
Abstract
The negatively charged nitrogen-vacancy (N-V−) color center in diamond is an important solid-state single photon source for applications to quantum communication and distributed quantum computation. Its full usefulness relies on sufficient radiative emission of the optical photons which requires realizable control to enhance emission into the zero-phonon line (ZPL) but until now is still a challenge. Detailed understanding of the associated excitation process would be of essential importance for such objective. Here we report a theoretical work that probes the spin-conserving optical excitation of the N-V− center. Using density-functional-theory (DFT) calculations, we find that the ZPL and the phonon-side band (PSB) depend sensitively on the axial strain of the system. Besides, we find a relatively small PSB appearing at about 100 GPa in the emission spectrum at low temperatures, which provides a means to enhance the coherent emission of the N-V− center in quantum optical networks.
Similar content being viewed by others
Introduction
The coherent coupling between quantum objects and optical photons is of crucial importance in quantum information science1,2,3,4,5,6,7,8,9,10,11. The associated quantum entangled states are widely used for applications to quantum information processings12,13. Among the solid-state hosts which are preferred for these applications because of scalability reasons, the N-V− center in diamond is a realizable solid-state quantum logic bit (qubit) as it offers long spin coherence time14,15 and the spin states can be optically initialized and read out16,17,18,19. Due to high photostability the N-V− center also presents a good candidate as a single-photon source used in quantum networks20. However, successful realization of long-distance spin-photon entanglement is still a challenge. One of the difficulties arises form the ZPL emission of this defect including very little part of the total emission21; while the frequency shifted PSB normally gives rise to deterioration of the spin-photon entanglement3.
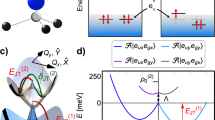
The N-V center in diamond consists of a N atom and a first-neighbor vacancy in the C site (shown in Fig. 1a). The electron paramagnetic resonance (EPR) experiments have revealed that the ground states and excited states involved in the optical excitations are both spin triplet (S = 1)22,23, implying an even number of electrons. Therefore, this defect is normally considered to be negatively charged and the extra electron is donated from the isolated N atom24. For the negatively charged N-V− center with trigonal (C3v) symmetry, there are two fully symmetric a1 states (labeled u and v) localized about the valance band maximum (VBM) and doubly degenerate e states (labeled ex and ey) deeply in the gap, with the occupation of six electrons25:


Where σ1–3 and σ4 are sp3 dangling bonds of the first-neighbor C and N atoms around the vacancy, respectively. λ is a parameter that determines the extent to which σ4 is mixed in the v states, S is the overlap integral.

Structure of the N-V center in diamond.
(a), Atomic structure of the N-V− center. The vacancy is indicated by a small yellow ball and the adjacent C and N by gray and blue balls, respectively (b), Electronic structure of the N-V− center: the calculated spin-polarized single-electron levels (with respect to the VBM), in 3A2 ground states and 3E excited states.
In the ground states, the a1 states are fully occupied and the e states are occupied by two electrons of parallel spin, as illustrated in Fig. 1b. Under the point group of C3V, the total wave function of the ground states has 3A2 symmetry. Upon optical excitation, as one electron in the spin-down  level is excited to the
level is excited to the  or
or  level, the total spin of the system is conserved (shown in Fig. 1b) and the resulting excited states is a spin triplet with the wavefunction of 3E symmetry. Importantly, the change of the electronic configuration is followed by ionic relaxations, which affect strongly the optical features as the relaxation of atoms varies the transition energy, leading to large PSB both in absorption and emission. The large PSB in optical spectrum of this defect has been observed since few decades ago21, but the theoretical understanding of the associated mechanism remains a problem26, as its interpretation is complicated by different results obtained for absorption and emission.
level, the total spin of the system is conserved (shown in Fig. 1b) and the resulting excited states is a spin triplet with the wavefunction of 3E symmetry. Importantly, the change of the electronic configuration is followed by ionic relaxations, which affect strongly the optical features as the relaxation of atoms varies the transition energy, leading to large PSB both in absorption and emission. The large PSB in optical spectrum of this defect has been observed since few decades ago21, but the theoretical understanding of the associated mechanism remains a problem26, as its interpretation is complicated by different results obtained for absorption and emission.
It is possible for there to be strain in diamond, namely, the axial strain and non-axial strain. The latter lowers the symmetry of the N-V− center, splits the excited states according to their orbital wavefunctions and affects consequently the selection rules for the optical transitions21,27,28,29. We don't discuss more here for the non-axial strain as it is not physically applicable to our objective. For the axial strain, it retains the N-V− center's C3V symmetry and is expected to shift the energy levels both in ground and excited states, varying the optical transition energies30. Furthermore, it is also possible to affect the oscillator strength of ZPL transitions, because of the strain changing the lattice parameter and lattice relaxation in the optical excitation. Until now, however, a complete picture for the dependence of the spin-conserving excitation on the axial strain of the lattice is still lacking. A detailed study to understand the excitation processes associated with the axial strain and lattice relaxation is therefore needed and is to be performed in this paper.
Our theoretical work was carried out using DFT, which is a standard method to investigate the properties of defect in solids. The geometry of the N-V center is modeled within a 216-atom cubic supercell of diamond. The axial strain of lattice was introduced by placing the N-V− center supercell in a hydrostatic pressure filed, while the strain was directly tuned through varying the pressure. For the excited states calculation, a preferred approach is to solve the Bethe-Salpeter Equation (BSE)31, together with employing a GW approximation31. However, this approach is computationally prohibitive for the calculations of geometric structures and of electronic structures of large supercells. A realizable solution to this problem is the application of constrained DFT (CDFT)32, which has been very successful in describing the excited states of solid-state system33,34 and was used here as the method to deal with the excited states of the N-V− center. Within CDFT, the calculated ZPL for the N-V− center in the absence of external strain is about 1.658 eV, which is 14.7% smaller than the experimentally measured 1.945 eV21 but consistent with the previous CDFT-LDA33 or CDFT-PBE results35. This can be understood as a common self-interaction error of DFT31. Recently, Gali et al.36 and Weber et al.37 have reported that using a screened Hartree-Fock hybrid density functional, more accurate ZPL for N-V− center can be obtained. However, our goal in this paper was to give approximately the excitation energies to predict and understand the dependence of the optical excitation on the axial strain of lattice. We chose here the traditional density functional since it has been very successful in predicting the electronic35 and vibration34 properties of N-V− center in diamond and the pressure dependences for excited-states-related properties of solids, such as pressure coefficients of band gaps38 and pressure-induced metallization39 etc.
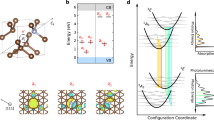
By minimizing the total energy of the N-V− system as a function of the ionic coordinates, we obtained the configuration coordinates (qg) for 3A2 ground states, then we placed the supercell in the stress field and within the same way we obtained ground states qg of the N-V− center with the lattice strain under high pressures. Based on CDFT, we set the orbital occupancies for 3E states and fixed the electronic configuration while relaxing the ionic structure for each system obtained above to find the configuration coordinates for 3E excited states (qe) and then we calculated the potential energy curves (PEC) of the 3E states and 3A2 states along the ionic relaxation paths, as shown in Fig. 2a. We note that the PEC of the 3E states becomes constantly steeper as the external pressure increases, while the PEC of 3A2 states flattens incipiently and then becomes steeper, revealing different changes of the potential energy surface (PES). As we will see, such difference gives rise to complicated changes of the excitation spectrum.

Potential energy curve and excitation spectrum of the N-V− center under high pressures.
(a), Potential energy curves of the N-V− center under zero and high pressures in the 3A2 ground states and 3E exited states along the ionic relaxation path in the 3E states. qg and qe correspond to the energy minima of the ground and excited states, respectively. (b), Calculated transition energies: vertical absorption and emission and ZPL energies of the N-V− center under zero and high pressures. (c), Calculated relaxation energies: Stokes Shift and Anti-Stokes Shift of the N-V− center under zero and high pressures.
Within the Franck-Condon approximation, the excitation spectrum of the N-V− center can be described as follows: assuming that the transition of the electron is very fast compared with the relaxation of ions in the lattice, the transitions A → B and C → D (see Fig. 2a) are processes corresponding to the vertical absorption and emission where the lattice structures do not change. After the lattice relaxation taking place with the participation of phonons, the transitions between the energy minima of the 3A2 states and 3E states (A↔C) can be realized, with a ZPL both in absorption and emission. The energy difference between the vertical absorption/emission and the ZPL corresponds to the relaxation energy, namely the Stokes-shift/anti-Stokes shift (SS/ASS), which in experiment associates with the frequency shifted PSB of the highest intensity in absorption/emission at low temperatures. The calculated SS and ASS at zero pressure are 0.206 and 0.177 eV, respectively, comparable with the 0.235 and 0.185 eV measured from experiment21. The calculated relaxation energies in the optical excitation are considered to be more accurate than the transition energies, as they are obtained as the energy difference between electronically similar conditions and therefore the self-interaction errors are expected to be smaller. The obtained transition energies, SS and ASS under external high pressures are shown in Fig. 2b and Fig. 2c, respectively.
The results show very clearly that the vertical absorption energy rises significantly as the external pressure increases, while the vertical emission energy versus pressure curve rises incipiently and then becomes flattened and decays at ultra-high pressure (about 300 GPa); meanwhile, the increasing of pressure also raises the energy of SS, but lowers the ASS in the range of 0–100 GPa and then raises it in higher range. The net result of these changes gives an increased ZPL. The obtained pressure coefficient for the shift of ZPL is about 5.75 meV/Gpa, in good agreement with the 5.5 meV/Gpa measured from experiment40 (by Kobayashi et al.), in which the photoluminescence (PL) spectrum of the N-V− center was studied in a small pressure range (0–8.8 GPa). One difference between their experiment and our theoretical study is that they found pressure-independent PSBs, which significantly contradict our results. The reason for this is that the ZPL and PSB data summarized in figure 2 of Ref. 40 are not consistent with the PL spectrums given in the same work (in figure 1). As a result, they failed to recognize the change of the PSB due to pressure. On the contrary of their claim, we have checked the change of the PL spectrum in figure 1 of their work and then concluded that, e.g., the peak separation between the ZPL and the first phonon line decreases by 8% and 11% when the pressure goes from 0 to 5.1 and 8.8 Gpa and this is in agreement with our calculated 9% and 15% decline of the ASS when going from 0 to 5 and 10 GPa, respectively. Furthermore, the calculated ASS also agrees well with the decreased Huang-Phys factor obtained by Kobayashi et al. in the same work, revealing relatively suppressed PSB in this pressure range.
To further understand the vertical absorption energies obtained, we calculated the spin-polarized single-electron levels of the N-V− defect in the 3A2 states (see Fig. 3a). With the lattice strain under high pressure, the  level which appears in the valence band, together with the
level which appears in the valence band, together with the  level localized in the gap, shift down in energy with respect to the valence band maximum (VBM), lowering the spin-splitting energies of the both. Simultaneously, the doubly degenerated e and
level localized in the gap, shift down in energy with respect to the valence band maximum (VBM), lowering the spin-splitting energies of the both. Simultaneously, the doubly degenerated e and  levels will shift up, resulting in larger energy range between the excitation-related levels.
levels will shift up, resulting in larger energy range between the excitation-related levels.

Defect levels and atomic configuration under external high pressures.
(a), Energy versus pressure curve for spin-polarized single-electron levels (with respect to the VBM) of the N-V− center in the 3A2 states. (b), Calculated C-C bond length (BC-C) in diamond; C-V (DC-V) and N-V (DN-V) distances of the N-V− center under high pressures in 3A2 and 3E states.
The defect levels depend not only upon the ionic environment but the electronic configuration as well. Therefore, either the change of the lattice arrangement or the varying of the orbital occupancies will give correspondingly orbital relaxations. This can be well verified by the calculated spin-polarized density of states (DOS, see Fig. 4) associated with the excitation: In the process of vertical absorption (A → B), as one electron is excited from the  level to the
level to the  level, the spin up e states shift up in energy, reducing the spin-splitting energy with respect to the
level, the spin up e states shift up in energy, reducing the spin-splitting energy with respect to the  states and the spin up v states shift down to a lower energy [comparable to the vertical absorption, inverse orbital relaxations of v and e states can be observed in the process of vertical emission (C → D)]. Subsequently, the ionic relaxation (B → C) will lower the e and
states and the spin up v states shift down to a lower energy [comparable to the vertical absorption, inverse orbital relaxations of v and e states can be observed in the process of vertical emission (C → D)]. Subsequently, the ionic relaxation (B → C) will lower the e and  states, while raise the v and
states, while raise the v and  states (we make no discussion for the u states as they are not sensitive to the ionic relaxation here). Because the
states (we make no discussion for the u states as they are not sensitive to the ionic relaxation here). Because the  level is unoccupied, the e level is occupied and the
level is unoccupied, the e level is occupied and the  level is partially occupied in the 3E states, such orbital relaxation consequently stabilizes the excited states of N-V− system; but the case is different for the deexcitation (D → A) as the ionic relaxation takes place in the electronic configuration of 3A2 states, in which the orbital occupancies differ from the 3E states as indicated in Fig. 1b: the
level is partially occupied in the 3E states, such orbital relaxation consequently stabilizes the excited states of N-V− system; but the case is different for the deexcitation (D → A) as the ionic relaxation takes place in the electronic configuration of 3A2 states, in which the orbital occupancies differ from the 3E states as indicated in Fig. 1b: the  level is occupied but the
level is occupied but the  level is unoccupied. Therefore, the energy difference associated with the orbital relaxation in the deexcitation would mostly cancel out and the obtained ASS is supposed to originate mostly from the incidental deformation of the delocalized states created from the back bonds of the center, which are delocalized in valence and conduction bands of the host. This interprets the observed difference of the PEC for 3A2 and 3E states under high pressures.
level is unoccupied. Therefore, the energy difference associated with the orbital relaxation in the deexcitation would mostly cancel out and the obtained ASS is supposed to originate mostly from the incidental deformation of the delocalized states created from the back bonds of the center, which are delocalized in valence and conduction bands of the host. This interprets the observed difference of the PEC for 3A2 and 3E states under high pressures.

The density of states (DOS) of the N-V− center.
(a), Calculated spin-polarized DOS of the N-V− center at 0 and 100 GPa. (b), Spin-polarized DOS at 300 and 500 GPa. A, B, C and D correspond to the ionic and electronic configurations indicated in Fig. 2a. The black and blue lines represent the spin-up and -down states; the vertical gray lines show the corresponding defect levels, where the solid, dot and dash lines indicate occupied, unoccupied and half occupied orbitals, respectively; the horizontal arrows represent the associated orbital shifts in the excitation/deexcitation processes. The zero of energy is set at the Fermi level of the ground states.
Once created in the bulk diamond, the N-V− center atoms will relax outward from the vacant site, the resulting extension strain retains the N-V− center's C3V symmetry and keeps the dangling bonds around the vacancy being highly localized. The calculated carbon-vacancy (C-V) and nitrogen-vacancy (N-V) distances are 1.645 Å and 1.698 Å compared with the 1.550 Å of the carbon-carbon (C-C) bond length in diamond (see Fig. 3b). Upon excitation, the C atoms further relax outward from the vacant site and the location of the N atom becomes less outward; the calculated displacement of C here is about 0.060 Å, which is comparable with that of N (0.061 Å). Because the defect center consists of three C but one N atom, the excitation will further lower the symmetry of the environmental tetrahedron (Td) crystal field and give even larger deformation of the delocalized states and this way the ASS is created. We note that under high pressures, the displacements of the C atoms remain almost constant in the excitation, while the displacement of the N atom gets actually larger. Due to the lattice shrinkage under high pressure, in the reciprocal space, the defect levels will become more sensitive to the excitation. Fig. 4 demonstrates that the orbital relaxations between the ground states and excited states (B → C) become more evident under higher pressures, which is responsible for the steeper PEC of the 3E states and the increasing SS obtained.
As the pressure rises, the C-V and N-V distances decrease more rapidly than the C-C bond length, as shown in Fig. 3b. This will lower the local strain of the center. For instance, at 100 GPa, the calculated C-V distances are 1.464 Å, which happens to be equal to the C-C bond length revealing almost no strain of the C, while the calculated N-V distance is 1.546 Å revealing also a suppressed outward strain of N (0.082 Å). This is because the interactions between the center's sp3 dangling bonds are not as strong as the bonding of the C atoms in diamond. Furthermore, the pressure will lower the relative strain between the ground and excited states if is not sufficiently high to induce an inward strain, lowering the ASS. This interprets the flattened PEC of the 3A2 states and the decreasing ASS in the pressure range of 0–100 GPa.
Note also that at 100 GPa, the calculated ASS converges very close to zero — −13 meV. The obtained negative value is due to mostly the static J-T distortion in the excited states, as the geometric structure of the N-V− system is fully relaxed without symmetry fixing. However, in most experimental cases (even at low temperatures), the breaking of the C3V symmetry due to J-T effect can not be observed41, as the potential well associated with the J-T distortion is of magnitude smaller than the zero-point energy34 and the minima of the potential surface are normally occupied by the zero-point vibrations or higher vibrational states. Thus, from this point of view, the negative value obtained here also rises from the zero-point vibration (ZPV) error within the adiabatic approximation in DFT. To exclude the associated error, here we have tested the ASS in the absence of J-T distortion, the obtained value is 2 meV which again reveals a small ASS. The reason that the N-V− center has significantly small ASS at this pressure can be understood as follows: In the excited states, as the ionic relaxation takes place, the C-V and N-V distances shift to about 1.525 and 1.461 Å (see Fig. 3b), corresponding to outward strain of C (0.061 Å) and very small inward strain of N (0.003 Å), respectively. That is, after excitation, the strain of the N atom is correspondingly removed accompanied with the presence of smaller strain of the three C atoms, which tends to cancel out the energy difference between qg and qe in the 3A2 states and thus lowers the ASS. This can be understood also as a result of redistribution of the localized phonon states, which lowers the oscillator strength of the phonon emission and improves emission into the ZPL. Therefore, one would expect the N-V− center to display a relatively small emission PSB under about this pressure at low temperatures. Although can not be avoided here, due to the flattened PEC of the 3A2 states, the intrinsic ZPV error is expected to be even smaller compared with the one at zero pressure and this also holds for a small PSB of the emission spectrum.
The inward strain of the center will take place and become evident as the pressure goes up further (see Fig. 3b) and consequently increases the relative strain between the ground and excited states. The DOS obtained here reveal different deformations of the delocalized states, which are noted to be relatively significant at 500 GPa and evidence the steep PEC of the 3A2 states and the large ASS obtained here. Note that the v levels become somewhat delocalized along with the inward strain. The calculated DOS show clear broadening of the v states at 300 and 500 GPa. This results from close interactions between the center's atoms as the distances between the neighboring C become relatively shorter and the overlap between the sp3 dangling bonds can be no more negligible. The v levels shift down in energy, couple strongly to the VBM states and become somewhat delocalized, lowering the energy. Indeed, we note that the delocalization of the v levels is not as significant in the excited states due to the ionic relaxation, which is also responsible for the large ASS and the inversely changed vertical emission energy starting at about 300 GPa.
In fact, the relaxation energy in the optical absorption/emission of the N-V− center arises from the orbital relaxation of the defect together with the deformation of the delocalized states, which are both correlated strongly with the local ionic arrangement. In the optical excitation, the relaxation energy associated with the orbital relaxation is relatively larger than that from the deformation of delocalized states and thus dominates the SS; whereas in the deexcitation, due to different electronic occupancies the ASS is derived mostly from the latter. This provides an understanding of how ionic relaxation affects the excitation/deexcitation of the N-V− system and interprets the experimental observation21. The obtained results also demonstrate a dependence of the excitation spectrum of the N-V− center on the axial strain of the lattice, which is directly tunable via the external pressure. We expect based on these results that pressure-control for the optical transitions of the N-V− center will be possible. Furthermore, the relatively small ASS obtained at 100 GPa demonstrates a suppressed PSB of the emission spectrum at low temperatures. This might prove useful as a means to improve the ZPL emission of the N-V− center for applications to quantum cryptography and quantum communication. Beyond these specific applications, the realizable control of the PSB emission also paves a pathway towards improving the efficiency of photon-spin coherent coupling, which is currently the key limitation in realization of long-distance photon-spin entanglement.
Methods
We optimized the geometry and calculated the charge density for the supercell structure of N-V− center employing spin polarized generalized gradient approximation (SGGA) with Perdrew-Burke-Enzerhof (PBE)42 exchange-correlation functional. Projected augmented wave (PAW) pseudopotentials43 for the C and N were used with a planewave basis-set cutoff energy of 520 eV, which is adequate for optimization of the geometry under high pressure. We used a 3 × 3 × 3 Monkhorst-Pack mesh for the k point samplings in the Brillouin zone. This provides a very well converged charge density. The 216-atom supercell of diamond was constructed by extending the conventional cubic cell three times along the lattice vector a, b and c, respectively; we then placed the N-V defect in the center of the supercell to model the defective system. The optimized lattice constant of the supercell is about 10.73 Å at 0 Pa and 9.11 Å at 500 GPa, which is appropriate for simulation of the isolated N-V defect and is also large enough to obtain accurately the associated spin density. Within CDFT, the electronic occupancies for the 3E excited states were obtained by setting zero occupation for the spin-down  level and one occupation for spin-down
level and one occupation for spin-down  or
or  level. In the geometric optimization, no symmetry restriction was applied, with all the atoms allowed to relax until the calculated Hellmann-Feynman forces became smaller than 1 meV/Å.
level. In the geometric optimization, no symmetry restriction was applied, with all the atoms allowed to relax until the calculated Hellmann-Feynman forces became smaller than 1 meV/Å.
References
Beveratos, A. et al. Single photon quantum cryptography. Phys. Rev. Lett. 89, 187901 (2002).
Babinec, T. M. et al. A diamond nanowire single-photon source. Nat. Nano. 5, 195–199 (2010).
Togan, E. et al. Quantum entanglement between an optical photon and a solid-state spin qubit. Nature 466, 730–734 (2010).
Jennifer, T. C. et al. Enhanced single-photon emission from a diamond–silver aperture. Nat. Photon. 5, 738–743(2013).
Childress, L. et al. Fault-tolerant quantum communication based on solid-state photon emitters. Phys. Rev. Lett. 96, 070504 (2006).
Bernien, H. et al. Heralded entanglement between solid-state qubits separated by three metres. Nature 497, 86–90 (2013).
Jiang, L., Taylor, J. M. & Lukin, M. D. Fast and robust approach to long-distance quantum communication with atomic ensembles. Phys. Rev. A 76, 062323 (2007).
Kok, P. et al. Linear optical quantum computing with photonic qubits. Rev. Mod. Phys. 79, 135–174 (2007).
Stute, A. et al. Tunable ion–photon entanglement in an optical cavity. Nature 485, 482–485 (2012).
Wilk, T., Webster, S. C., Kuhn, A. & Rempe, G. Single-atom single-photon quantum interface. Science 317, 488–490 (2007).
Matsukevich, D. N. et al. Entanglement of a photon and a collective atomic excitation. Phys. Rev. Lett. 95, 040405 (2005).
Gao, W. B. et al. Observation of entanglement between a quantum dot spin and a single photon. Nature 491, 426–430 (2012).
Blinov, B., Moehring, D. & Duan, L. Observation of entanglement between a single trapped atom and a single photon. Nature 428, 153–157 (2004).
Gaebel, T. et al. Room-temperature coherent coupling of single spins in diamond. Nat. Phys. 2, 408–413 (2006).
Balasubramanian, G. et al. Ultralong spin coherence time in isotopically engineered diamond. Nature Mater. 8, 383–387 (2009).
Jelezko, F. et al. Observation of coherent oscillation of a single nuclear spin and realization of a two-qubit conditional quantum Gate. Phys. Rev. Lett. 93, 130501 (2004).
Jelezko, F. et al. Observation of coherent oscillations in a single electron spin. Phys. Rev. Lett. 92, 076401 (2004).
Doherty, M. W. et al. The nitrogen-vacancy colour centre in diamond. Phys. Rep. 528, 1–45 (2013).
Robledo, L. et al. Control and coherence of the optical transition of single nitrogen vacancy centers in diamond. Phys. Rev. Lett. 105, 177403 (2010).
Moehring, D. L. et al. Entanglement of single-atom quantum bits at a distance. Nature 449, 68–71 (2007).
Davies, G. et al. Optical studies of 1.945 eV vibronic band in diamond. Proc. R. Soc. A 348, 285–298 (1976).
Reddy, N. R. S., Manson, N. B. & Krausz, E. R. 2-Laser spectral hole burning in a color center in diamond. J. Lumin. 38, 46–47 (1987).
Redman, D. A. et al. Spin dynamics and electronic states of N-V centers in diamond by EPR and four-wave-mixing spectroscopy. Phys. Rev. Lett. 67, 3420 (1991).
Mita, Y. Change of absorption spectra in type-Ib diamond with heavy neutron irradiation. Phys. Rev. B 53, 11360 (1996).
Lenef, A. & Rand, S. C. Electronic structure of the N-V center in diamond: Theory. Phys. Rev. B 53, 13441 (1996).
Davies, G. Vibronic spectra in diamond. J. Phys. C: Solid State Phys. 7, 3797 (1974).
Doherty, M. W., Manson, N. B., Delaney, P. & Hollenberg, L. C. L. The negatively charged nitrogen-vacancy centre in diamond: the electronic solution. New J. Phys. 13, 025019 (2011).
Maze, J. R. et al. Properties of nitrogen-vacancy centers in diamond: the group theoretic approach. New J. Phys. 13, 025025 (2011).
Manson, N., Harrison, J. & Sellars, M. Nitrogen-vacancy center in diamond: model of the electronic structure and associated dynamics. Phys. Rev. B 74, 104303 (2006).
Doherty, M. W. et al. Electronic properties and metrology applications of the diamond NV− Center under Pressure. Phys. Rev. Lett. 112, 047601 (2014).
Onida, G., Reining, L. & Rubio, A. Electronic excitations: density-functional versus many-body Green's-function approaches. Rev. Mod. Phys. 74, 601–659 (2002).
Wu, Q. & Van Voorhis, T. Constrained density functional theory and its application in long-range electron transfer. J. Chem. Theo. Comp. 2, 765–774 (2006).
Gali, A., Fyta, M. & Kaxiras, E. Ab initio supercell calculations on nitrogen-vacancy center in diamond: Electronic structure and hyperfine tensors. Phys. Rev. B 77, 155206 (2008).
Zhang, J. H. et al. Vibrational modes and lattice distortion of a nitrogen-vacancy center in diamond from first-principles calculations. Phys. Rev. B 84, 035211 (2011).
Ma, Y., Rohlfing, M. & Gali, A. Excited states of the negatively charged nitrogen-vacancy color center in diamond. Phys. Rev. B 81, 041204R (2010).
Gali, A. et al. Theory of Spin-conserving excitation of the N-V− center in diamond. Phys. Rev. Lett. 103, 186404 (2009).
Weber, J. R. et al. Quantum computing with defects. PNAS 107, 8513–8518 (2010).
Fahy, S. et al. Pressure coefficients of band gaps of diamond. Phys. Rev. B 35, 5856–5859 (1986).
Wei, S.-H. & Krakauer, H. Local-density-functional calculation of the pressure-induced metallization of BaSe and BaTe. Phys. Rev. Lett. 55, 1200–1203 (1985).
Kobayashi, M. & Nisida, Y. High pressure effects on photoluminescence spectra of color centers in diamond. Jpn. J. Appl. Phys. 32 Suppl. 32–1, 279–281 (1993).
Bersuker, I. B. & Polinger, Z. Vibronic interactions in molecules and crystals (Springer-Verlag, Berlin, Heidelberg, 1989).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Acknowledgements
We are thankful to Michel A. Van Hove, R.B. Liu and D. Han for encouraging discussions. B.D. acknowledges support from the High Performance Cluster Computing Centre, Hong Kong Baptist University, which receives funding from the Research Grants Council, University Grants Committee of the HKSAR and Hong Kong Baptist University.
Author information
Authors and Affiliations
Contributions
B.D. and R.Q.Z. wrote the main manuscript text, B.D. prepared figures 1–4 and X.Q.S. provided technical support for the ab initio calculations. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Deng, B., Zhang, R. & Shi, X. New insight into the spin-conserving excitation of the negatively charged nitrogen-vacancy center in diamond. Sci Rep 4, 5144 (2014). https://doi.org/10.1038/srep05144
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05144
This article is cited by
-
Electronic and magnetism properties of two-dimensional stacked nickel hydroxides and nitrides
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
