
Abstract
Mutations in the Tsc1 or Tsc2 genes cause tuberous sclerosis complex (TSC). Tsc1 and Tsc2 proteins form a complex that inhibits mammalian target of rapamycin complex 1 (mTORC1) signalling through Rheb-GTPase. We found that Tsc2+/− neurons showed impaired spine synapse formation, which was resistant to an mTORC1 inhibitor. Knockdown of mTOR also failed to restore these abnormalities, suggesting mTORC may not participate in impaired spinogenesis in Tsc2+/− neurons. To address whether Rheb activation impairs spine synapse formation, we expressed active and inactive forms of Rheb in WT and Tsc2+/− neurons, respectively. Expression of active Rheb abolished dendritic spine formation in WT neurons, whereas inactive Rheb restored spine synapse formation in Tsc2+/− neurons. Moreover, inactivation of Rheb with farnesyl transferase inhibitors recovered spine synapse morphogenesis in Tsc2+/− neurons. In conclusion, dendritic spine abnormalities in TSC neurons may be caused through activation of Rheb, but not through of mTORC1.
Similar content being viewed by others
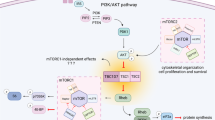


Introduction
In the mammalian brain, dendritic spines make most excitatory synapses. During development, synapse formation and elimination are accompanied by changes in dendritic spine number and morphology, allowing the establishment and remodelling of neuronal circuits. Structural plasticity of dendritic spines is tightly correlated with synaptic function and plasticity; even subtle changes in dendritic spines may have marked effects on synaptic plasticity. In particular, a large number of brain disorders, including developmental disorders are accompanied by disease-related changes in dendritic spine density and/or morphology.
Tuberous sclerosis complex (TSC) is a systemic genetic disease that causes the growth of hamartomas in the brain and other organs1. The neurological symptoms of TSC include mental retardation, intractable epilepsy and autism1. TSC results from inactivating mutations in either Tsc12 or Tsc23, which encode hamartin and tuberin, respectively. Hamartin and tuberin form a complex that exhibits guanosine triphosphatase (GTPase)-activating protein (GAP) activity toward Rheb, a Ras-like small G-protein4,5,6. Inactivating mutations in Tsc1 or Tsc2 result in the accumulation of the GTP-bound form of Rheb, which activates mammalian target of rapamycin complex 1 (mTORC1)7. The mTOR is a serine/threonine protein kinase that stimulates translation initiation processes involving both eIF4E/4E-binding protein (4EBP) and p70S6 kinase/ribosomal S6 protein.
Mouse models that harbor a mutation in the Tsc1 or Tsc2 genes have been extensively investigated to clarify the mechanisms of the pathogenesis of TSC. Several lines of evidence suggest that these TSC model mice exhibit impairments in dendritic spine formation8, long-term depression and cognitive behaviors9, suggesting that activation of mTORC1 in TSC neurons may cause memory disturbance probably through dendritic spine abnormalities. However, the mTOR signaling has been demonstrated to be activated during learning and is required for spatial memory formation10. This controversy may suggest that another mechanism other than the mTOR signaling could be involved in memory impairment in TSC.
In the present study, we first confirmed that dendritic spine formation was abrogated and also excitatory spine synapses were drastically reduced in cultured rat Tsc2+/− neurons. This observation was consistent with the finding that filopodia-like protrusions were increased in Tsc2+/− rat brain. We treated cultured Tsc2+/− neurons with rapamycin, a specific mTORC1 inhibitor; however, it did not restore spine morphology but rather elongated dendritic protrusions. Knockdown of mTOR did not restore spine synapse formation either, suggesting that the mTORC pathway may not participate in spine synapse morphogenesis. We therefore focused on Rheb upstream of mTORC1 and expressed the GDP-bound form of Rheb in Tsc2+/− neurons, resulting in restoration of spine synapse formation. Conversely, expression of the GTP-bound Rheb abolished spine formation in WT neurons. Furthermore, treatment with farnesyl transferase inhibitors (FTIs) lonafarnib and tipifarnib also rescued spine synapse formation in Tsc2+/− neurons. Taken together, these results suggest that spine synapse deformity in TSC neurons may be dependent on the state of Rheb, but not on mTORC activation.
Results
Tsc2+/− neurons show impaired spine synapse formation
In this study, we used spontaneous Tsc2-mutated Eker rats as animal models of TSC11. We first compared dendritic spine morphology of cultured hippocampal neurons of wild type (WT) and heterozygous Eker (Tsc2+/−) rats. At 21 days in vitro (DIV), the dendritic spines of WT neurons displayed mushroom-type spines (Fig. 1a)(top), whereas the dendrites of Tsc2+/− neurons possessed long, thin filopodia (Fig. 1a)(bottom). Quantification showed the Tsc2 mutation reduced the width of dendritic protrusions (Fig. 1a, b) and increased their length (Fig. 1a, c). To assess the influence of Tsc2 haploinsufficiency on spine synapse formation, we immunolabelled neurons with vesicular glutamate transporter 1 (vGlut1), a presynaptic excitatory marker. In Tsc2+/− neurons, dendritic vGlut1 puncta were rarely located on protrusions but preferentially localized to dendritic shafts (Fig. 1a, d, e), indicating that Tsc2+/− growth cones form synapses with dendritic shafts.

Tsc2 mutation suppresses dendritic spine synapse formation.
(a) Images of dendrites from wild-type (top) and Tsc2+/− (bottom) pyramidal neurons in hippocampal culture. Neurons were transfected with EGFP and immunostained with anti-vGlut1 antibody. Scale bar, 10 μm. (b, c) Cumulative probability plots of protrusion width and length shown in (a). Tsc2+/− neurons showed a decrease in protrusion width (D = 0.2515, P < 0.001) and an increase in protrusion length (D = 0.4127, P < 0.001) compared with WT neurons (Kolmogorov-Smirnov test). (d) Densities of vGlut1-positive spine synapses in WT and Tsc2+/− neurons (***P < 0.001, unpaired, two-tailed t-test). (e) Percentage of spine synapses in WT and Tsc2+/− neurons (***P < 0.001, unpaired, two-tailed t-test). For each experiment (a–e), a total of 1,060/15/5 (WT) and 1,007/15/5 (Tsc2+/−) protrusions/neurons/experiments were analysed, respectively. (f) Representative DiI-labelled dendrites of hippocampal CA1 neurons from WT and Tsc2+/− rats. Scale bar, 10 μm. (g, h) Cumulative probability plots of the protrusion width (g) and length (h) shown in (f). Tsc2+/− neurons showed a decrease in the protrusion width (D = 0.2950, P < 0.001) and an increase in the protrusion length (D = 0.2256, P < 0.001) compared with WT neurons (n = 1,001/12/6 (WT) and 1,034/12/6 (Tsc2+/−) protrusions/neurons/experiments (Kolmogorov-Smirnov test).
Next, to confirm whether dendritic protrusions of in vivoTsc2+/−neurons exhibit similar morphological changes to those in cultured Tsc2+/−neurons, we labeled hippocampal CA1 neurons in WT and Tsc2+/−fixed brain tissues with biolistic-DiI (Fig. 1f). We observed similar morphological changes in spine width and length in Tsc2+/− neurons in vivo (Fig. 1g, h) to those observed in cultured Tsc2+/− neurons.
The mTORC1 inhibitor rapamycin does not restore spine synapse morphogenesis in Tsc2+/− neurons
To address whether the effects of Tsc2 haploinsufficiency on neuronal morphology are mediated by elevated mTORC1 activity, we examined the ability of rapamycin to reverse defects in neuronal morphology. WT and Tsc2+/− hippocampal neurons were treated with rapamycin and maintained for 6 days. Application of rapamycin to Tsc2+/− neurons completely abolished the phosphorylation of S6 ribosomal protein, which is a substrate of the mTORC1 downstream target S6 kinase12 (Fig. 2a, Supplementary Fig. S4a, b). Although rapamycin did not affect spine morphology in WT neurons (Fig. 2b, c, d), it rather induced the growth of long, thin protrusions in Tsc2+/− neurons (Fig. 2b, c, d) without restoration of spine synapse formation (Fig. 2b, e, f). To test whether mTORC2 participates in spine synapse formation, we next knocked down mTOR in WT and Tsc2+/− neurons by genetic silencing with siRNA (Supplementary Fig. S1). However, mTOR siRNA did not reverse spine synapse formation but rather elongated dendritic protrusions in Tsc2+/− neurons, like rapamycin treatment (Fig. 2g–k). The mTOR inhibition by either rapamycin or siRNA did not significantly change the number of spine synapses (Fig. 2e, f, j, k). Thus, neither mTORC1 nor mTORC2 may participate in the impaired spine synapse morphogenesis in TSC neurons.

Rapamycin fails to reverse spine synapse formation in Tsc2+/− neurons.
(a) Rapamycin treatment completely abolished the phosphorylation of S6 in both WT and Tsc2+/− neurons. These blots are cropped and full-length blots are presented in Supplementary Figure 1. (b) High-power images of the dendrites of DMSO- or 100 nM rapamycin-treated WT and Tsc2+/− neurons. Scale bar, 10 μm. (c, d) Cumulative probability plots of dendritic protrusion width (c) and length (d) in shown in (b). Note that rapamycin treatment increases dendritic protrusion lengths compared with DMSO treatment in Tsc2+/− neurons (D = 0.1717, P < 0.001, Kolmogorov-Smirnov test). (e) The densities of the vGlut1-positive spine synapses shown in (b). (f) The percentage of spine synapses in (b) (n = 400/7/3 (DMSO, WT), 400/7/3 (Rapa, WT), 1091/12/6 (DMSO, Tsc2+/−) and 1020/12/6 (Rapa, Tsc2+/−) protrusions/neurons/experiments). Note that rapamycin treatment did not significantly change the number of spine synapses. (g) Dendrites from control-, or mTOR siRNA-transfected WT and Tsc2+/− neurons. Scale bar, 10 μm. (h, i) Cumulative probability plots of protrusion width (h) and length (i) shown in (g). Note that mTOR siRNA expression causes increases in dendritic protrusion lengths in both WT and Tsc2+/− neurons, compared with scrambled siRNA (D = 0.1684, P < 0.01 (WT), D = 0.3676, P < 0.001 (Tsc2+/−), Kolmogorov-Smirnov test). (j) The densities of the vGlut1-positive spine synapses shown in (g). (k) The percentage of spine synapses in (g) (n = 400/7/3 protrusions/neurons/experiments). Note that treatment with mTOR siRNA did not significantly change the number of spine synapses.
Inactivation of Rheb recovers spine synapse formation in Tsc2+/− neurons
We therefore focused on Rheb, a GTPase located downstream of the Tsc1/2 complex and upstream of mTORC1 and examined whether Rheb activation is directly involved in spine synapse abnormalities in Tsc2+/− neurons. We first asked whether activation of Rheb in WT neurons causes morphological changes similar to those in Tsc2+/− neurons. To address this question, we expressed two kinds of Rheb mutants Gln64Val (RhebQ64V) and Asp60Ile (RhebD60I) in WT or Tsc2+/− neurons (Supplementary Fig. S2a, b). Gln64 mutant of Rheb displays a higher basal GTP level5, whereas RhebD60I mutation interferes selectively with GTP binding13. Expression of the RhebQ64V completely abolished spine synapse formation in WT neurons, whose dendritic morphology was similar to that of Tsc2+/− neurons (Fig. 3a–e). In contrast, RhebQ64V expression did not affect filopodia-like morphology of dendritic protrusions in Tsc2+/− neurons (Fig. 3a–c). Conversely, the expression of RhebD60I restored spine synapse formation in Tsc2+/− neurons by increasing the ratio of spine synapses (Fig. 3f, i, j). RhebD60I expression also rescued spine width in Tsc2+/− neurons to a similar extent to those in WT neurons (Fig. 3f, g). However, overexpression of RhebD60I in WT neurons differentially modified their spine structure from those of Tsc2+/− neurons. RhebD60I expression unexpectedly caused narrower and longer dendritic protrusions (Fig. 3f, g, h) and reduced the ratio of spine synapses in WT neurons (Fig. 3f, i, j). These results suggest that appropriate temporal activation of Rheb may be required for normal spine development in WT neurons. Thus, impaired spine synapse morphogenesis in Tsc2+/− neurons may be mediated by constitutively active state of Rheb and could be rescued by inactivation of Rheb.

Rheb regulates spine synapse formation.
(a) Dendrites of EGFP- and mock- or FLAG-RhebQ64V-expressing hippocampal neurons prepared from WT and Tsc2+/− rats at DIV 19. Neurons were co-immunostained with anti-GFP and vGlut1 antibodies. Scale bar, 10 μm. (b, c) Cumulative probability distributions of protrusion width (b) and length (c) in (a). RhebQ64V-expressing WT neurons showed narrower (D = 0.2690, P < 0.001) and longer (D = 0.2391, P < 0.001) dendritic protrusions than mock transfectants (Kolmogorov-Smirnov test). (d) Quantification of the spine synapse densities shown in (a). (e) Percentage of spine synapses shown in (a). FLAG-RhebQ64V-expressing WT neurons showed few vGlut1-positive spines compared with mock-transfected neurons. The histograms show the mean values ± SEM (***P < 0.001, Two-Way ANOVA, post-hoc Tukey test). A total of 803/10/5 (WT, mock), 1,029/12/6 (WT, RhebQ64V), 501/6/3 (Tsc2+/−, mock), 320/6/3 (Tsc2+/−, RhebQ64V) protrusions/neurons/experiments, respectively, were analysed. (f) Dendrites of EGFP- and mock- or FLAG-RhebD60I-expressing hippocampal neurons prepared from WT and Tsc2+/− rats at DIV 19. Neurons were co-immunostained with anti-GFP and vGlut1 antibodies. Scale bar, 10 μm. (g, h) Cumulative probability plots of dendritic protrusion width (g) and length (h) in mock- or RhebD60I-expressing WT and Tsc2+/− neurons in (f). Note that RhebD60I increased dendritic protrusion width (D = 0.2903, P < 0.001) and decreased length (D = 0.4306, P < 0.001) in Tsc2+/− neurons, whereas RhebD60I decreased protrusion width (D = 0.638, P < 0.001) and increased length (D = 0.509, P < 0.001) in WT neurons, compared with mock transfectants (Kolmogorov-Smirnov test). (i) Quantitative analysis of the spine synapse density shown in (f). (j) The percentage of spine synapses in (f) (n = 300/6/3 (WT, mock), 300/6/3 (WT, RhebD60I), 988/12/6 (Tsc2+/−, mock) and 1,044/12/6 (Tsc2+/−, RhebD60I) protrusions/neurons/experiments, *P < 0.05, ***P < 0.001, Two-Way ANOVA, post-hoc Tukey test).
FTI treatment restores spine synapse morphogenesis in Tsc2+/− neurons
To address whether pharmacological inactivation of Rheb recovers spine synapse formation in Tsc2+/− neurons, we utilized two kinds of FTIs, lonafarnib and tipifarnib. FTIs have been shown to reverse growth and actin filaments distribution in Tsc1−/− and Tsc2−/− cells in a rapamycin-sensitive and -insensitive manners14. Therefore, we treated Tsc2+/− neurons with lonafarnib or tipifarnib15 for 3 days. These FTI treatments resulted in recuperation of F-actin accumulation in dendritic spines (Fig. 4a, b) and increases in spine synapse ratio (Fig. 4c–g) in Tsc2+/− neurons. On the other hand, either lonafarnib or tipifarnib treatment did not affect spine synapse formation in WT neurons (Fig. 4c, f, g). In conclusion, persistent activation of Rheb may abolish dendritic spine morphogenesis and that inactivation of Rheb with FTI may restore spine synapse formation in TSC neurons.

FTI treatment restores spine synapse formation in Tsc2+/−neurons.
(a) Confocal images of cultured EGFP-expressing neurons from WT or Tsc2+/− hippocampi. Neurons were treated with DMSO, 2 μM lonafarnib or 2 μM tipifarnib for 3 days. F-actin was visualized with Alexa647-coupled phalloidin. Arrows denote positive puncta that are associated with dendritic spines. Scale bars, 10 μm. (b) Densities of the F-actin -positive spines shown in (a). (c) Immunodetection of vGlut1 (red)-positive presynaptic boutons in EGFP (green)-expressing WT and Tsc2+/− neurons treated with DMSO, lonafarnib or tipifarnib. (d, e) Cumulative probability distributions of protrusion width (d) and length (e) in (c). Note that lonafarnib or tipifarnib treatment increased dendritic protrusion width (D = 0.545, P < 0.001 (lonafarnib), D = 0.416, P < 0.001 (tipifarnib)) and decreased dendritic protrusion length (D = 0.367, P < 0.001 (lonafarnib), D = 0.242, P < 0.001 (tipifarnib)) compared with DMSO treatment in Tsc2+/− neurons (Kolmogorov-Smirnov test). (f) Quantification of vGlut1-positive spines in (c). (g) The percentage of spine synapses in (c) (n = 300/6/3 (WT, DMSO), 286/6/3 (WT, lonafarnib), 321/6/3 (WT, tipifarnib), 300/6/3(Tsc2+/−, DMSO), 376/6/3(Tsc2+/−, lonafarnib), 363/6/3 (Tsc2+/−, tipifarnib) spines/neurons/experiments, ***P < 0.001, **P < 0.01, Two-Way ANOVA, post hoc Tukey test).
Finally, to reveal the functional differences in excitatory synaptic transmission between WT and Tsc2+/−neurons, we measured evoked EPSC amplitude, mEPSC amplitude/frequency, readily releasable pool size and vesicular release probability from both cultured neurons. However, unexpectedly, synaptic transmission was sufficiently observed in cultured Tsc2+/− neurons without any significant difference in presynaptic or postsynaptic features between WT and Tsc2+/− neurons (Supplementary Fig. S3a–f). These results suggest that basal synaptic transmission may be preserved in Tsc2+/− neurons as WT neurons, irrespective of spine synapse abnormalities. Other electrophysiological properties, such as long-term depression, might be affected by impaired spine morphogenesis in Tsc2+/− neurons. Further studies are necessary for understanding the effects of morphological abnormalities of dendritic spines on the pathogenesis of TSC.
Discussion
In this paper, we have demonstrated activation of Rheb, not of mTORC1, is a main cause of dendritic spine synapse abnormality in Tsc2+/− neurons. We showed that active Rheb mutant abolished spine formation in WT neurons, whereas the inactive mutant of Rheb restored spine synapse morphogenesis in Tsc2+/− neurons. Furthermore, we demonstrated that FTI treatment, but not rapamycin treatment, rescued spine synapse morphology in Tsc2+/− neurons. These results, taken together, suggest that Rheb inhibition by FTI may be necessary for recovery of spine synapse formation in the TSC brain.
Mouse models of TSC have demonstrated dendritic spine abnormalities that are resistant to rapamycin treatment8,16. For example, Tsc1-deficient mice develop several pathological features observed in human TSC patients. Rapamycin treatment improved neurofilament abnormalities, myelination and cell enlargement in Tsc1-ablated neurons, but it did not reverse dendritic spine morphology17. Additional studies have revealed that the application of rapamycin to Tsc1- or Tsc2-reduced neurons did not restore spine formation8. F-actin is the major cytoskeletal component in dendritic spines and has been thought to provide the structural basis for dendritic spines18. However, rapamycin has been shown to rather suppress F-actin reorganization in an mTOR-dependent manner18,19,20. Therefore, inhibition of F-actin reorganization by rapamycin might cause an elongation of existing filopodial protrusions, consistent with our results. Taken together, the impaired spine formation in TSC neurons may not be mediated by mTORC1.
Our results indicate that FTI treatment could restore morphological abnormalities in Tsc2+/− neurons. Farnesyl transferase (FT) recognizes the carboxyl-terminal CAAX box of small GTP-binding proteins and catalyses the transfer of lipid onto the cysteine residue. Following carboxymethylation of the farnesyl cysteine renders the carboxyl terminus more hydrophobic21. Membrane localization is essential for Rheb activity as for other small GTP-binding proteins22. To prevent Rheb farnesylation and membrane localization, FTIs have been used14,23. Unlike Ras, Rheb is not a substrate for geranylgeranyltransferases and can only undergo farnesylation14. Therefore, FTIs may suppress the Rheb activity mainly. On the other hand, another small GTP-binding protein RhoB has been shown to be an additional target of FTIs24,25. Because FTI treatment induces apoptosis in the endothelial cells of sprouting and developing blood vessels via inhibition of RhoB25, pharmaceutical inactivation of RhoB could cause possible side effects as cardiovascular disorders where the angiogenesis would be desirable.
The finding that FTI was capable of restoring dendritic spine formation suggests that there are two different pathways downstream of Rheb in neurons, one signalling to neuronal growth and the other regulating neuronal morphology. The pathway affecting growth is likely to be mTORC1-dependent, whereas the pathway regulating neuronal morphology is rapamycin-insensitive. This mTORC1-dependent signalling is activated during learning and plays a crucial role in the spatial learning10. By contrast, the TSC mice develop significant cognitive deficits9. This discrepancy might be explained by the notion that activation of the mTORC1-independent signalling may impair spine synapse formation in TSC and dominantly disturb memory acquisition or retention despite the mTORC1 activation.
What could be involved in the mTORC1-independent pathway to control neuronal morphology? We initially considered the possibility of the mTORC2 pathway; however, this seems unlikely, because mTOR knockdown fails to reverse spine synapse formation in Tsc2+/− neurons (Fig. 2g–k). Further experiments may provide important insights into how Rheb regulates neuronal morphology. As Rheb-mediated regulation of cytoskeleton has not been reported so far, we are currently searching Rheb-binding protein(s) in neurons. Clarification of this mechanism could extend our understanding of the prevailing role of Rheb in memory impairments in TSC patients.
Methods
Reagents
Rapamycin and FTIs (lonafarnib and tipifarnib) were purchased from LKT Laboratories and Toronto Research Chemicals, respectively. The used siRNAs are as follows: mTOR siRNA (CAGCAGCUGGUACAUGACAAGUACU and AGUACUUGUCAUGUACCAGCUGCUG) and Stealth RNAi siRNA negative control kit. Oligonucleotides were synthesized and duplexed by Invitrogen.
Animals
The Eker (Tsc2+/−) and wild-type (WT) genotypes were determined using PCR26. All animal experiments were approved by the Committee on the the Animal Care and Use Committees of the Tokyo Metropolitan Institute of Medical Science and Fukuoka University and were performed according to their recommendation.
Primary culture and transfection
Primary culture and transfection of hippocampal neurons from Tsc2+/− and WT littermates (E20) have been previously described27,28,29. Cultured neurons were maintained in Neurobasal Medium supplemented with B27, gentamicin and GLUTAMAX. The neurons were transfected at DIV11–12 using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cultured hippocampal neurons were treated with 100 nM rapamycin for 6 days, 2 μM lonafarnib or 2 μM tipifarnib for 3 days. All experiments comparing WT and Tsc2+/− neurons were performed in parallel.
Spine and/or shaft synapse density analysis were performed as described previously30. Spines were defined as any protrusions from the dendritic shaft <8 μm in length. The number of vGlut1 puncta on spine and shaft synapses of the dendritic segment is determined by counting the number of vGlut1 puncta that are partially overlapping to the GFP-positive shaft or spines. Any vGlut1 puncta that are not touching the GFP-positive dendrite are not included.
Immunostaining
Rat cultured hippocampal neurons were fixed with 4% paraformaldehyde, blocked, permeabilized with BL solution (3% normal goat serum or 3% bovine serum albumin with 0.1% Triton X-100 in PBS) and incubated overnight at 4°C with one of the following primary antibodies: anti-GFP (chicken, Millipore), anti-phospho S6 (Ser235/236) (rabbit, CST), anti-phospho-Akt (Ser473) (rabbit, CST) or anti-vGlut1 (guinea pig, Millipore). The immunoreactivity was visualized using species-specific, fluorochrome-conjugated secondary antibodies. Neuronal F-actin was visualized by staining with Alexa Fluor 647 phalloidin (Invitrogen).
Image acquisition and quantification
Confocal images of neurons were obtained using a Zeiss 63× lens (NA 1.4) with sequential acquisition settings at the resolution of the confocal microscope (512 × 512 pixels). Each image was a composite constructed from a series of images captured throughout the z aspect of each cell. The parameters of each composite image were optimized for the particular lens and pinhole setting. Identical confocal microscope settings were maintained for all scans in each experiment. All morphometric analyses and quantifications were performed using MetaMorph image analysis software (Universal Imaging). For the spine morphology studies, individual dendritic protrusions were manually traced and the maximum length and head width of each dendritic protrusion were measured and recorded in Microsoft Excel.
Diolistic labelling
Eight-week-old rats were deeply anesthetized with 0.1 mg/kg phenobarbital perfused transcardially with 0.1 M PB, followed by 50 ml of 2% PFA. The brains were dissected and post-fixed in the same fixative for 4 hr. Subsequently, sagittal brain sections were obtained using a vibratome and collected in PBS. Twenty-five milligrams of gold particles (Bio-Rad) were coated with 7.0 mg of the lipophilic dye DiI (Invitrogen) and injected into Tefzel tubing, which was subsequently cut into bullets. These particles were delivered diolistically at 80 psi using a Helios gene-gun system (Bio-Rad). The slices were fixed again with 4% PFA at 4°C for 1 hr and mounted on slides in VectaShield Mounting Medium (Vector Laboratories).
Western blot analysis
Western blot analyses of rat hippocampal neurons with anti-phospho S6 (Ser235/236) (rabbit, CST) antibody31, anti-S6 (rabbit, CST) antibody32 or HEK 293T cells with anti-mTOR antibody33 were performed as described previously29.
Autaptic culture
Astrocytes were obtained from cerebral cortices of newborn (P0) ICR mice (Kyudo). Cerebral cortices were removed from the brains, placed in cold Hank's Balanced Saline Solution (HBSS, Invitrogen) and dissociated with 0.05% trypsin-EDTA (Wako). The cells were plated in plating medium composed of Dulbecco's Modified Eagle Medium with GLUTAMAX and pyruvate (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen) and 0.1% MITO+ Serum Extender (BD Biosciences). Two weeks later, astrocytes adhered to the bottom of the culture bottle were trypsinized and plated onto microdot-coated coverslips at a density of 6,000 cells/cm2.
The autaptic cultures of E18–19 hippocampal neurons were generated in accordance with previous reports34,35,36. Briefly, CA1-CA3 hippocampi were isolated from embryo rats and enzymatically dissociated in papain (2 units/ml, Worthington) in DMEM. The dissociated hippocampal neurons were plated at a density of 1,500 cells/cm2 onto the above astrocyte island plates. Before plating the dissociated neurons, the astrocyte-conditioned media was exchanged for Neurobasal-A medium (Invitrogen) supplemented with 2% B27 supplement (Invitrogen) and GLUTAMAX.
Electrophysiology
Synaptic responses from WT and Tsc2+/− autaptic neurons were recorded in the whole-cell configuration under the voltage-clamp mode at a holding potential (Vh) of −70 mV (MultiClamp 700B, Molecular Devices) and room temperature. The patch pipette resistance was 3–5 MΩ and a series resistance was compensated by 70–90%. The AMPA receptor-mediated evoked glutamatergic synaptic transmission was recorded in response to an action potential elicited through a brief (2 ms) somatic depolarization pulse (to +0 mV) from the patch pipette. Spontaneous glutamatergic mEPSCs were recorded in the presence of 0.5 μM tetrodotoxin (TTX). The size of the readily releasable pool (RRP) of synaptic vesicles was estimated using a synaptic transient charge elicited through a 10 sec application of 0.5 M sucrose. The number of synaptic vesicles in the RRP was subsequently calculated by dividing the RRP charge by an individual average mEPSC charge. The vesicular release probability (Pvr) was calculated as the ratio of the charge of the evoked EPSC to the RRP charge successfully obtained from the same neuron. The synaptic responses were recorded at a sampling rate of 20 kHz and filtered at 10 kHz. The template for the mEPSC data analysis was set as a rise time constant of 0.5 ms, a decay time constant of 4 ms, a baseline of 5 ms and a template length of 10 ms. The data were analysed off-line using Axograph X 1.2 software (AxoGraph Scientific). Recordings of synaptic responses were performed at DIV 7–9 for immature synapses and at DIV 19–22 for mature synapses.
The standard extracellular solution contained (in mM) NaCl 140, KCl 2.4, HEPES 10, glucose 10, CaCl2 2 and MgCl2 1, with pH 7.4 and an adjusted osmotic pressure of 315–320 mOsm. Patch pipettes were filled with an intracellular solution containing (in mM) K-gluconate 146.3, MgCl2 0.6, ATP-Na2 4, GTP-Na2 0.3, phosphocreatine 12, EGTA 1 and HEPES 17.8, as well as creatine phosphokinase 50 U/ml (pH 7.4). The extracellular solutions were applied using a fast-flow application system (Warner Instruments). All chemicals were purchased from Sigma-Aldrich unless otherwise specified.
Statistical analysis
The data were statistically analysed using Student's t-test, two-way ANOVA or the Kolmogorov-Smirnov test. Differences were considered significant when p < 0.05.
References
Crino, P. B., Nathanson, K. L. & Henske, E. P. The Tuberous Sclerosis Complex. New Engl J Med 355, 1345–1356 (2006).
van Slegtenhorst, M. et al. Identification of the Tuberous Sclerosis Gene TSC1 on Chromosome 9q34. Science 277, 805–808 (1997).
Kandt, R. S. et al. Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker for polycystic kidney disease. Nat Genet 2, 37–41 (1992).
Zhang, Y. et al. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 5, 578–581 (2003).
Inoki, K., Li, Y., Xu, T. & Guan, K.-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17, 1829–1834 (2003).
Zou, J. et al. Rheb1 Is Required for mTORC1 and Myelination in Postnatal Brain Development. Dev Cell 20, 97–108 (2011).
Manning, B. D. & Cantley, L. C. Rheb fills a GAP between TSC and TOR. Trends Biochemi Sci 28, 573–576 (2003).
Tavazoie, S. F., Alvarez, V. A., Ridenour, D. A., Kwiatkowski, D. J. & Sabatini, B. L. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 8, 1727–1734 (2005).
Goorden, S. M. I., van Woerden, G. M., van der Weerd, L., Cheadle, J. P. & Elgersma, Y. Cognitive deficits in Tsc1+/− mice in the absence of cerebral lesions and seizures. Ann Neurol 62, 648–655 (2007).
Qi, S., Mizuno, M., Yonezawa, K., Nawa, H. & Takei, N. Activation of mammalian target of rapamycin signaling in spatial learning. Neurosci Res 68, 88–93 (2010).
Yeung, R. S. et al. Predisposition to renal carcinoma in the Eker rat is determined by germ-line mutation of the tuberous sclerosis 2 (TSC2) gene. Proc Natl Acad Sci USA 91, 11413–11416 (1994).
Peterson, R. T. & Schreiber, S. L. Translation control: Connecting mitogens and the ribosome. Curr Biol 8, R248–R250 (1998).
Tabancay, A. P. et al. Identification of Dominant Negative Mutants of Rheb GTPase and Their Use to Implicate the Involvement of Human Rheb in the Activation of p70S6K. J Biol Chem 278, 39921–39930 (2003).
Gau, C.-L. et al. Farnesyltransferase inhibitors reverse altered growth and distribution of actin filaments in Tsc-deficient cells via inhibition of both rapamycin-sensitive and -insensitive pathways. Mol Cancer Ther 4, 918–926 (2005).
Appels, N. M. G. M., Beijnen, J. H. & Schellens, J. H. M. Development of Farnesyl Transferase Inhibitors: A Review. Oncologist 10, 565–578 (2005).
Meikle, L. et al. Response of a Neuronal Model of Tuberous Sclerosis to Mammalian Target of Rapamycin (mTOR) Inhibitors: Effects on mTORC1 and Akt Signaling Lead to Improved Survival and Function. J Neurosci 28, 5422–5432 (2008).
Meikle, L. et al. A Mouse Model of Tuberous Sclerosis: Neuronal Loss of Tsc1 Causes Dysplastic and Ectopic Neurons, Reduced Myelination, Seizure Activity and Limited Survival. J Neurosci 27, 5546–5558 (2007).
Hotulainen, P. & Hoogenraad, C. C. Actin in dendritic spines: connecting dynamics to function. J Cell Biol 189, 619–629 (2010).
Liu, L., Chen, L., Chung, J. & Huang, S. Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene 27, 4998–5010 (2008).
Liu, L. et al. Rapamycin Inhibits Cytoskeleton Reorganization and Cell Motility by Suppressing RhoA Expression and Activity. J Biol Chem 285, 38362–38373 (2010).
Gutierrez, L., Magee, A. I., Marshall, C. J. & Hancock, J. F. Post-translational processing of p21ras is two-step and involves carboxyl-methylation and carboxy-terminal proteolysis. EMBO J 8, 1093–8 (1989).
Clark, G. J. et al. The Ras-related Protein Rheb Is Farnesylated and Antagonizes Ras Signaling and Transformation. J Biol Chem 272, 10608–10615 (1997).
Basso, A. D. et al. The Farnesyl Transferase Inhibitor (FTI) SCH66336 (lonafarnib) Inhibits Rheb Farnesylation and mTOR Signaling: ROLE IN FTI ENHANCEMENT OF TAXANE AND TAMOXIFEN ANTI-TUMOR ACTIVITY. J Biol Chem 280, 31101–31108 (2005).
Prendergast, G. C. Farnesyltransferase inhibitors: antineoplastic mechanism and clinical prospects. Curr Opin Cell Biol 12, 166–173 (2000).
Adini, I., Rabinovitz, I., Sun, J. F., Prendergast, G. C. & Benjamin, L. E. RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev 17, 2721–2732 (2003).
Rennebeck, G. et al. Loss of function of the tuberous sclerosis 2 tumor suppressor gene results in embryonic lethality characterized by disrupted neuroepithelial growth and development. Proc Natl Acad Sci USA 95, 15629–15634 (1998).
Irie, Y. et al. Molecular Cloning and Characterization of Amida, a Novel Protein Which Interacts with a Neuron-specific Immediate Early Gene Product Arc, Contains Novel Nuclear Localization Signals and Causes Cell Death in Cultured Cells. J Biol Chem 275, 2647–2653 (2000).
Sugiura, H. et al. Inhibitory Role of Endophilin 3 in Receptor-mediated Endocytosis. J Biol Chem 279, 23343–23348 (2004).
Yasuda, S. et al. Activity-Induced Protocadherin Arcadlin Regulates Dendritic Spine Number by Triggering N-Cadherin Endocytosis via TAO2[beta] and p38 MAP Kinases. Neuron 56, 456–471 (2007).
Aoto, J. et al. Postsynaptic EphrinB3 Promotes Shaft Glutamatergic Synapse Formation. J Neurosci 27, 7508–7519 (2007).
Fonseca, B. D. et al. Pharmacological and Genetic Evaluation of Proposed Roles of Mitogen-activated Protein Kinase/Extracellular Signal-regulated Kinase Kinase (MEK), Extracellular Signal-regulated Kinase (ERK) and p90RSK in the Control of mTORC1 Protein Signaling by Phorbol Esters. J Biol Chem 286, 27111–27122 (2011).
Possemato, R. et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–50 (2011).
Gao, D. et al. mTOR Drives Its Own Activation via SCFβTrCP-Dependent Degradation of the mTOR Inhibitor DEPTOR. Mol Cell 44, 290–303 (2011).
Wojcik, S. M. et al. A Shared Vesicular Carrier Allows Synaptic Corelease of GABA and Glycine. Neuron 50, 575–587 (2006).
Pyott, S. J. & Rosenmund, C. The effects of temperature on vesicular supply and release in autaptic cultures of rat and mouse hippocampal neurons. J Physiol 539, 523–535 (2002).
Burgalossi, A. et al. Analysis of neurotransmitter release mechanisms by photolysis of caged Ca2+ in an autaptic neuron culture system. Nat. Protocols 7, 1351–1365 (2012).
Acknowledgements
We are grateful to K. Kohyama and Y. Matsumoto for gene gun; T. Yamauchi for invaluable advice. This work was supported by JSPS KAKENHI Grant Number 24659093, 25293239, MEXT KAKENHI Grant Number 23110525, 25110737 and a grant from the Naito Foundation.
Author information
Authors and Affiliations
Contributions
K.Y., S.Y. and H.S. designed research; S.Y., H.S., S.K., T.S., K.T., K.I., T.K., O.H. and K.Y. performed experiments; H.T. analysed data; and K.Y., S.Y. and H.S. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Yasuda, S., Sugiura, H., Katsurabayashi, S. et al. Activation of Rheb, but not of mTORC1, impairs spine synapse morphogenesis in tuberous sclerosis complex. Sci Rep 4, 5155 (2014). https://doi.org/10.1038/srep05155
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05155
This article is cited by
-
Dysregulation of protein synthesis and dendritic spine morphogenesis in ASD: studies in human pluripotent stem cells
Molecular Autism (2020)
-
Autism spectrum disorder: neuropathology and animal models
Acta Neuropathologica (2017)
-
Rheb activation disrupts spine synapse formation through accumulation of syntenin in tuberous sclerosis complex
Nature Communications (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
