
Abstract
Hotspots of genetic diversity are regions of utmost importance for species survival and conservation and their intimate link with the geographic location of glacial refugia has been well established. Nonetheless, the microevolutionary processes underlying the generation of hotspots in such regions have only recently become a fervent field of research. We investigated the phylogeographic and population genetic structure of the agile frog, Rana dalmatina, within its putative refugium in peninsular Italy. We found this region to harbour far more diversity, phylogeographic structure and lineages of ancient origin than that by the rest of the species' range in Europe. This pattern appeared to be well explained by climate-driven microevolutionary processes that occurred during both glacial and interglacial epochs. Therefore, the inferred evolutionary history of R. dalmatina in Italy supports a view of glacial refugia as ‘factories’ rather than as repositories of genetic diversity, with significant implications for conservation strategies for hotspots.
Similar content being viewed by others

Introduction
Hotspots of genetic diversity, defined as geographic areas harbouring a major portion of a species' genetic diversity, have been observed for most species studied to date. The discovery of hotspots and the causes of their formation have become vigorous areas of research1,2,3,4,5,6,7,8,9,10. Indeed, hotspots are increasingly seen as excellent opportunities for the study of long-debated themes in ecology and evolutionary biology, including the microevolutionary processes leading to the formation of latitudinal patterns of biodiversity, the role of divergence with or without gene flow in moulding current biodiversity, the role of hybridization in evolution and the geographic mosaic of coevolutionary interactions5,6,11,12. On the other hand, these areas are also of the utmost importance for long-term survival and conservation of species, since they frequently harbour most species' resources for coping with natural and anthropogenic environmental changes13,14,15.
Two main evolutionary scenarios have been described to date for the generation of hotspots of intraspecific biodiversity in temperate species16. Under each of these scenarios, past climatic oscillations are seen as the major distal cause of the process and the location of hotspots is seen as intimately linked to the so-called ‘glacial refugia’. Under the first scenario, a hotspot would testify to the long-term persistence of demographically stable, large populations within a glacial refugium. In contrast, under the second scenario the high genetic diversity found in glacial refugia would be the outcome of climate-driven cycles of allopatric differentiation within sub-refugia, which is possibly followed by a secondary admixture of divergent lineages. In spite of some fossil evidence suggesting that refugial populations could have been small, isolated and at low density17,18, the first scenario was considered as prevalent in early literature, particularly for wide-ranging species8,19,20. However, mounting data from phylogeographic studies focused on populations within glacial refugia identify the second scenario as relevant for many temperate species and geographic regions1,5,7,9,21,22.
Identifying the geographic locations of hotspots of genetic diversity, the genetic structure of populations therein and the microevolutionary processes that have led to their formation are important not only for the advancement of knowledge in ecology, biogeography and evolutionary biology, but also have major practical implications. For instance, regional networks of protected areas and management plans for species conservation are often designed under the assumption of homogeneous distribution of diversity within species' populations, even when there is inadequate knowledge of the geography and depth of the population genetic structure14,15. Such regional networks and plans would be appropriate only in scenarios where a single evolutionary unit (ESU; sensu23) is present, whereas they should be carefully re-evaluated in cases where multiple ESUs are found, deserving separate conservation efforts, or in cases where major hotspots of genetic diversity fall outside such networks.
The agile frog, Rana dalmatina Fitzinger in Bonaparte, 1839, is a brown frog that is widely distributed in western, central and southern Europe, where it inhabits a variety of landscapes, mostly within mixed deciduous forests from sea level to 2000 m a.s.l. R. dalmatina spends most of its life on land, breeding in a variety of natural and anthropogenic water bodies, including flooded meadows, ponds, lakes, temporary pools and slow-running waters24. Although this species is not considered to be under imminent conservation risk, population declines and disappearances have recently been observed25. In a recent survey of the genetic variation throughout the species' range, R. dalmatina populations have appeared substantially homogeneous, with the single exception of a strongly differentiated population in southern Italy, which was considered as evidence for a glacial refugium of this species being located within this area26.
In this study, we further investigate the population genetic structure and evolutionary history of R. dalmatina, focusing on its putative refugial range in peninsular Italy. This geographic area has recently emerged as a hotspot of genetic diversity and as a site of multiple refugia for many temperate animal species, including amphibians6,21,27,28,29,30,31. Here we adopted a dense sampling strategy within this area, with the following aims: i) to assess the phylogeographic and population genetic structure and the historical demographic trends of R. dalmatina within its prospected glacial refugium, ii) to investigate the microevolutionary processes that have moulded its genetic diversity within this area; and iii) to evaluate the main implications of our results for the conservation of this species.
Results
Mitochondrial DNA
The final dataset included 741 bp of the mitochondrial cytochrome oxidase subunit I gene (COI; 46 variable positions, 37 parsimony informative) and 653 bp for the mitochondrial cytochrome B gene (CYTB; 55 variable positions, 42 parsimony informative), for all the 244 individuals analysed. Forty-nine composite haplotypes were found (see Table 1), showing a sequence divergence (p-distance) ranging between 0.1% and 4.5%.
The statistical parsimony analysis did not connect all haplotypes into a single phylogenetic network (Figure 1A). Instead, 2 main groups of haplotypes were observed (haplogroups N and S) having an ML-corrected net sequence divergence of 4.0% (0.5% S.E.), each showing a discrete geographic distribution. Haplogroup N was found in north-central and northern Italy, Croatia and Slovenia, (samples 1–19), whereas haplogroup S was distributed in southern Italy (samples 20–40). Both of these haplogroups were further divided into 2 sub-haplogroups, the distribution of which is presented in Figure 1C. While haplogroups N and S were never found in syntopy, the respective sub-haplogroups (NI-NII and SI-SII) showed overlapping distributions mostly limited to geographically intermediate populations.

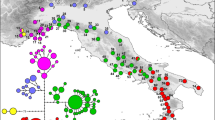
(A) Phylogenetic networks of the 49 mtDNA haplotypes found among Rana dalmatina populations in peninsular Italy, based on the statistical parsimony procedure implemented in TCS. Circle sizes are proportional to haplotype frequency (see inset, lower left); missing intermediate haplotypes are shown as open dots. (B) Haplotype genealogy yielded using HaplotypeViewer, based on the ML phylogenetic tree of the 49 haplotypes found. (C) Geographic locations of the 40 sampled populations of R. dalmatina and frequency distribution of the main haplogroups among populations, shown as pie diagrams. Populations are numbered as in Table 1. The map was drawn using the software Canvas 11 (ACD Systems of America, Inc.).
The haplotype genealogy yielded by HaplotypeViewer showed the same groupings as the statistical parsimony networks, with minor topological differences that did not affect the general pattern (Figure 1B). Within this genealogy, haplogroups N and S were separated by 49 mutational steps.
Estimates of genetic diversity for the 4 haplogroups are shown in Table 2. Particularly low values of h and π were observed for the northernmost haplogroup (NI), whereas high values of both indices were found for the southernmost haplogroup (SII) and to a lesser extent, for SI.
The AMOVA analysis was carried out by separating populations according to the geographic and frequency distribution of the 4 main haplogroups ([1–18], [19–20], [21–27], [28–40]). This analysis indicated that the amount of genetic variance explained by the among-group level of variation was 91.6% (FCT = 0.92), the amount explained by the variation among populations within groups was 2.7% (FSC = 0.32), while the within-population level explained 5.7% (FST = 0.94). All variance components and fixation indices were highly significant (P < 0.001).
The analysis of the divergence time dating with BEAST yielded effective sample size (ESS) values that largely exceeded 200 for all parameters of interest and full convergence for parameter estimates between runs. The divergence time between haplogroups N and S was estimated at 2.109 My (95% HPD: 1.433–2.929 My). On the other hand, the divergence between haplogroups NI–NII and SI–SII was estimated to have occurred at 0.381 My (95% HPD: 0.172–0.594 My) and 0.346 My (95% HPD: 0.197–0.519 My), respectively.
The historical demographic trends for all the main haplogroups were investigated by means of a bayesian skyline plot (BSP) analysis, except for NII because of its low sample size (n = 10). The BSPs estimated for the remaining haplogroups are shown in Figure 2. The time to the most recent common ancestors (TMRCAs) was estimated as 23 000 years (95% HPD: 8300–45 600), 84 500 years (95% HPD: 27 700–158 800) and 155 000 years (95% HPD: 63 200–266 400) for haplogroups NI, SI and SII, respectively. For haplogroup NI, a marked demographic expansion was inferred that began shortly after the TMRCA and lasted until recent times. In contrast, the BSPs for haplogroups SI and SII showed no marked demographic changes; the median estimates of effective population size for these groups never fell outside the 95% HPDs estimated along the entire time axis.

Bayesian skyline plots showing the historical demographic trends of 3 of the main mtDNA haplogroups identified within Rana dalmatina.
The x-axis is scaled in years (bp). The y-axis is scaled in units of effective population size per generation length.
The estimated values of the neutrality test statistics are given in Table 2. In agreement with the BSP analysis, large negative values of FS and D and a small positive value of R2 (all suggesting recent and marked population growth) were only observed for haplogroup NI (all P < 0.05). On the other hand, none of the estimated values of the statistics for the haplogroups NII, SI and SII was significant.
Microsatellites
A total of 170 individuals from 38 locations were genotyped at 6 microsatellite loci (see Table 1). Among these individuals, none was scored at less than 5 loci and the overall proportion of missing data in the final dataset was 0.2%.
Evidence of Hardy–Weinberg or linkage disequilibria was not found across samples or loci. For all populations, the observed heterozygosity ranged from 0.69 (H1 locus) to 0.89 (G12 locus), the expected heterozygosity ranged from 0.84 (H1 locus) to 0.96 (E8 locus) and the number of alleles observed at each locus was as follows: F5 = 19, G12 = 28, G11 = 16, E8 = 38, H1 = 16 and B5 = 22. Consequently, the total number of alleles found within the study area (n = 139) was 12.9% higher than that previously reported for the same loci throughout the species range26.
The analysis of population genetic structure by using TESS indicated K = 4 as the best grouping option for our data, since only a minor decrease in DIC values was observed at higher K values (Figure 3A). As previously observed for the mtDNA data, 2 groups were found in south-central and southern Italy (samples 21–40; see Figure 3B). However, unlike the mtDNA data, the 2 population groups were largely admixed in most of the areas (Figures 3B and 3C), with some exceptions in marginal samples. Two groups were observed in the north-central and northern portion of the study area. However, the geographic distribution of the 2 groups did not conform to the geographic pattern observed in the mtDNA data. Indeed, the area of admixture between the 2 groups was not located in central Italy (samples 17–20), but on the north-eastern side of the Italian Peninsula (samples 8–10, 14). Finally, some instances of unidirectional gene exchange from south to north were observed among samples located in central Italy (samples 17–20), which otherwise belonged to the north-central group (Figure 3C). Finally, the mean expected heterozygosity estimated for the 4 groups was 0.90 (0.01 SE), 0.92 (0.01 SE), 0.88 (0.03 SE), 0.88 (0.02 SE) for the southern, south-central, north-central and northern groups respectively.

Genetic structure of Rana dalmatina populations in Italy, which were estimated using the Bayesian clustering method implemented in TESS.
(A) Mean values of the DIC statistics (averaged over 100 runs) estimated for models with the number of genetic clusters (K) ranging from 2 to 7; (B) Posterior predictive map of the admixture proportions generated from the spatial interpolation procedure implemented in TESS; (C) Admixture proportions of each sampled individual for the 4 recovered genetic clusters. The map was drawn using the software TESS Ad-Mixer.
Discussion
Our analysis of genetic variation in the agile frog in peninsular Italy allowed us to identify this area as not only a site of the most divergent lineage but also as the major hotspot of genetic diversity within this species. Indeed, populations from this area harbour far more genetic variation than that found in the central, eastern and southern European populations combined26. Further, most of the genetic variation was accounted for by differences between 4 genetically divergent and geographically well-defined lineages, suggesting a long-lasting and multi-episode evolutionary history for R. dalmatina within this area. In turn, the occurrence of 4 lineages with relatively ancient divergence clearly indicates that peninsular Italy did not act as a single refugium for the species, but rather as a mosaic of refugia, at least during the last Pleistocene glacial-interglacial cycles.
The most ancient event inferred in the history of R. dalmatina populations was the divergence between the N and S mtDNA haplogroups, which has been roughly dated at 2.1 million years (MY). The precise dating of this divergence was prevented by the large 95% CI for the time estimate (1.4–2.9 My), our use of an externally calibrated substitution rate, along with the many uncertainties of the molecular clock32,33. Nevertheless, it is worth noting that the phylogeographic discontinuity between the N and S clades is located close to the northern edge of a mid-peninsular region that was repeatedly subjected to marine-flooding during the Plio-Pleistocene and where phylogeographic discontinuities have been repeatedly reported6,28,29,34,35. Thus, it is plausible that a marine transgression that occurred in the early Pleistocene might have primed the observed divergence. This divergence is conspicuous (sequence divergence was 4.0% in the mtDNA data), syntopy was not observed between the 2 mtDNA clades and the admixture of nuclear genomes appeared both geographically and quantitatively limited (see Figure 3C). Therefore, further study is required to locate the secondary contact zone between both clades and to investigate what microevolutionary processes occurred following the formation of this zone.
The transition zone between the NI and NII lineages in the mtDNA data (Figure 2) was not reflected by the transition zone between the north-central and the northern population groups in the microsatellite data (Figure 3). Although not unusual, it may be difficult to reconcile the discordance between nuclear and mitochondrial data to infer the history of populations, given the diversity of microevolutionary processes that are potentially involved36. However, while the northern Apennine is a well known suture zone between both intra- and interspecific lineages27,37,38,39,40, a contact zone in the area close to our sites 17–19 would not have any analogues in the phylogeographic literature of co-distributed temperate species. In addition, no environmental features (present or past) were present that could explain the observed pattern. This observation led us to speculate that, after the secondary contact between the 2 lineages in an area close to the northern Apennines, the NI mitochondrial lineage incurred massive introgression into the NII range, resulting in the former replacing the latter throughout most of central Italy. On the other hand, unfavourable climatic conditions along the central Apennines chain during the glacial stages41,42 might provide a viable explanation for the divergence between the 2 lineages. During these stages, the NI and NII lineages might have persisted in temperate lowland areas along the eastern Adriatic and the central Tyrrhenian coasts, respectively42.
The phylogeographic discontinuity observed between the SI and SII mtDNA clades was located in central Calabria. Although a more admixed picture emerged at the nuclear microsatellites, central Calabria was also the main area of transition between the south-central and the southern groups of populations (marked as purple and green, respectively, in Figure 3). Interestingly, several phylogeographic breaks and secondary contact zones were clustered in this area6,21,27,28,29,30, which was cyclically inundated by marine transgressions until the late-Middle/Late Pleistocene43. Therefore, it seems plausible that these transgressions also primed the divergence between the 2 R. dalmatina groups found in south-central and southern Italy.
Despite the relative geographic contiguity of the alleged past ranges of the divergent R. dalmatina lineages found, the lineages showed substantially different demographic trends during the Late Pleistocene. The star-like structure of the haplotype genealogy for lineage NI, its BSP reconstruction and the estimated values of the neutrality test statistics jointly indicated that, after a marked demographic bottleneck occurred at the last glacial maximum (LGM; as revealed by the estimated TMRCA around 23 000 years ago), NI underwent a substantial and abrupt demographic expansion. Since NI corresponds to the widespread lineage found by Vences and colleagues26 (2013; data not shown) throughout most of R. dalmatina's range in Europe and in light of its Middle-Pleistocene divergence from NII (0.38 [0.17–0.59] My), we suggest that lineage NI re-colonized most of its current range after the LGM from a glacial refugium located somewhere between the western Balkans and the wide coastal plain that occupied what is now the Adriatic Sea42,44. In contrast, lineages SI and SII showed the genetic imprints of demographic stability, lasting for most of the Late Pleistocene (SI), or before (SII). Similar trends of prolonged demographic stability have also been hypothesized for intraspecific lineages of other temperate species studied to date in southern Italy6,29. Thus, contrary to what is frequently observed farther north, the most striking effect of glacial–interglacial cycles on the history of populations from south-central and southern Italy seems to be driven not by climate forcing on range/demographic size variations, but by glacio-eustatic oscillations in the sea level and the resulting allopatric fragmentation of populations imposed by the intervening seaways. Consequently, this area could be viewed as a ‘glacial refugium’ (‘southern refugium’ sensu45) from the worsening climate during the ice ages and as a set of ‘interglacial refugia’ from the rising sea levels during interglacial epochs.
Among wide-ranging species in Western Europe, geographic patterns of genetic diversity as observed for R. dalmatina (more diversity to the south and less diversity to the north, i.e. the ‘southern-richness, northern-purity’ pattern16) are not unusual4,7,46. In most of these species (including R. dalmatina26), the ‘southern-richness’ component of this pattern, i.e. the existence and location of hotspots, has been interpreted as the outcome of the long-term persistence of ancestral populations within southern peninsulas2,3. In contrast with this view for wide-ranging species and in parallel with recent observations in species endemic to these peninsulas5,6,8,47, our results emphasize the key role played by the ‘diversity-enhancing’ effects of the cycles of allopatric divergence within these areas. Since a sampling scheme specifically focused on the putative refugial ranges is crucial for disentangling the above scenarios, we anticipate that as more studies adopt such sampling schemes, our view of the diversity of processes involved in the formation of hotspots of intraspecific biodiversity will continue to change.
Finally, our results have at least 3 major implications for the conservation of R. dalmatina populations, both within our study area and at the whole species' range. First, in order to preserve the species' resources for coping with future natural and anthropogenic environmental changes, peninsular Italy should receive special attention since most of R. dalmatina's genetic diversity is apportioned there and in light of the historical role played by this area in the species' survival. This appears especially important when considering the declining trend recently observed for R. dalmatina populations from several portions of its range, including the Italian peninsula25. Second, since the deep genetic structure of the populations indicates the occurrence of at least two ESUs (sensu23) within this area, specific conservation efforts should be developed to optimize the amount of genetic variation preserved. Finally, a study of the genetic structure of the presumed secondary contact zone between the N and S lineages in central Italy should be carried out to define the patterns of past and present interaction among them, their most appropriate taxonomic arrangement and the best strategy for their conservation.
Methods
Sampling and laboratory procedures
We collected tissue samples from 244 individuals of R. dalmatina from 40 localities, 33 of which were located along the Italian peninsula south of the Alps, 3 from Slovenia and 4 from Croatia. The geographical position and number of individuals sampled at each location are given in Table 1 and Figure 1. Tissue samples were obtained through toe-clipping and all individuals were released at the place of collection immediately after tissue removal. Samples were transported to the laboratory in liquid nitrogen and were stored at −80°C until subsequent analyses.
We extracted genomic DNA by using the standard phenol–chloroform protocol48. Two partial fragments of the mitochondrial DNA (mtDNA) genes encoding CYTB and COI were amplified and sequenced. The same PCR protocol was used for both mtDNA fragments. Amplifications were performed in a final volume of 25 μl containing MgCl2 (2.5 mM), reaction buffer (1×; Promega), 4 dNTPs (0.2 mM each), 2 primers (0.2 μM each), Taq polymerase (2-unit; Go-Taq Promega) and 2 μl of template DNA. PCR cycles were as follow: 94°C for 5 min followed by 36 cycles of 94°C for 45 s, annealing temperature (50°C for CYTB and 52°C for COI) for 45 s, 72°C for 90 s and a final step at 72°C for 10 min. The primers used for amplification and sequencing of the CYTB fragment were 49449 and MVZ1650; primers for the COI fragment were Amp-P3r and Amp-P3f51. Sequencing reactions were carried out by Macrogen Inc. (www.macrogen.com) by using an ABI PRISM 377 DNA sequencer (PE Applied Biosystems) following the ABI PRISM BigDye Terminator Cycle Sequencing protocol. All individuals analysed were double sequenced.
Seven microsatellite loci (F5, G12, G11, E8, H1, B5 and F3) were amplified using previously published protocols52. One locus (F3) was subsequently removed from the analysis because a large percentage of data were missing (>50%).
Fragments were sized using the GeneScan 400HD internal size standard and alleles were scored using PEAKSCANNER 1.0 (Applied Biosystems).
We used the method implemented in the software MICROCHECKER53 to check for the presence of null alleles and large allele dropout; however, there was no evidence of such problems.
Mitochondrial DNA data analysis
Sequence electropherograms were checked by eye, by using ChromasPro (Technelysium Ltd.) and multiple sequence alignments were carried out using ClustalX54. Sequences of the CYTB and COI fragments were concatenated using Concatenator 1.1.055, after a partition homogeneity test56 conducted in Paup*4.0b10 did not reject the hypothesis of homogeneity in the phylogenetic signal carried by both fragments. The optimal model of sequence evolution for both fragments separately and the concatenated dataset were selected among 88 possible models using Jmodeltest 0.1.157. The best-fit models selected using the Akaike information criterion were TrN + Γ58 (gamma distribution shape parameter = 0.032) for COI, TrN + I (proportion of invariable sites = 0.073) for CYTB and TrN + I (proportion of invariable sites = 0.824) for the concatenated dataset. Therefore, the latter was used in all downstream analyses on the unpartitioned dataset.
Sequences were collapsed into unique haplotypes and their nucleotide variation was evaluated with the software DnaSP 559. The same software was used to estimate the haplotype (h) and nucleotide (π) diversity60 among major groups of haplotypes as identified by phylogenetic analyses; Mega 561 was then used to compute the net sequence divergence among haplotype groups.
The genealogical relationships among the identified haplotypes were investigated using 2 approaches. First, we estimated phylogenetic networks using the statistical parsimony procedure of Templeton62, implemented in the software TCS 1.2163 using the 95% limit for a parsimonious connection. Second, to account for recent concerns about the accuracy of network-building methods64, we further investigated the genealogical relationships among haplotypes by using the approach suggested by64. A phylogenetic tree topology was estimated using the maximum likelihood algorithm as implemented in PhyML65. We used the default setting in PhyML for all parameters except the substitution model (TrN + I) and the type of tree improvement (SPR & NNI66). The estimated tree topology was then converted into a haplotype genealogy by using the software HaplotypeViewer65.
Estimates of the divergence time between identified haplogroups were obtained using BEAST 1.7.567 using the Bayesian Skyline as coalescent tree prior, which imposes minimal assumptions on the data67. Preliminary analyses were run using a relaxed molecular clock model, assuming an uncorrelated lognormal distribution. However, since the standard deviation of the uncorrelated lognormal relaxed clock (the parameter ucld.stdev) was close to zero, subsequent analyses were run enforcing a strict clock model. Since we had neither internal calibration points nor previous estimates of the substitution rates for the 2 mtDNA fragments studied in R. dalmatina, this and subsequent demographic analyses were carried out using a uniform prior for the substitution rate, bounded at 0.012 and 0.014 substitutions/site/My, to reflect the range of substitution rates previously estimated for CYTB and COI fragments in other European brown frogs68,69. The final analysis was run in triplicate to check for consistency among runs, each with a Markov chain Monte Carlo (MCMC) run for 20 million generations and sampled every 2000 generations. Further, satisfactorily high ESS values for all parameters in the analysis were obtained using these settings. Convergence, stationarity and the appropriate number of steps to be discarded as burn-in were assessed using TRACER 1.6.
A hierarchical analysis of molecular variance (AMOVA70) was carried out by using ARLEQUIN 3.171, in order to estimate the amount of variation attributable to differences among population groups, among populations within groups and within populations. This analysis was run using the TrN model, with population groups defined according to the geographical distribution of the main haplogroups.
The historical demography of the main haplogroups was inferred using the Bayesian skyline plot method (BSP) implemented in BEAST72,73. Five preliminary replicates of the BSP analysis were carried out to fine-tune the parameters of the MCMC using the auto-optimization option. Then, we ran the final analyses in triplicates for each haplogroup to check for consistency. The number of groups (m) was set to 10 (but setting different values in preliminary analyses did not affect the resulting demographic trends); the MCMCs were run for 30 million generations and sampled every 1000 generations, with the first 10% discarded as burn-in. BSP analyses were run using the TrN + I, a strict clock model and a uniform prior for the substitution rate, bounded at 0.012 and 0.014 substitutions/site/My. The results were analysed and summarised using TRACER 1.6. Finally, the occurrence of past demographic changes was evaluated by computing the neutrality test statistics FS74, R275 and D76, by using DNaSP. The significance of these statistics was assessed using 1000 coalescent simulations.
Microsatellites data analysis
We tested for deviations from the expected Hardy–Weinberg and linkage equilibria using FSTAT 2.9.3. The software GENETIX was used to obtain estimates of genetic diversity based on the observed and the expected heterozygosity and the number of alleles per locus.
The extent of population genetic structure across the study area was investigated by the Bayesian clustering algorithm implemented in TESS 2.377. This method is advantageous in that it takes the spatial distribution of sampled individuals into account and has been shown to outperform similar methods, particularly in cases of shallow population structures77. The TESS analysis was carried out by modelling admixture using the conditional autoregressive (CAR) model. A preliminary analysis with 20 000 runs (of which the first 5 000 were discarded as burn-in) and 10 replicates for each K value (i.e. the number of clusters) between 2 and 20, was carried to assess model performance, the need to fine-tune the model parameters and to reduce the range of plausible K values. The final analysis contained 100 replicates with K = 2 to 7, each with 50 000 runs (of which the first 20 000 were discarded as burn-in). The spatial interaction parameter was set to 0.6 (i.e. the default value) and the option to update this parameter was activated. The model that best fit the data was selected using the deviance information criterion (DIC). DIC values were averaged over the 100 replicates for each K value and the most likely K value was selected as the one at which the average DIC reached a plateau. The estimated admixture proportions of the 10 runs with the lowest DIC values were averaged using CLUMPP 1.1.278. Finally, to obtain an easily interpretable representation of the estimated admixture proportions, we generated posterior predictive maps by the spatial interpolation procedure available in TESS and with the maps being visualised by TESS Ad-Mixer79. Finally, we used the posterior estimate of the allele frequencies at each locus in each cluster to compute the mean expected heterozygosity of each cluster.
Additional information
GenBank accession numbers: KJ789621–KJ789718.
References
Riddle, B. R. et al. Cryptic vicariance in the historical assembly of a Baja California peninsular desert biota. Proc. Natl. Acad. Sci. USA. 97, 14438–14443 (2000).
Widmer, A. & Lexer, C. Glacial refugia: sanctuaries for allelic richness, but not for gene diversity. Trends Ecol. Evol. 16, 267–269 (2001).
Petit, R. et al. Glacial refugia: hotspots but not melting pots of genetic diversity. Science. 300, 1563–1565 (2003).
Hewitt, G. M. The structure of biodiversity – insights from molecular phylogeography. Front. Zool. 1, 4 (2004).
Gómez, A. & Lunt, D. H. [Refugia within refugia: patterns of phylogeographic concordance in the Iberian Peninsula]. Phylogeography in Southern European refugia: evolutionary perspectives on the origins and conservation of European biodiversity [Weiss, S., Ferrand, N. (eds)] [155–188] (Dordrecht, Springer, (2007).
Canestrelli, D., Aloise, G., Cecchetti, S. & Nascetti, G. Birth of a hotspot of intraspecific genetic diversity: notes from the underground. Mol. Ecol. 19, 5432–5451 (2010).
Hewitt, G. M. [Mediterranean peninsulas – the evolution of hotspots]. Biodiversity hotspots [Zachos, F. E., Habel, J. C. (eds)] (Springer, Amsterdam 2011).
Nieto Feliner, G. Southern European glacial refugia: A tale of tales. Taxon. 60, 365–372 (2011).
Byrne, M. Evidence for multiple refugia at different time scales during Pleistocene climatic oscillations in southern Australia inferred from phylogeography. Quaternary Sci. Rev. 27, 2576–2585 (2008).
Carnaval, A. C., Hickerson, M. J., Haddad, C. F. B., Rodrigues, M. T. & Moritz, C. Stability predicts genetic diversity in the Brazilian Atlantic forest hotspot. Science. 323, 785–789 (2009).
Martin, P. R. & McKay, J. K. Latitudinal variation in genetic divergence of populations and the potential for future speciation. Evolution. 58, 938–945 (2004).
Thompson, J. D. The geographic mosaic of coevolution (University of Chicago Press, Chicago, 2005).
Hampe, A. & Petit, R. J. Conserving biodiversity under climate change: the rear edge matters. Ecol. Lett. 8, 461–467 (2005).
Vandergast, A. G., Bohonak, A. J., Hathaway, S. A., Boys, J. & Fisher, R. N. Are hotspots of evolutionary potential adequately protected in southern California? Biol. Conserv. 141, 1648–1664 (2008).
Thomassen, H. A. et al. Mapping evolutionary process: a multi-taxa approach to conservation prioritization. Evol. Appl. 4, 397–413 (2011).
Hewitt, G. M. Some genetic consequences of ice ages and their role in divergence and speciation. Biol. J. Linnean. Soc. 58, 247–276 (1996).
Tzedakis, P. C. [The Balkans as prime glacial refugial territory of European temperate trees]. Balkan biodiversity [Griffiths, H. I., Krystufek, B. & Reed, J. M. (eds.)] [49–68] (Dordrecht, Boston,London: Kluwer, 2004).
Magri, D. Patterns of post-glacial spread and the extent of glacial refugia of European beech (Fagus sylvatica). J. Biogeogr. 35, 450–463 (2008).
Taberlet, P., Fumagalli, L., Wust-Saucy, A. G. & Cosson, J. F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 7, 453–464 (1998).
Hewitt, G. M. The genetic legacy of the Quaternary ice ages. Nature. 405, 907–913 (2000).
Canestrelli, D., Cimmaruta, R., Costantini, V. & Nascetti, G. Genetic diversity and phylogeography of the Apennine yellow-bellied toad Bombina pachypus, with implications for conservation. Mol. Ecol. 15, 3741–3754 (2006).
Hewitt, G. M. Quaternary phylogeography: the roots of hybrid zones. Genetica. 139, 617–638 (2011).
Moritz, C. Defining ‘evolutionary significant units’ for conservation. Trends Ecol. Evol. 9, 373–375 (1994).
Gasc, J. P. et al. Atlas of amphibians and reptiles in Europe (Collection Patrimoines Naturels, 29, Societas Europaea Herpetologica, Muséum National d'Histoire Naturelle & Service du Petrimone Naturel, Paris, 1997).
Uğur Kaya, U. et al. Rana dalmatina. In: IUCN 2013. IUCN Red List of Threatened Species. Version 2013.1. <www.iucnredlist.org>. Downloaded on 07 February 2013.
Vences, M. et al. Radically different phylogeographies and patterns of genetic variation in two European brown frogs, genus Rana. Mol. Phylogenet. Evol. 68, 657–670 (2013).
Canestrelli, D., Cimmaruta, R. & Nascetti, G. Phylogeography and historical demography of the Italian treefrog, Hyla intermedia, reveals multiple refugia, population expansions and secondary contacts within peninsular Italy. Mol. Ecol. 16, 808–4821 (2007).
Canestrelli, D., Cimmaruta, R. & Nascetti, G. Population genetic structure and diversity of the Apennine endemic stream frog, Rana italica – insights on the Pleistocene evolutionary history of the Italian peninsular biota. Mol. Ecol. 17, 3856–3872 (2008).
Canestrelli, D., Sacco, F. & Nascetti, G. On glacial refugia, genetic diversity and microevolutionary processes: deep phylogeographic structure in the endemic newt Lissotriton italicus. Biol. J. Linn. Soc. 105, 42–55 (2012).
Podnar, M., Mayer, W. & Tvrtkovíc, N. Phylogeography of the Italian wall lizard, Podarcis sicula, as revealed by mitochondrial DNA sequences. Mol. Ecol. 14, 575–588 (2005).
Colangelo, P., Aloise, G., Franchini, P., Annesi, F. & Amori, G. Mitochondrial DNA reveals hidden diversity and an ancestral lineage of the bank vole in the Italian peninsula. J. Zool. 287, 41–52 (2012).
Ayala, F. J. Vagaries of the molecular clock. Proc. Natl. Acad. Sci. USA. 94, 7776–7783 (1997).
Ho, S. Y., Shapiro, B., Phillips, M., Cooper, A. & Drummond, A. J. Evidence for time dependency of molecular rate estimates. Syst. Biol. 56, 515–522 (2007).
Canestrelli, D., Zangari, F. & Nascetti, G. Genetic evidence for two distinct species within the Italian endemic Salamandrina terdigitata Bonnaterre, 1789 (Amphibia: Urodela: Salamandridae). Herpetol. J. 16, 221–227 (2006).
Barbanera, F. et al. Molecular phylogeography of the asp viper Vipera aspis (Linnaeus, 1758) in Italy: evidence for introgressive hybridization and mitochondrial DNA capture. Mol. Phylogenet. Evol. 52, 103–114 (2009).
Toews, D. P. L. & Brelsford, A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 21, 3907–3930 (2012).
Canestrelli, D. & Nascetti, G. Phylogeography of thepool frog Rana (Pelophylax) lessonae in the Italian peninsula and Sicily: multiple refugia, glacial expansions and nuclear–mitochondrial discordance. J. Biogeogr. 35, 1923–1936 (2008).
Canestrelli, D., Salvi, D., Maura, M., Bologna, M. A. & Nascetti, G. One species, three Pleistocene evolutionary histories: phylogeography of the Italian crested newt, Triturus carnifex. PLoS ONE. 7, e41754 (2012).
Di Giovanni, M. V., Vlach, M. R., Giangiuliani, G., Goretti, E. & Torricelli, R. Genetic analysis of the species of Sigara s str (Heteroptera, Corixidae) in the Italian Peninsula. Ita. J. Zool. 65, 393–397 (1998).
Stefani, F., Galli, P., Crosa, G., Zaccara, S. & Calamari, D. Alpine and Apennine barriers determining the differentiation of the rudd (Scardinius erythrophthalmus L) in the Italian peninsula. Ecol. Fresh. Fish. 13, 168–175 (2004).
Giraudi, C. [The Appennine glaciations in Italy]. Developments in quaternary science 2 [Ehlers, J. & Gibbard, P. L. (eds.)][251–260] (Amsterdam, The Netherland,2004).
Marta, S., Mattoccia, M. & Sbordoni, V. Modelling landscape dynamics in a glacial refugium–or the spatial and temporal fluctuations of tree line altitudes. J. Biogeogr. 40, 1767–1779 (2013).
Tansi, C., Muto, F., Critelli, S. & Iovine, G. Neogene-Quaternary strike-slip tectonics in the central Calabrian Arc (southern Italy). J. Geodyn. 43, 393–414 (2007).
Shackleton, J. C., Van Andel, T. H. & Runnels, C. N. Coastal paleogeography of the central and western Meditterranean during the last 125,000 years and its archeological implications. J. Field. Archaeol. 11, 307–314 (1984).
Stewart, J. R., Lister, A. M., Barnes, I. & Dalén, L. Refugia revisited: individualistic responses of species in space and time. Proc. R. Soc. A. 277, 661–671 (2010).
Schmitt, T. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front. Zool. 4, 11 (2007).
Ursenbacher, S. et al. Molecular phylogeography of the nose-horned viper (Vipera ammodytes, Linnaeus (1758)): Evidence for high genetic diversity and multiple refugia in the Balkan peninsula. Mol. Phylogenet. Evol. 46, 1116–1128 (2008).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd Edition (Cold Spring Harbor Laboratory Press, 1989).
Kocher, T. D. et al. Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc. Natl. Acad. Sci. USA. 86, 6196–6200 (1989).
Moritz, C., Schneider, C. J. & Wake, D. B. Evolutionary relationships within the Ensatina eschscholtzii complex confirm the ring species interpretation. Syst. Biol. 41, 273–291 (1992).
San Mauro, D., Gower, D. J., Oommen, O. V., Wilkinson, M. & Zardoya, R. Phylogeny of caecilian amphibians (Gymnophiona) based on complete mitochondrial genomes and nuclear RAG1. Mol. Phylogenet. Evol. 33, 413–427 (2004).
Hauswaldt, J. S., Fuessel, J., Guenther, J. & Syeinfartz, S. Eight new tetranucleotide microsatellite loci for the agile frog (Rana dalmatina). Mol. Ecol. Resour. 8, 1457–1459 (2008).
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P. & Shipley, P. Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes. 4, 535–538 (2004).
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 (1997).
Pina-Martins, F. & Paulo, O. S. CONCATENATOR: sequence data matrices handling made easy. Mol. Ecol. 8, 1254–1255 (2008).
Farris, J. S., Kallersjo, M., Kluge, A. G. & Bult, C. Testing significance of incongruence. Cladistics. 10, 315–319 (1994).
Posada, D. JModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256 (2008).
Tamura, K. & Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526 (1993).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 25, 1451–1452 (2009).
Nei, M. Molecular Evolutionary Genetics (Columbia University Press, New York, 1987).
Tamura, K. et al. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance and Maximum Parsimony Methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Templeton, A. R., Crandall, K. A. & Sing, C. F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data III Cladogram estimation. Genetics. 132, 619–633 (1992).
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1660 (2000).
Salzburger, W., Ewing, G. B. & Von Haeseler, A. The performance of phylogenetic algorithms in estimating haplotype genealogies with migration. Mol. Ecol. 20, 1952–1963 (2011).
Guindon, S., Lethiec, F., Duroux, P. & Gascuel, O. PHYML Online–a web server for fast maximum likelihood-based phylogenetic inference. 33 (suppl 2): W557–W559 (2004).
Guindon, S. et al. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 30. Syst. Biol. 59, 307–21 (2010).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Veith, M., Kosuch, J. & Vences, M. Climatic oscillations triggered post-Messinian speciation of Western Palearctic brown frogs (Amphibia, Ranidae). Mol. Phylogenet. Evol. 26, 310–327 (2003).
Stefani, F. et al. Refugia within refugia as a key to disentangle the genetic pattern of a highly variable species: the case of Rana temporaria Linnaeus, 1758 (Anura, Ranidae). Mol. Phylogenet. Evol. 65, 718–726 (2012).
Excoffier, L., Smouse, P. E. & Quattro, J. M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics. 131, 479–491 (1992).
Excoffier, L., Laval, G. & Schneider, S. ARLEQUIN (version 3.0): An integrated software package for population genetic data analysis. Evol. Bioinform. Online. 1, 47–50 (2005).
Drummond, A., Rambaut, A., Shapiro, B. & Pybus O. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192 (2005).
Drummond, A., Ho, S., Phillips, M. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88 (2006).
Fu, Y. X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 147, 915–925 (1997).
Ramos-Onsins, S. E. & Rozas, J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 19, 2092–2100 (2002).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 123, 585–595 (1989).
Chen, C., Durand, E., Forbes, F. & Francois, O. Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol. Ecol. Notes. 7, 747–756 (2007).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 23, 1801–1806 (2007).
Mitchell, M. W., Rowe, B., Sesink Clee, P. R. & Gonder, M. K. TESS Ad-Mixer: A novel program for visualizing TESS Q matrices. Conserv. Genet. Resour. 5, 1075–1078 (2013).
Acknowledgements
We are grateful to Gaetano Aloise, Francesco Paolo Caputo and Francesco Spallone for their help with sample collection and to Jacek Methody Szymura for the kind discussions during early phases of manuscript preparation. This work has been funded with grants by the Italian Ministry of Environment, the Agenzia Regionale dei Parchi–Lazio (ARP) and the Italian Ministry of Education, University and Research (PRIN project 2012FRHYRA).
Author information
Authors and Affiliations
Contributions
D.C. and G.N. conceived and designed the study. D.C., R.B. and F.S. performed the experiments. D.C., R.B. and F.S. analysed the data. D.C. and R.B. wrote the paper. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Canestrelli, D., Bisconti, R., Sacco, F. et al. What triggers the rising of an intraspecific biodiversity hotspot? Hints from the agile frog. Sci Rep 4, 5042 (2014). https://doi.org/10.1038/srep05042
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05042
This article is cited by
-
Reconstructing hotspots of genetic diversity from glacial refugia and subsequent dispersal in Italian common toads (Bufo bufo)
Scientific Reports (2021)
-
Genetic Divergence Across Glacial Refugia Despite Interglacial Gene Flow in a Crested Newt
Evolutionary Biology (2021)
-
Hybridization and extensive mitochondrial introgression among fire salamanders in peninsular Italy
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
