
Abstract
Ancient Jian wares are famous for their lustrous black glaze that exhibits unique colored patterns. Some striking examples include the brownish colored “Hare's Fur” (HF) strips and the silvery “Oil Spot” (OS) patterns. Herein, we investigated the glaze surface of HF and OS samples using a variety of characterization methods. Contrary to the commonly accepted theory, we identified the presence of ε-Fe2O3, a rare metastable polymorph of Fe2O3 with unique magnetic properties, in both HF and OS samples. We found that surface crystals of OS samples are up to several micrometers in size and exclusively made of ε-Fe2O3. Interestingly, these ε-Fe2O3 crystals on the OS sample surface are organized in a periodic two dimensional fashion. These results shed new lights on the actual mechanisms and kinetics of polymorphous transitions of Fe2O3. Deciphering technologies behind the fabrication of ancient Jian wares can thus potentially help researchers improve the ε-Fe2O3 synthesis.
Similar content being viewed by others


Introduction
The Jian kilns in the Jianyang county of the Fujian province of China had a long history of producing black glazed Jian wares, particularly during the Song dynasty (960 AD–1279 AD). The crown jewels of this period were the black-glazed Jian tea bowls1,2,3. On one hand, these Jian tea bowls were highly prized by the cultivated few during the Song dynasty for their thick and lustrous black glaze, which provides a perfect visual contrast to the white milky tea during tea contest extravaganza (Cha Lu ‘Record of Tea’ by Cai Xiang)1,2. On the other hand, they were also produced in large quantities to serve the need of ordinary people. The Jian ware's simplicity and beauty have been appreciated by generations of people hereafter. Beside its rich black glaze, Jian bowls are also known for their spectacular streaked or mottled patterns (Figure 1). The most common and famous variety, called “Hare's fur” (HF), is a shining black glaze showing fine radial rust-colored streaks. Other less common features, such as “oil spot” (OS) and “partridge spot” have also been reported1,2,4,5,6. These patterns are believed to be the result of iron oxide crystal precipitation at the glaze surface during the firing process in the dragon kiln. Some of the finest Jian tea bowls were preserved and admired in Japan, where they are known as Yohen Tenmoku.

a) Surface of the HF Jian sherd analyzed in this paper. Brown/yellow stripes over a black glaze are clearly observed; b) OS sample from this study. The spots with a silver color are spread over the black glaze; c) Cross section of the HF sample observed with an optical microscope. Crystals and bubbles are dispersed in the thick semi-opaque glaze. d). Schematic drawing of Jian bowl surface pattern formation process. The surface patterns of the Jian wares such as Hare's Fur (HF) and Oil Spot (OS) are believed to be linked to the crystallization of iron oxides.
Previous studies indicated that the bodies of the Jian wares are made from the local iron rich (5–9% wt.) clay. Other main constituents are SiO2 (65–70% wt.), Al2O3 (19–23% wt.) and K2O (2–3% wt.). Small amount of MgO (0.5–1% wt.) and TiO2 (0.5–0.9% wt.) were also detected2,4,5. The recipe for the glaze preparation is assumed to use a mixture of the local clay, limestone and wood ashes. This is consistent with the chemical analysis results, which show higher concentrations of CaO (5–8% wt.) and MnO (0.5–0.8%) than in the body2,4,5. More detailed background information of Jian Ware can be found in the Supporting Information.
The surface patterns of the Jian wares such as Hare's Fur and Oil Spot are believed to originate from the crystallization of iron oxides (Figure 1d)2,4,5,6. During the high temperature firing process, the glaze starts to melt and the iron oxides in the glaze are thermally reduced and release oxygen once the temperature reach 1240°C2. As the oxygen bubbles grow in size, more and more iron will accumulate at the gas-liquid interface near the bubbles. Eventually, the large bubbles move out within the molten glaze and carry iron towards the surface. Depending on the temperature, the surface iron-rich (flux) spots can start to flow down along the sides of the bowl. Upon cooling, the iron oxides crystals will form on the surface and give the streaks that characterize the HF patterns. Under lower firing temperatures and a reducing environment, the more viscous glaze will prevent the formation of streaks and circular and/or oval shape “oil spot” patterns will form instead (OS samples). The control of the atmosphere during the firing process (oxidizing and/or reducing) will determine the oxidation state of iron in the body, inside the glaze and at the top surface. It is obvious that the precipitation of iron oxides at the surface plays a key role in the pleasing appearance of the Jian tea bowls. It is currently believed that hematite (α-Fe2O3) crystals are formed on the HF surface and mixtures of magnetite (Fe3O4) and hematite crystals are precipitated on the OS surface4,6. However, such a theory cannot fully explain the highly reflective nature and the silvery appearance of OS patterns. Nevertheless, few mineralogical studies have been carried out to confirm this commonly accepted theory.
To reveal the crystallographic nature of the iron oxides and their formation mechanism, we have investigated the microstructure and local chemical composition of ancient Jian bowl samples through a combination of X-ray based and electron based characterization techniques. Remarkably, the rare metastable ε-Fe2O3 phase7,8 (instead of hematite and magnetite as inferred in previous studies) was identified as the phase for the Fe oxide precipitates on the OS pattern surface using micro-X-ray diffraction. These crystals in OS samples are up to several micrometers in size which is significantly larger than the nano-sized ε-Fe2O3 crystals that can be currently synthesized. Furthermore, the majority of these ε-Fe2O3 crystals on the OS sample surface are organized in an interesting two dimensional (2D) periodic fashion. This ε-Fe2O3 phase has also been found on the HF surface along with hematite. These findings are substantiated by SEM and micro-Raman studies conducted on the same samples. The fact that significant amounts of ε-Fe2O3 are found in both HF and OS sample suggests that the formation of ε-Fe2O3 may not be a rare event in Jian ware making. We believe that the iron enrichment at the Jian glaze surface and a reducing firing condition are keys to obtain pure large size ε-Fe2O3 crystals.
To our knowledge, ε-Fe2O3 has not been previously found in ancient Jian ware and ancient pottery in general. The large size and the 2D arrangement of these crystals on the Jian glaze surface are also unique among synthesized ε-Fe2O37,8. Understanding the chemical compositions and firing procedure of ancient Jian ceramics can provide researchers with valuable information on how to optimize the growth conditions of the single phase ε-Fe2O3, an industrially attractive compound for magnetic applications7,8, with a high yield, desired size, morphology and stability.
Results
Optical Microscopy and Scanning Electron Microscopy (SEM)
Samples were collected from Jian bowl sherds provided by the museum of the Fujian province2. As shown in Figure 1, one has the typical brown HF pattern (Fig 1a) and the other shows silvery OS pattern (Fig 1b). Part of the analyses was performed on polished cross-sections. The sherds were cut with a diamond saw and the resulting sections were mounted in epoxy and mechanically polished. Figure 1c shows a cross-section sample observed under an optical microscope. The 500 μm thick semi-opaque glaze, in which crystals and bubbles are dispersed, is clearly visible. The surface patterns of the Jian wares were also studied directly without any sample preparation.
Examination of both HF and OS samples under an optical microscope (light is polarized in the horizontal x direction) in reflection mode reveals the presence of crystal clusters of different sizes from sub-micron to few hundred microns (Figure 2). They are mainly concentrated in the brown-yellow Fe-rich streaks of the HF sample and in the iron-rich silver spots of OS sample. For the HF sample, the majority of the crystals on the surface appear to be brownish, which is consistent with the appearance of hematite crystals. Nevertheless, some “white” crystals are also clearly visible (point “B” in Figure 2a). For the tested OS sample, the crystal clusters exhibit a pyramidal shape (Figure 2b, point “C” and “D”). The faces of these pyramids strongly reflect the incident light of the microscope, giving them their “white” appearance. Under particular orientations relative to the incident light, rainbow colors are visible. There are also small submicron “white” crystals around the large crystal clusters across the OS surface as seen in Figure 2b at point “E”.

(a). Optical images of the HF sample and (b) the OS sample. “White” and “brown” crystals are visible for the HF sample. Only “white” crystals are visible at the surface of the OS sample, the big crystal clusters showing a blue-red cross in the middle. All the crystals investigated at the surface of the OS sample correspond to the epsilon-Fe2O3 phase. (c) SEM image (10,000×) of a part of pyramidal shape crystal cluster found on OS surface. It shows a dendritic 2D network of crystals arranged in a periodic fashion. (d). A zoom in view of a selected region.
SEM images of the OS surface in Figure 2 (c) and (d) reveal a stunning 2D network of dendritic crystals within one of the “white” crystal clusters. All crystal branches are parallel to each other and ordered in a periodic fashion. The longest branches are more than 5 μm in length. This 2D morphology of surface crystals are in stark contrast to those reported in the previous SEM study on HF samples2, which illustrates the different mineralogical origins for the OS surface crystals. Furthermore, the 2D periodic nature of these surface crystals can also explain some of the optical properties possessed by OS sample.
TEM/EDX study of iron oxide crystals
Using TEM, we examined the OS glaze cross-section samples across OS patterns to obtain high resolution information on the crystal shape, distribution and most importantly the chemical composition of sub-surface crystallites by EDX analyses. A typical TEM image of a cross-section sample is shown in Figure 3. Two main kinds of crystal morphologies were found. The first is a dendritic leave-like shaped crystal spreading from the surface over several hundred of nanometers inside the glaze. These crystals look quite similar to the ones organized as a 2D dentritic network at the surface of the glaze. The second type consists of smaller crystals with sizes around 20 nm, randomly dispersed within the glaze below the surface. The EDX chemical mappings of Fe, Al and Ca for each type at one given region are shown in Figure 3c–3e and Figure 3g–3i respectively. From all the collected EDX maps, we find that iron is clearly the only cationic element present in these crystals. Additionally, no Al, Ca and Si (not shown) enrichments are found in any of the crystals tested within the EDX detection limit. From these TEM results, we can narrow down the probable phases of these OS crystals as belonging to the iron oxide family.

Transmission Electronic Microscopy images from a region close to the surface of the OS sample.
Two types of crystals were found (star shape and dendritic shape). EDX analyses revealed that Fe is the only cationic element present in these crystals.
XRD Phase identification of ε-Fe2O3 surface crystals
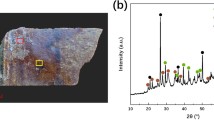
To unambiguously identify the iron oxide phases, we performed powder X-ray diffraction (XRD) on several regions of HF and OS samples. A Bruker D8 Advance diffractometer equipped with a 15 cm long and 100 μm in diameter optical fiber and a fast LynxEye detector, was used. The XRD patterns were collected from both the black glaze region and the surface pattern region. For both samples, no diffraction signal can be found in the low iron regions of black glaze (labeled as “HF-Surf-G” and “OS-Surf-G”). This shows that the black glaze body is of glassy nature within the detection depth of the XRD. Diffraction rings can only be observed while focusing the X-ray beam on the HF and OS Fe-rich regions (HF-Surf-Fe and OS-Surf-Fe). Therefore, we assign the diffraction pattern to iron oxide crystals at the surface. In Figure 4a and 4b, we show the XRD results collected from HF sample and OS sample respectively. The broad bumps within the 20–30° (2θ; CuKα) range in both diagrams come from the amorphous glaze. A Le Bail fit was performed, after identification of the main phases for both samples. In Figure 4a, peaks at 33.15° and 36.28° (2θ CuKα) for the HF sample were found to correspond to the hematite α-Fe2O3 phase. Interestingly, a second phase diffracting at 18.84°, 35.39°, 36.78°, 38.23° and 40.48° (2θ CuKα) was also obtained for both samples. However, those peaks do not match with any of the common iron oxide phases such as Fe3O4 (magnetite), α-Fe2O3 (hematite) and γ-Fe2O3 (maghemite). This phase has in fact been indexed with an orthorhombic cell (space group Pna21, a = 5.0810 Å, b = 8.7411 Å, c = 9.4083 Å) and corresponds to a rare variety of ferric oxide, called ε-Fe2O37,9. The high intensity of the peak at 18.84° (2θ CuKα), attributed to the 002 reflection, is an indication of a preferred orientation, with the c-axis of the crystal perpendicular to the surface of the sample.

Powder XRD analyses of the surface of the HF and OS samples.
a) Le Bail fit of the HF Fe-rich surface region, showing the presence of hematite and ε-Fe2O3; b) Le Bail fit of the OS Fe-rich surface region, showing the presence of ε-Fe2O3. For a) and b), powder patterns were recorded with a Bruker D8 Advanced and a beam of ~100 μm in diameter. The high intensity of the 002 reflection at 18.84° (2θ CuKα) indicates preferred orientation. c) 2D diffraction pattern obtained from the surface of the HF sample, from a low content Fe zone (HF-Surf-G) and from a Fe-rich region (HF-Surf-Fe). Theoretical positions for the ε-Fe2O3 are indicated in red; d) 2D diffraction pattern obtained from 2 Fe-rich regions at the surface of the OS sample. Theoretical positions for the ε-Fe2O3 are indicated in red. For c) and d), 2D patterns were obtained at ALS beamline 12.3.2 using a ~15 μm*15 μm microbeam. While the 2D diffraction rings look homogeneous for HF-Surf-Fe, they are highly textured for OS-Surf-Fe.
To substantiate this surprising discovery of the ε-Fe2O3 phase, additional XRD analyses were carried out on ALS Beamline12.3.2 using a 15 μm*15 μm synchrotron x-ray beam and an area detector. Corresponding 2D diffraction patterns for HF-Surf-Fe and OS-Surf-Fe, along with the positions of the theoretical rings expected for the ε-Fe2O3 phase are shown in Figure 4c and Figure 4d. In Figure 4c, the HF-Surf-G (top panel) and HF-Surf-Fe (bottom panel) patterns were collected from a black glaze region and brown strip region respectively. No diffraction ring is observable in the top panel. This is consistent with the previous XRD experiment that the amorphous black glaze body does not contribute to XRD signal. In the bottom panel, we observe clear uniform XRD rings at the expected positions for the ε-Fe2O3 phase at the brown strip regions. This is another evidence that ε-Fe2O3 indeed exists in the HF region. Due to the small size of the x-ray beam, only randomly distributed particles with dimensions inferior to ~100 nm would produce homogeneous and non-spotty diffraction rings. Therefore, the uniformly distributed intensity of the diffraction rings for the HF sample indicates that ε-Fe2O3 there consists of randomly oriented sub-100 nm size crystals.
For the OS sample, two diffraction patterns are shown in Figure 4d. They are taken from two different Fe-rich oil spot zones. The diffracted rings collected once again match those positions of the ε-Fe2O3 phase. Unlike the continuous diffraction rings from the HF sample shown in Figure 4c, we find that the diffraction rings of the OS sample are fragmented in the azimuthal direction indicative of a preferred crystal orientation (texture). The textured nature of the diffraction patterns on the OS sample indicates that ε-Fe2O3 crystals are co-oriented in a particular direction in consistency with the 2D arrangement of dendrites observed with SEM. The presence of some intense and sharp diffraction spots along the rings was also noted, indicating diffraction from bigger crystals (>1 μm). The larger crystal size of the ε-Fe2O3 phase in the OS is furthermore confirmed by the fact that a Laue diffraction pattern can be obtained using a 1 μm size beam (see Supporting Information).
Micro-Raman Study of ε-Fe2O3 crystals
In addition, Raman spectra collected at different spots on the HF and OS surfaces, Point “A–E” labeled in Figure 2a and 2b, are shown in the Figure 5a. For the HF sample, the spectrum taken at the brown colored region, point “A”, was identified to belong to the α-Fe2O3 phase10,11. The second spectrum collected at the “white” crystal region, point “B”, shows a different set of peaks that do not belong to either α-Fe2O3, γ-Fe2O3 or Fe3O4. Furthermore, Raman spectra obtained from OS sample on top of the large crystal clusters (“C” and “D”) and on the smaller crystal cluster (“E”) are shown in Figure 5b. Interestingly, all three spectra show similar features. The majority of the Raman peak positions found at “white” crystal regions of the OS also match those of the “white” crystal clusters in HF sample. Only a few of the peaks are slightly shifted in position. The similarities between the Raman spectra taken at “B” (HF), “C”, “D” and “E” strongly suggest that the white crystals found on the surface of both HF and OS samples are mineralogically very similar. This result is consistent with the XRD observation. We believe that the white crystals on both HF and OS are made of ε-Fe2O3. Detailed information of Micro-Raman results can be found in the supporting information.

Raman spectra collected at different spots on the HF and OS surfaces are shown in (a) and (b) respectively.
Discussion
The ε-Fe2O3 phase was first mentioned by Forestier and Guillain in 193412,13. However, the first description of the crystallographic structure was only reported in 1998 by Tronc et al.9 and was later refined by Kelm and Mader14. This ferric oxide phase is a rare and metastable polymorph. It is structurally considered to be an intermediate between hematite α-Fe2O3 and maghemite γ-Fe2O315. Recently, it has attracted a lot of attention due to its giant coercivity (20 kOe) at room temperature, an appealing property for future magnetic and spintronic applications7,8,16,17. ε-Fe2O3 also exhibits significant ferromagnetic resonance18,19 and coupled magnetoelectric properties20. These properties and its giant room temperature coercivity do not exist in any other simple metal oxides. Given the earth abundance of iron oxides, this phase has been considered as a new potential material for applications in recording media and telecommunications7,8.
Due to its metastable nature, the ε-Fe2O3 phase is known to be difficult to grow and can only be synthesized as nanoscale objects such as nanoparticles9,15,21,22, nanowires23 and thin films24. Many of these synthesis methods use the γ-Fe2O3 → ε-Fe2O3 → α-Fe2O3 phase transformation pathway. However, most of these synthetic routes yield α-Fe2O3 and/or γ-Fe2O3 impurities7. The nanoscale nature and growth mechanism of ε-Fe2O3 is generally explained by its low surface energy21,22,25. Thermodynamically, the contribution of the surface to the whole system increases as the ferric oxide particles size is reduced. The free energy (G) per volume (V) of an iron oxide particle can be expressed as:

where the Gbi and σi are the bulk free energy and surface/interface free energy of different i- Fe2O3 (i = α, β, γ, ε) respectively. c is a shape dependent geometric constant and d is the diameter of the nanoparticle.
A low surface/interface energy (σε) can therefore stabilize the formation of ε-Fe2O3, a metastable ferric oxide, when the particle dimensions d is in the nanoscale regime21,22,25. Assuming Gbγ > Gbε > Gbα and σα > σε > σγ, ε-Fe2O3 would form at intermediate sizes between maghemite and hematite, resulting in the γ-Fe2O3 → ε-Fe2O3 → α-Fe2O3 phase transformation pathway. Several techniques have been developed to modify the surface/interface energy to increase the yield of ε-Fe2O3 as well as controlling the size and morphology of crystal formed. So far the purest ε-Fe2O3 samples prepared are synthesized using nanoparticles enclosed in a silica matrix with addition of group IIA metal ions (eg. Ba2+, Ca2+)8,17,21. But these techniques still rely on the γ-Fe2O3 → ε-Fe2O3 → α-Fe2O3 phase transformation pathway7. It has also been suggested that a successive γ-Fe2O3 → ε-Fe2O3 → β-Fe2O3 → α-Fe2O3 polymorphous transformation26,27 and structural evolution from Fe3O4 → ε-Fe2O323 can happen. But so far, no consensus has been reached on these different pathways.
In Jian ware, the iron oxide crystallites are precipitated in the molten glaze during the cooling phase. The fact that γ-Fe2O3 phase is not observed by XRD in the samples tested could suggest that ε-Fe2O3 can be the initial phase precipitating from the molten glass. However, we do recognize XRD is not sensitive to diluted quantities of ultra-small nanoscale crystals even though micro-XRD was used here. Although additional investigations will have to be carried out to determine the presence of γ-Fe2O3 phase, a traditional ε-Fe2O3 → α-Fe2O3 phase transformation pathway is sufficient to understand the HF results: due to the unique chemical compositions of Jian glaze (high Ca and Mn)14,15,20 and firing temperature (1200°C–1300°C), the interface energy of ε-Fe2O3 inside a glass matrix can be different from previous studies7,8,9,14,17,21,22,23,24,25,28,29. It is possible that the free energy per volume of ε-Fe2O3 is lower than both γ-Fe2O3 and α-Fe2O3 under these conditions for small crystals. Therefore, only the ε-Fe2O3 crystallites can be detected. They will transform into α-Fe2O3 as the crystallites grow larger. This can explain why only homogeneous 2D diffraction rings of ε-Fe2O3 are detected in Figure 4c and the co-existence of both ε-Fe2O3 and α-Fe2O3 phases in the HF sample (Figure 3a). We believe that small ε-Fe2O3 crystallites (<100 nm) belong to the early phases of the precipitation of ferric oxide at the HF sample surface and the α-Fe2O3 phase starts to dominate as the particle size increases.
However, this theory falls short in explaining the OS results where only the ε-Fe2O3 phase is detected and grows in a 2D dendritic network (Figure 2). It is quite remarkable that such a large size pure ε-Fe2O3 phase crystallite network could be produced in such an appealing way more than one thousand years ago. To our knowledge, all the ε-Fe2O3 crystals synthesized today are in nano scale and no micron-sized ε-Fe2O3 crystals has been reported7,8,9,14,15,17,21,22,23,24,25,27,28,29,30,31. Thus, identifying and understanding the underlying mechanisms of the iron oxide crystal transformation in ancient Jian ware and, OS in particular, are highly relevant for producing high quality ε-Fe2O3 material and can potentially help researchers to fully understand its extraordinary yet surprising magnetic properties, which is a key step for this material to realize its application potential. As we know, the chemical composition of OS and HF glaze are very similar. This leads us to believe that the differences in the ε-Fe2O3 phase transformation observed in OS and HF samples are due to their different firing conditions.
The epitaxial growth of ε-Fe2O3 on mullite was reported by Kusano et al. through a reproduction study of creating “Hidasuki” red patterns29,31. In their studies, they also found that the partial oxygen pressure during firing had significant influence on the formation of ε-Fe2O3. The firing temperature used, 1250°C, is very close to that of Jian ware. Under identical temperature treatment, ε-Fe2O3 was produced only for an oxygen to nitrogen ratio between 1:99 and 2:98. Once the oxygen to nitrogen ratio increased to 5:95, no ε-Fe2O3 could be detected. Furthermore, the oxygen partial pressure can also change the crystal shape of ε-Fe2O3. Under an oxygen to nitrogen ratio of 1:99, the ε-Fe2O3 crystals attached to mullite exhibit a square columnar shape. Interestingly, ε-Fe2O3 crystals change to a dendritic fin-like crystals under an oxygen to nitrogen ratio of 2:98. In addition, these fin-like crystals seem to have preferred orientation as well (Figure 12 in reference 2731).
To understand the firing conditions of Jian ware, XRD analysis and Raman spectroscopy were carried out on the Jian ware body to reveal the mineral composition of the body of the HF and OS samples. The two XRD patterns, with corresponding Le Bail fit, are shown in Figure 6. Main phases, space groups and refined cell parameters are listed in Table 1 and Table 2. Mullite (alumino-silicate) and cristobalite (silicon dioxide), characteristic of a high temperature firing process, are found in both samples. Quartz was also identified. More importantly, hematite Fe2O3 and magnetite Fe3O4 are found in the HF sample and the OS sample, respectively. The presence of hematite exclusively composed of Fe(III), in the HF sample is an indication of an oxidizing atmosphere. The presence of hematite in the body of HF sample suggests that it was cooled under at least partially oxidizing conditions. The porous unvitrified Jian body is permeable to oxygen and allows Fe phases to oxidize to hematite. On the contrary, formation of magnetite, in which both Fe(II) and Fe(III) co-exist, is related to a process in more reduced conditions. Raman investigation on the HF sample also revealed the presence of rutile (TiO2) and pseudobrookite (Fe2TiO5) crystals (not shown).

Le Bail fitting of the body of the Jian ceramics for the a) HF sample: (1) Mullite, (2) Hematite, (3) Cristobalite, (4) Quartz and b) OS sample: (1) Mullite, (2) Magnetite, (3) Cristobalite, (4) Quartz.
Red: experimental points, Black: calculated diagram, Green: Bragg positions, Blue: difference curve.
Because of its principally amorphous nature, the Jian glaze was investigated using NEXAFS. Using the centroid energy positions of the Fe K pre-edge peaks, the iron oxidation states of both HF glaze and OS glaze can be extracted32. For the OS sample, we find that the glaze away from the surface seems to be fully reduced while the glaze surface is more oxidized (see Supporting Information). For HF sample, the surface of the glaze is also more oxidized than the glaze body. The Fe in the HF glaze are mainly Fe(III), which is more oxidized than that of the OS glaze. The complete information about the NEXAFS study can be found in the Supporting Information. Combined with the XRD results above, we conclude that the OS samples are fired and cooled under more reducing conditions than that of the HF sample, which is also in agreement with previous literature2,4,5,6. In addition, the difference between iron oxidation state in the HF glaze and OS glaze also shows that there is more oxygen dissolved in the HF glaze upon firing.
Based on all the experimental observations, we propose the following hypothesis on the ε-Fe2O3 growing mechanism in Jian ware. For a given temperature, the precipitation and growth of ferric oxide crystals under the glaze surface are influenced by the high iron concentration, the presence of other elements (such as Ca, Mn) and oxygen concentration dissolved in the glaze. From the bubble formation and elimination processes, the iron within the Jian glazes is redistributed and there is significant iron enrichment at the glaze surface. Upon cooling, iron oxides crystallize and grow at the near surface region. The HF sample is fired and cooled in an oxidizing environment, which is evident from the XRD study of the body. As shown in the NEXAFS, the HF glaze are mainly Fe(III) rich. There is more dissolved oxygen and oxygen penetrates deeper into the surface, thus ferric oxide can be crystallized and grown in a deeper region. This higher oxygen concentration in the glaze also helps to promote the expected and previously discussed ε-Fe2O3 → α-Fe2O3 transformation. For OS samples, they are fired under more reduced conditions (Figure 6b). As demonstrated in the NEXAFS, the dissolved oxygen concentration in the glaze is lower and will have a shallower distribution below the surface, which limits the ferric oxide crystallization and growth to a much shallower region. Therefore, ferric oxide crystallite growth is confined at the surface within a small 2D region. Such a reducing environment seems to contribute to the prolonged growth (stabilization) of ε-Fe2O3 as well.
It is quite remarkable that this “Jian” method can produce 2D periodic array of micro sized ε-Fe2O3 crystals that have not been produced by modern synthesis techniques. This synthetic method, i.e. high iron concentration glass, high temperature and reducing gaseous environments, can be potentially of use for modern material synthesis. Further systematic study is clearly needed to make it a reality. Moreover, the vast amount of dentritic 2-D networks of these ε-Fe2O3 also offer clue for us to understand the highly reflective nature of some of the OS patterns and the origins of their silvery appearance, which shall be addressed in a future study.
In summary, using optical microscopy, SEM, TEM/EDX, micro-XRD and Raman spectroscopy, we have identified the iron oxide crystallites in HF brown strip as a mixture of α-Fe2O3 (hematite) and the metastable ε-Fe2O3 phases. For the OS sample, the crystallites responsible to the “oil spot” appearance are mainly ε-Fe2O3. The fact that significant amount of ε-Fe2O3 are found in both HF and OS sample suggests that the formation of ε-Fe2O3 may not be a rare event in Jian ware making. The micro-XRD results also indicate that there are differences between ε-Fe2O3 crystallites in the HF sample and the OS sample. The ε-Fe2O3 crystallites in HF are nanometer size and randomly oriented, while the ε-Fe2O3 crystallites in the OS sample grow to larger micron size and self-organize in 2D periodic structures. This is a direct indication that the firing conditions of ceramics significantly affect the formation of iron oxide crystals. OS firing conditions are clearly more favorable to the formation and stabilization of ε-Fe2O3 crystallites. We believe that the bubble formation and elimination processes at high temperature, from which the iron within the glazes is redistributed, cause a significant iron enrichment at the glaze surface. This surface iron enrichment and a firing process under reducing condition are keys to obtain pure ε-Fe2O3 phase. The study of the chemical compositions and firing of these ancient ceramics provide us with valuable information on the growth conditions of ε-Fe2O3.
This work also highlights the importance of advanced characterization tools in cultural heritage material research in general. Through this type of studies, we can understand and preserve our history better, in so doing not only penetrate the mysteries of ancient society but also sometimes advance the current technology through learning from our own past (Archaeomimetism)33.
Methods
Micro X-ray fluorescence (μ-XRF) measurements were performed at Beamline 2–3 of the Stanford Synchrotron Radiation Lightsource (SSRL) (Stanford, USA) and at Beamline 12.3.2 of the Advanced Light Source of the Lawrence Berkeley National Laboratory (Berkeley, USA) both using the Vortex-EM single element Si-drift fluorescence detector.
Micro X-ray Absorption Near Edge Spectroscopy (μ-XANES) experiments were also performed at SSRL-BL2.3. Based on the μ-XRF maps, several spots were selected for Fe K-edge micro-XANES analyses. Energy calibration was performed using a Fe metal foil, with the Fe K-edge set at 7110.75 eV34. All micro-XANES spectra were collected in fluorescence mode, calibrated, deadtime corrected, pre-edge background subtracted and post-edge normalized using standard procedures. SSRL data were analyzed using the software package SMAK (Sam's microprobe analysis kit)35.
Micro X-ray powder diffraction experiments (μ-XRD) on the glaze surfaces were conducted at ALS BL12.3.236. The energy of the incident beam was set to 8 keV. Powder diffraction patterns were collected using a 2D DECTRIS Pilatus 1 M X-ray area detector. The detector was positioned at around 150 mm and 45° (2θ) with respect to the incident beam and the sample at 2.5° incident angle (reflection geometry). 2D patterns were processed with the XMAS software37. Laue μ-XRD measurements using a polychromatic 1 μm size beam with an energy range of 5 to 24 keV has been performed on the same beamline. For this experiment, the sample was positioned at 45° relative to the incident beam and the detector was placed at 90° with a sample to detector distance of ~140 mm.
The second part of the μ-XRD measurements were carried out at the CEMES Laboratory on a Bruker D8 Advance device in Bragg–Brentano geometry, equipped with 15 cm long and 100 μm in diameter optical fiber and a fast LynxEye detector. A Cu point focus anode was used to produce X-rays at the characteristic Cu Kα fluorescence lines. Diffraction patterns were acquired with a step size of 0.03° (2θ) and a counting time of 200 s/step. Le Bail fitting of powder diffraction patterns was performed with the Fullprof package38.
Micro Raman spectroscopy
Raman spectra were recorded using the XploRA (HORIBA Jobin Yvon) spectrometer (CEMES Laboratory). A 532 nm laser excitation was used and the laser power on the sample was limited to 1 mW. A 100× microscope objective was used to focus the laser beam down to a 1 μm (in diameter) spot. Wavenumber calibration was regularly checked by measuring the position of the 520 cm−1 Raman peak from a silicon reference crystal.
Transmission electron microscopy (TEM) was performed at the CEMES Laboratory. Imaging and EDX mapping were carried out using a Philips CM20-FEG electron microscope operating at 200 kV and equipped with a Microanalyse QUANTAX detector XFlash 30 mm2 127 eV. TEM specimens were prepared by mechanical polishing and ion-beam thinning (Gatan PIPS). The detail of this type of sample preparation can be found elsewhere39.
Scanning electron microscope (SEM) images were carried out using a FEI DualBeam Helios NanoLab 600i.
References
Wu, M. P. S. Black-glazed Jian ware and tea drinking in the Song Dynasty. Vol. 29 22–31 (Orientations, 1998).
Li, W., Luo, H., Li, J., Li, J. & Guo, J. Studies on the microstructure of the black-glazed bowl sherds excavated from the Jian kiln site of ancient China. Ceramics International 34, 1473–1480, 10.1016/j.ceramint.2007.04.004 (2008).
Mowry, R. D., Farrell, E., Rousmaniere, N. C. & Arthur, M. Hare's Fur, Tortoiseshell and Partridge Feathers: Chinese Brown-and Black-Glazed Ceramics, 400–1400. (Harvard University Art Museums, 1996).
Wood, N. Chinese glazes: their origins, chemistry and recreation. (Univ of Pennsylvania Press., 1999).
Chen, X., Chen, S., Huang, R., Zhou, X. & Ran, M. A Study on Song Dynasty Jian Bowl (in Chinese). Chinese Ceramics 1, 58–66 (1983).
Ling, Z. D. A Preliminary Study of the Ancient “TIAN-MU” (TENMOKU) of Our Country (in Chinese). J. Chinese Ceramics Society 7, 190 (1979).
Machala, L., Tuček, J. & Zbořil, R. Polymorphous Transformations of Nanometric Iron(III) Oxide: A Review. Chem.Mater. 23, 3255–3272, 10.1021/cm200397g (2011).
Tuček, J., Zbořil, R., Namai, A. & Ohkoshi, S.-i. ε-Fe2O3: An Advanced Nanomaterial Exhibiting Giant Coercive Field, Millimeter-Wave Ferromagnetic Resonance and Magnetoelectric Coupling. Chem.Mater. 22, 6483–6505, 10.1021/cm101967h (2010).
Tronc, E., Chanéac, C. & Jolivet, J. P. Structural and Magnetic Characterization of ε-Fe2O3 . J. Solid State Chem. 139, 93–104, 10.1006/jssc.1998.7817 (1998).
Leon, Y. et al. Micro-Raman investigation of terra sigillata slips: a comparative study of central Italian and southern Gaul productions. J. Raman Spectrosc. 41, 1550–1555, 10.1002/jrs.2678 (2010).
Zoppi, A., Lofrumento, C., Castellucci, E. M., Dejoie, C. & Sciau, P. Micro-Raman study of aluminium-bearing hematite from the slip of Gaul sigillata wares. J. Raman Spectrosc. 37, 1131–1138, 10.1002/jrs.1597 (2006).
Forestier, H. & Guiot-Guillain, G. New ferromagnetic variety of ferric oxide. C. R. Acad. Sci. (Paris) 199, 720 (1934).
Schrader, R. & Büttner, G. Eine neue Eisen(III)-oxidphase: ε-Fe2O3 . Zeitschrift für anorganische und allgemeine Chemie 320, 220–234, 10.1002/zaac.19633200503 (1963).
Kelm, K. & Mader, W. Synthesis and Structural Analysis of ε-Fe2O3 . Zeitschrift für anorganische und allgemeine Chemie 631, 2383–2389, 10.1002/zaac.200500283 (2005).
Zboril, R., Mashlan, M. & Petridis, D. Iron(III) Oxides from Thermal ProcessesSynthesis, Structural and Magnetic Properties, Mössbauer Spectroscopy Characterization and Applications. Chem.Mater. 14, 969–982, 10.1021/cm0111074 (2002).
Gich, M. et al. High- and Low-Temperature Crystal and Magnetic Structures of ε-Fe2O3 and Their Correlation to Its Magnetic Properties. Chem.Mater. 18, 3889–3897, 10.1021/cm060993l (2006).
Jin, J., Ohkoshi, S. & Hashimoto, K. Giant Coercive Field of Nanometer- Sized Iron Oxide. Adv. Mater. 16, 48–51, 10.1002/adma.200305297 (2004).
Namai, A. et al. Synthesis of an Electromagnetic Wave Absorber for High-Speed Wireless Communication. J. Am. Chem. Soc. 131, 1170–1173, 10.1021/ja807943v (2009).
Ohkoshi, S.-i. et al. A Millimeter-Wave Absorber Based on Gallium-Substituted ε-Iron Oxide Nanomagnets. Angew. Chem., Int. Ed. 46, 8392–8395, 10.1002/anie.200703010 (2007).
Gich, M. et al. Magnetoelectric coupling in ε-Fe2O3 nanoparticles. Nanotechnology 17, 687 (2006).
Ohkoshi, S.-i., Sakurai, S., Jin, J. & Hashimoto, K. The addition effects of alkaline earth ions in the chemical synthesis of ε-Fe2O3 nanocrystals that exhibit a huge coercive field. J. Appl. Phys. 97, 10K312, 10.1063/1.1855615 (2005).
Kurmoo, M. et al. Formation of Nanoparticles of ε-Fe2O3 from Yttrium Iron Garnet in a Silica Matrix: An Unusually Hard Magnet with a Morin-Like Transition below 150 K. Chem.Mater. 17, 1106–1114, 10.1021/cm0482838 (2005).
Ding, Y., Morber, J. R., Snyder, R. L. & Wang, Z. L. Nanowire Structural Evolution from Fe3O4 to ε-Fe2O3 . Adv. Funct. Mater. 17, 1172–1178, 10.1002/adfm.200601024 (2007).
Gich, M. et al. Epitaxial stabilization of ε-Fe2O3 (00l) thin films on SrTiO3 (111). Appl. Phys. Lett. 96, 112508, 10.1063/1.3360217 (2010).
Gich, M. et al. Stabilization of metastable phases in spatially restricted fields: the case of the Fe2O3 polymorphs. Faraday Discuss. 136, 345–354, 10.1039/B616097B (2007).
Ninjbadgar, T., Yamamoto, S. & Takano, M. Thermal properties of the γ-Fe2O3/poly(methyl methacrylate) core/shell nanoparticles. Solid State Sci. 7, 33–36, 10.1016/j.solidstatesciences. 2004.10.030 (2005).
Sakurai, S., Namai, A., Hashimoto, K. & Ohkoshi, S.-i. First Observation of Phase Transformation of All Four Fe2O3 Phases (γ → ε → β → α-Phase). J. Am. Chem. Soc. 131, 18299–18303, 10.1021/ja9046069 (2009).
Popovici, M. et al. Optimized Synthesis of the Elusive ε-Fe2O3 Phase via Sol−Gel Chemistry. Chem.Mater. 16, 5542–5548, 10.1021/cm048628m (2004).
Kusano, Y. et al. Epitaxial Growth of ε-Fe2O3 on Mullite Found through Studies on a Traditional Japanese Stoneware. Chem.Mater. 20, 151–156, 10.1021/cm7023247 (2008).
Laurent, S. et al. Magnetic Iron Oxide Nanoparticles: Synthesis, Stabilization, Vectorization, Physicochemical Characterizations and Biological Applications. Chem. Rev. 108, 2064–2110, 10.1021/cr068445e (2008).
Kusano, Y. et al. Science in the Art of the Master Bizen Potter. Acc. Chem. Res. 43, 906–915, 10.1021/ar9001872 (2010).
Wilke, M., Farges, F., Petit, P.-E., Brown, G. E. & Martin, F. Oxidation state and coordination of Fe in minerals: An Fe K-XANES spectroscopic study. Am. Mineral. 86, 714–730 (2001).
Dejoie, C. et al. Revisiting Maya Blue and Designing Hybrid Pigments by Archaeomimetism. arXiv:1007.0818v1 [cond-mat.mtrl-sci] (2010).
Kraft, S., Stümpel, J., Becker, P. & Kuetgens, U. High resolution x-ray absorption spectroscopy with absolute energy calibration for the determination of absorption edge energies. Rev. Sci. Instrum. 67, 681–687, 10.1063/1.1146657 (1996).
Webb, S. (Stanford Synchrotron Radiation Laboratory, Melo Park, CA., 2006).
Kunz, M. et al. A dedicated superbend x-ray microdiffraction beamline for materials, geo- and environmental sciences at the advanced light source. Rev. Sci. Instrum. 80, 035108, 10.1063/1. 3096295 (2009).
Tamura, N. in Microdiffraction Analysis of Local and Near Surface Hierarchical Organization of Defects [Barabash, R. & Ice, G (ed.)] (Imperial College Press, in the press, 2014).
Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Physica B: Condensed Matter 192, 55–69, 10.1016/0921-4526(93)90108-I (1993).
Ayache, J., Beaunier, L., Boumendil, J., Ehret, G. G. & Laub, D. Guide de préparation des échantillons pour la microscopie électronique en transmission. Vol. 2 (PUSE, Saint-Etienne, 2007).
Acknowledgements
This work is supported by key project of Natural Science Foundation of China (No. 51232008). The authors would like to thank Robin Cours, Sébastien Joulié, Sébastien Moyano, Frédéric Neumayer and Nicolas Ratel-Ramond from technical Poles of CEMES Lab for support during the experimental measurements. The Advanced Light Source at the Lawrence Berkeley National Laboratory is supported by the Office of Science, Office of Basic Energy Sciences, Scientific User Facilities Division of the US Department of Energy under contract No. DE-AC02-05CH11231. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. Z.L. acknowledges the support of National Natural Science Foundation of China under Contract No. 11227902.
Author information
Authors and Affiliations
Contributions
C.D., W.D.L. and Z.L. conceived the experimental research; C.D., Z.L., N.T., P. S. and M.K., Performed XRD measurements; C.D., Z.L., A.M. and K.C. performed μ-XRF and μ-XANES measurements; P.S. conducted Micro Raman spectroscopy experiments; L.N. and P.S. carried out TEM, EDX and SEM experiments; W.D.L. and H.J.L. processed and provided samples for this study. C.D. and Z.L. wrote the manuscript with contribution from all authors; everyone participated in discussions and analysis of the results.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supporting Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Dejoie, C., Sciau, P., Li, W. et al. Learning from the past: Rare ε-Fe2O3 in the ancient black-glazed Jian (Tenmoku) wares. Sci Rep 4, 4941 (2014). https://doi.org/10.1038/srep04941
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04941
This article is cited by
-
The earliest known artificial synthesized ε-Fe2O3 in the Deqing Kiln ceramic ware of Tang Dynasty
Heritage Science (2023)
-
Characterizing ancient materials
Nature Materials (2023)
-
The microstructure of multicolor hare’s fur glaze: the correlation between morphological and compositional characteristics and glaze color
Heritage Science (2021)
-
Stability and nature of the volume collapse of ε-Fe2O3 under extreme conditions
Nature Communications (2018)
-
Epitaxially stabilized thin films of ε-Fe2O3 (001) grown on YSZ (100)
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
