaya, N., Graule, T. & Kuebler, J. Microstructure and mechanical properties of Ni/10 mol% Sc2O3–1 mol% CeO2–ZrO2 cermet anode for solid oxide fuel cells. J. Eur. Ceram. Soc. 33, 557–564 (2013).
Chen, D., Yang, G., Ciucci, F., Tadé, M. O. & Shao, Z. 3D core–shell architecture from infiltration and beneficial reactive sintering as highly efficient and thermally stable oxygen reduction electrode. J. Mater. Chem. A. 2, 1284–1293 (2014).
Li, M. et al. A family of oxide ion conductors based on the ferroelectric perovskite Na0.5Bi0.5TiO3 . Nature Mater. 13, 31–35 (2014).
Woolley, R. J. & Skinner, S. J. Novel La2NiO4+δ and La4Ni3O10−δ composites for solid oxide fuel cell cathodes. J. Power Sources. 243, 790–795 (2013).
Minh, N. Q. Ceramic fuel cells. J. Am. Ceram. Soc. 76, 563–588 (1993).
Jiang, S. P. Development of lanthanum strontium manganite perovskite cathode materials of solid oxide fuel cells: a review. J. Mater. Sci. 43, 6799–6833 (2008).
Tsai, T. & Barnett, S. A. Effect of LSM-YSZ cathode on thin-electrolyte solid oxide fuel cell performance. Solid State Ionics. 93, 207–217 (1997).
Leng, Y. J., Chan, S. H., Khor, K. A. & Jiang, S. P. Development of LSM/YSZ composite cathode for anode-supported solid oxide fuel cells. J. Appl. Electrochem. 34, 409–415 (2004).
Brugnoni, C., Ducati, U. & Scagliotti, M. SOFC cathode/electrolyte interface. Part I: Reactivity between La0.85Sr0.15MnO3 and ZrO2-Y2O3 . Solid State Ionics. 76, 177–182 (1995).
Kenjo, T. & Nishiya, M. LaMnO3 air cathodes containing ZrO2 electrolyte for high temperature solid oxide fuel cells. Solid State Ionics. 57, 295–302 (1992).
Yamamoto, O., Takeda, Y., Kanno, R. & Noda, M. Perovskite-type oxides as oxygen electrodes for high temperature oxide fuel cells. Solid State Ionics. 22, 241–246 (1987).
Clausen, C., Bagger, C., Bilde-Sorensen, J. B. & Horsewell, A. Microstructural and microchemical characterization of the interface between La0.85Sr0.15MnO3 and Y2O3-stabilized ZrO2 . Solid State Ionics. 59, 70–71 (1994).
Ostergard, M. J. L., Clausen, C., Bagger, C. & Mogensen, M. Manganite-zirconia composite cathodes for SOFC: Influence of structure and composition. Electrochim. Acta. 40, 1971–1981 (1995).
Mitterdorfer, A. & Gauckler, L. J. La2Zr2O7 formation and oxygen reduction kinetics of the La0.85Sr0.15MnyO3, O2(g)|YSZ system. Solid State Ionics. 111, 185–218 (1998).
Mori, M. et al. Reaction mechanism between lanthanum manganite and yttria doped cubic zirconia. Solid State Ionics. 123, 113–119 (1999).
Chen, M., Liu, Y. L., Hagen, A., Hendriksen, P. V. & Poulsen, F. W. LSM-YSZ Reactions in Different Atmospheres. Fuel Cells. 9, 833–840 (2009).
Cai, L., Liu, Z., An, K. & Liang, C. Unraveling structural evolution of LiNi0.5Mn1.5O4 by in situ neutron diffraction. J. Mater. Chem. A. 1, 6908–6914 (2013).
Wang, S. et al. Novel chemically stable Ba3Ca1.18Nb1.82−xYxO9−δ proton conductor: improved proton conductivity through tailored cation ordering. Chem. Mater. (2014) 10.1021/cm403684b.
Wiik, K. Reactions between Strontium-Substituted Lanthanum Manganite and Yttria-Stabilized Zirconia: I, Powder Samples. J. Am. Ceram. Soc. 82, 721–28 (1999).
Backhaus-Ricoult, M. Interface chemistry in LSM–YSZ composite SOFC cathodes. Solid State Ionics. 177, 2195–2200 (2006).
Kawada, T., Sakai, N., Yokokawa, H. & Dokiya, M. Reaction between solid oxide fuel cell materials. Solid State Ionics. 50, 189–196 (1992).
Jiang, S. P., Zhang, J. P. & Föger, K. Chemical interactions between 3 mol% yttria-zirconia and Sr-doped lanthanum manganite. J. Eur. Ceram. Soc. 23, 1865–1873 (2003).
Taimatsu, H., Wada, K., Kaneko, H. & Yamamura, H. Mechanism of Reaction between Lanthanum Manganite and Yttria-Stabilized Zirconia. J. Am. Ceram. Soc. 75, 401–405 (1992).
Chen, M., Grundy, A. N., Hallstedt, B. & Gauckler, L. J. Thermodynamic modeling of the La-Mn-Y-Zr-O system. Calphad 30, 489–500 (2006).
Jiang, S. P., Zhang, J. P., Ramprakash, Y., Milosevic, D. & Wilshier, K. An investigation of shelf-life of strontium doped LaMnO3 materials. J Mater Sci. 35, 2735–2741 (2000).
Zhang, J. P., Jiang, S. P., Love, J. G., Föger, K. & Badwal, S. P. S. Chemical interactions between strontium-doped praseodymium manganite and 3 mol% yttria-zirconia. J. Mater. Chem. 8, 2787–2794 (1998).
Chen, Y. et al. Residual stress and biaxial strength in Sc2O3–CeO2–ZrO2/Y2O3–ZrO2 layered electrolytes. Fuel Cells. 13, 1068–1075 (2013).
van Roosmalen, J. A. M. & Cordfunke, E. H. P. Chemical reactivity and interdiffusion of (La,Sr)MnO3 and (Zr,Y)O2 solid oxide fuel cell cathode and electrolyte materials. Solid State Ionics. 52, 303–312 (1992).
Wang, X. L. et al. First results from the VULCAN diffractometer at the SNS. Mater. Sci. Forum. 652, 105–110 (2010).
Ma, D. et al. Observation of a transient texture during the α → β phase transformation in titanium. Metall. Mater. Trans. A. 42, 1444–1448 (2011).
An, K. et al. First in situ lattice strains measurements under load at VULCAN. Metall. Mater. Trans. A. 42, 95–99 (2011).
Toby, B. H. EXPGUI, a graphical user interface for GSAS. J. Appl. Crystallogr. 34, 210–213 (2001).
Acknowledgements
This work was supported by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Division of Materials Sciences and Engineering. Neutron scattering was carried out at the Spallation Neutron Source, DOE user facilities at Oak Ridge National Laboratory, which is sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. The authors thank Dr. J. Kiggans for help on the sample preparation and Dr. L. Cai, Mrs. R. A. Mills, Mr. D.P. Armitage and Mr. H.D. Skorpenske from SNS for their technical support of the neutron experiments. The authors thank Mrs. Gumin Zhu for the technical support.
Author information
Authors and Affiliations
Contributions
K.A. designed and guided the project. L.Y. prepared the materials and performed the neutron diffraction experiments. F.R. collected the SEM images. Y.C., L.Y. and K.A. conducted the data analysis. Y.C. and K.A. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
SUPPLEMENTARY INFORMATION
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Chen, Y., Yang, L., Ren, F. et al. Visualizing the Structural Evolution of LSM/xYSZ Composite Cathodes for SOFC by in-situ Neutron Diffraction. Sci Rep 4, 5179 (2014). https://doi.org/10.1038/srep05179
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05179
This article is cited by
-
Spring Based Connection of External Wires to a Thin Film Temperature Sensor Integrated Inside a Solid Oxide Fuel Cell
Scientific Reports (2019)
-
Progress and outlook for solid oxide fuel cells for transportation applications
Nature Catalysis (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
