
Abstract
Advances in the field of Multipotent Mesenchymal Stromal cell (MSC) biology have demonstrated that MSCs can improve disease outcome when ‘activated’ to exert immunomodulatory effects. However, the precise mechanisms modulating MSC-immune cells interactions remain largely elusive. In here, we activated MSC based on a recent polarization paradigm, in which MSCs can be polarized towards a pro- or anti-inflammatory phenotype depending on the Toll-like receptor stimulated, to dissect the mechanisms through which MSCs physically interact with and modulate leukocytes in this context. Our data show that MSCs activated through the Toll-like receptor (TLR) 4 pathway increased VCAM-1 and ICAM-1 dependent binding of leukocytes. On the other hand, TLR3 stimulation strongly increases leukocytes affinity to MSC comparatively, through the formation of cable-like hyaluronic acid structures. In addition, TLR4 activation elicited secretion of pro-inflammatory mediators by MSCs, whereas TLR3-activated MSCs displayed a milder pro-inflammatory phenotype, similar to inactivated MSCs. However, the differently activated MSCs maintained their ability to suppress leukocyte activation at similar levels in our in vitro model and this immunomodulatory property was shown here to be partially mediated by prostaglandin. These results reinforce the concept that alternate activation profiles control MSC responses and may impact the therapeutic use of MSCs.
Similar content being viewed by others
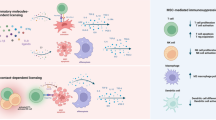


Introduction
Advances in the Mesenchymal Stem cell (MSC) field have resulted in the concept that MSCs react dynamically to the surrounding microenvironment with potent immunomodulatory effects1,2,3,4. Recent studies have shown that MSCs, like immune cells, can respond to different ‘danger’ signals that are generated at injured sites as result of inflammation, even in cases when inflammation occurs aseptically, as in the case of ischemia and autoimmune diseases5. MSCs accomplish this feature by expressing different Toll-like receptors (TLR), whose activation leads to significant cellular changes on MSCs6,7,8. Based on these observations, a recent MSC1/MSC2 polarization paradigm relying on TLR4/TLR3 priming has been suggested, in which MSC1s displays a pro-inflammatory phenotype, with secretion of inflammatory mediators such as IL-6 and IL-8, while MSC2s secretes anti-inflammatory mediators like IP-10 and IL-1RA and can inhibit T lymphocyte proliferation through expression of PGE2 and IDO9.
Noteworthy, many immunomodulatory factors expressed by MSCs, including PGE210,11, IDO12 and NO13 require close proximity for effectiveness. Therefore it is not surprising that, when exposed to inflammatory cytokines, MSCs express known adhesion molecules such as VCAM-1 and ICAM-113, which enables MSCs to sequester and modulate immune cells. Interestingly, reports involving smooth muscle cells, another cell of mesenchymal origin, describe the deposition of Hyaluronic Acid (HA) structures on the cell body following TLR3 stimulation by Poly (I:C). Classically, leukocyte adhesion is mediated by interactions between integrins (CD11/CD18) expressed on leukocytes and adhesion molecules (such as VCAM-1 and ICAM-1)14. However, smooth muscle cells that accumulated HA formed cable-like structures that significantly increased smooth muscle cells adhesiveness towards mononuclear leukocytes, a process that was implicated in the progression of inflammatory bowel disease15,16. Although functional myogenic differentiation of MSCs is thought to constitute a rare event17, in here we sought to further determine the impact of MSC polarization on leukocyte-MSC interaction and inflammatory responses based on the hypothesis that activation of bone marrow-derived human MSCs with Poly (I:C) would lead to HA accumulation on MSCs.
Results
TLR3 activated MSCs bind more leukocytes than TLR4-α
Based on the hypothesis that the polarization regimens would differently affect MSC-leukocyte interactions, we investigated the effects of TLR4 or TLR3 activation on the adhesion properties of MSCs and leukocytes. It is known that upon activation with inflammatory cytokines (IL-1, TNF-α and IFN-γ), MSCs express adhesion molecules like ICAM-1 and VCAM-1 to increasingly bind leukocytes18. However, the effects of TLR3 and TLR4 activation on the binding property of MSCs are currently unknown. To discern net changes in leukocyte adhesion, we activated adherent cultures of MSC with lipopolysaccharide (LPS) to activate TLR4 signaling (L-MSC), PIC to activate TLR3 pathway in MSC (P-MSC) or a media control (N-MSC). We then introduced non-adherent fluorescently-labeled leukocytes of various origins (PBMCs or monocytic leukemia cell lines THP-1 and U937) to the activated MSC and incubated the co-culture for 1 h at 4°C to prohibit any physiological changes initiated by cell-cell contact. Unbound cells were then washed away and the remaining mix of bound leukocytes and MSC were lysed and red fluorescence was used to count leukocytes compared to a standard curve.
Overall, binding of PBMCs, THP-1 and U937 cells to the P-MSCs was increased when compared to N-MSCs at 24 h after MSC activation (Fig. 1a–c). Among the cell types tested, U937 cells bound more efficiently to P-MSCs when compared to PBMCs or THP-1 cells (2 × 105 versus 6 × 104 and 6.3 × 104 respectively) (Fig. 1a–c). Sufficient activation time was critical for strong U937 cell adhesion depended on activation time, as 6 h of activation did not significantly alter leukocyte binding within the MSC groups (data not shown). After 24 h of activation more U937 cells bound to L-MSC (1.01 × 105) when compared to N-MSCs (4.42 × 104), however, P-MSC bound most U937 cells (2.01 × 105) (Fig. 1c). Therefore, all subsequent adhesion assays were performed using U937 cells and a 24 h activation period to study the effects of activation on MSC-leukocyte binding mechanics. Noteworthy, this effect was not due to changes in MSC proliferation or viability as a result of activation, as the number of MSCs remained constant across treatment groups (Fig. 1d).

Mononuclear leukocytes differentially bind activated MSCs.
MSCs (2 × 104 cells/well, n = 6) were activated with Poly (I:C) (P-MSCs) or LPS (L-MSCs) for 24 h in 24 well plates and fluorescently-labeled bound (a) PBMCs, (b) THP-1 were allowed to bind for 1 h at 4°C. (c) MSCs (2 × 104 cells/well, n = 6) were activated with Poly (I:C) (P-MSCs) or LPS (L-MSCs) for 24 h in 24 well plates and fluorescently-labeled bound U937 cells (5 × 105 cells/well) were allowed to bind for 1 h at 4°C. Plates were washed and bound cells were quantified based on standard curves of known number of cells. (d) MSCs were plated at 5 × 103 cells/well (n = 3) in 96 well plates and changes in MSC proliferation were assessed 30 h after activation with an MTT assay. No changes in cell number were observed due to activation. Values are mean ± SD. **p < 0.01 by ANOVA followed by Tukey's test for multiple comparisons.
Different MSC activation cues result in different binding mechanisms
Previous studies have shown that U937 cells bind to several cell types, including activated arterial and microvascular endothelial cells, using the adhesion molecule VCAM-119,20. In addition, a recent study has shown that only VCAM-1, but not L-selectin, ICAM-1 or VLA-4, was upregulated by TNF-α treatment in MSCs21 and this effect has been associated with MSC's ability to adhere to endothelial cells22. Therefore, we sought to determine the role of adhesion molecules in the binding of U937 to activated MSC populations.
Our results show that MSCs activated by TLR4 increase surface VCAM-1 and ICAM-1 compared to TLR3. In a representative data set, flow cytometry analysis found that VCAM-1 was expressed in 16.5% of L-MSCs and 10% of P-MSCs, compared to 4% on N-MSCs (Fig. 2a). ICAM-1 was expressed on 35.2% on L-MSC and 19.4% in P-MSC, compared to 1.9% in N-MSCs (Fig. 2a). Similar changes were observed with two other MSC donors (data not shown). As expected, the presence of VCAM-1 and ICAM-1 binding inhibitors or a combination of both during the adhesion assay resulted in a significant reduction of U937 cell binding to L-MSC (Fig. 2b). The specific inhibition of P-selectin with KF 38789 had no significant effects on adhesion (data not shown). These treatments did not effect U937 binding to P-MSC (Fig. 2b). This strongly indicates that although VCAM-1 and ICAM-1 are involved in the U937 binding to L-MSCs, it is not predominantly responsible for the increased adhesion seen in P-MSCs.

Activated MSCs utilize different leukocyte binding mechanisms.
MSCs (105 cells/well, n = 6) were activated, as before, in 24 well plates for 24 h. (a) Cells were harvested and stained with anti-ICAM-1 and anti VCAM-1 for detection of surface expression by flow cytometry. ICAM-1 and VCAM-1 expression and side scatter (SSC) were used to determine positive expression compared to unstained cells (not shown). Data are representative of 2 independent experiments with 3 MSC donors. (b) Fluorescent-labeled U937 (5 × 105 cells/well) were allowed to bind for 1 h at 4°C to activated MSCs. In some experiments, U937 cells were incubated with specific inhibitors of LFA-1 (50 μM) and VLA-4 (40 nM) prior to the binding phase for the inhibition of ICAM-1- and VCAM-1-mediated adhesion respectively. Plates were washed and quantification of bound cell was based on standard curves of known number of cells. Legends, C: no treatment; I: ICAM-1 binding inhibition, V: VCAM-1 binding inhibition. Values are mean ± SD. *p < 0.05, n.s. not significant, by ANOVA followed by Tukey's test for multiple comparisons.
Hyaluronic acid is responsible for the affinity of leukocytes to P-MSCs
As previously mentioned, smooth muscle cells can be activated specifically by TLR3 agonists to create HA cables with high affinity for mononuclear leukocytes15,16. We investigated if HA plays a role in the adhesiveness of TLR3 stimulated MSC. MSCs activated with PIC were fixed and HA was visualized using fluorescently conjugated HA binding protein (HABP). Our results show that P-MSCs have a significant deposition of HA structures outside the cell in cable-like structures that span multiple cell lengths (Fig. 3a). A common pattern of high intensity, punctate, perinuclear staining is shared by N-MSCs, L-MSCs. This staining profile was highly consistent between MSCs derived from several different donors (Supplemental Fig. 1).

Differential HA deposition following MSC activation.
MSCs (2 × 104 cells/well, n = 6) were activated, as before, in 24-well plates. (a) Representative HA staining of N-, L- or P-MSCs detected by immunofluorescence microscopy using a biotin-conjugated HA binding protein followed by a FITC-conjugated streptavidin. (b) Representative MSC-leukocyte interactions using fluorescence microscopy analysis following adhesion assay. Leukocytes were pre-stained with CellTracker dye (c) Effects of Poly (I:C) dose, exposure time and the presence of serum on the binding properties of P-MSC. (d) Testicular hyaluronidase was added to plates prior to the adhesion assay. Quantification of bound U937 cells was based on standard curves of known number of cells. Values are mean ± SD. *p < 0.05, **p < 0.01, n.s. not significant, by ANOVA followed by Tukey's test for multiple comparisons.
To visualize the interactions between HA and U937 cells, red fluorescently labeled U937 cells were added to activated MSC cultures, allowed to bind and then stained for HA and visualized by fluorescence microscopy as above. As expected, fluorescent-labeled U937 cells were found often associated to HA structures on P-MSCs (Fig. 3b) whereas U937 primarily bound directly to MSC cell body in N-MSC and L-MSC in reduced numbers. Further characterization of the interaction between P-MSCs and U937 through HA revealed that this phenomenon is dependent on Poly (I:C) dose and time of exposure and the presence of serum in culture media (Fig. 3c).
The direct contribution of HA to retain U937 cells was then quantified by enzymatically degrading HA on activated MSCs using hyaluronidase (100 μg/ml) prior to the start of the adhesion assay. We observed a significant 87.5% decrease in U937 binding to P-MSCs, demonstrating that HA is intimately involved in the binding mechanism triggered by TLR3 activation (Fig. 3d). Moreover, the same hyaluronidase treatment only reduced binding by 14.51% in L-MSC cultures. This indicates that P-MSCs almost exclusively use HA to bind leukocytes, while L-MSC mainly utilizes VCAM-1 and ICAM-1 to increase adhesion.
Deposition of HA on P-MSCs is not due to changes in HAS and TSG-6 gene expression
Stimuli such as growth factors and cytokines can regulate the expression of HA synthase (HAS) isoforms and TSG-6 in vitro23,24. To address potential sources of the observed HA, we sought to determine if the different activation pathways altered expression of any of the three HAS or TSG-6, a known HA crosslinker expressed by MSCs24. MSCs were activated and RNA collected at 8 h for assessment of TSG-6 and HAS 1, 2 and 3 gene expressions.
Our results show that changes to HAS and TSG-6 expression do not correlate with the changes observed in HA between the activated MSC cultures. There was a slight decrease in the gene expression of HAS1 and HAS2 and no difference in TSG-6 expression on P-MSCs compared to N-MSCs (Fig. 4). A similar gene expression pattern between N- and P-MSCs was observed at 4 h and 18 h (data not shown). In L-MSC, expression of HAS1 and TSG-6 increased while HAS2 decreased (Fig. 4). HAS3 expression was not detected in any of the MSC samples (data not shown). Despite a recent study indicating that the ribosomal house-keeping gene RPL13A displayed superior stability when compared to beta-actin or GAPDH during MSCs differentiation25, we did not observe significant changes on GAPDH expression when compared to RPL13A upon MSC activation (Supplemental Fig. 3). Collectively, these data indicate the deposition of HA is not due to changes in HAS and TSG-6 expression, suggesting another mechanism is responsible for the increased HA deposition from TLR3 activation.

HA accumulation does not correlate with changes in HAS or TSG-6 gene expression on MSCs.
MSCs (1 × 105 cells/well, n = 3) were activated as before in 6-well plates. Gene expression for HAS1, HAS2, or TSG-6 was assessed by Real-Time RT-PCR at 8 h.
Despite altering secretome profile, different activators do not affect immunomodulatory capacity of MSCs
Previous studies indicate MSCs have the ability to secrete several immune mediators depending on the activation of discrete pathways, often resulting in polarizing effects on immune responses9,26. To further investigate how different activators could affect the ability of MSCs to modulate immune responses, we first measured changes to the MSC secretome following TLR activation using a cytokine detection array (Fig. 5a). Of interest, the concentration of a number of cytokines commonly considered pro-inflammatory (CXCL1, IL-6, IL-8 and CCL2) were increased when MSCs were activated with LPS. In contrast, after Poly (I:C) exposure, there was only a modest increase in IL-6 and MIF. Levels of CXCL1, IL-8 and CCL2 did not differ between N-MSCs and MSCs activated with Poly (I:C), confirming that TLR3 activation leads to a less inflammatory phenotype.

Effects of activation on the immunosuppressive properties of MSCs.
(a) Cytokine and chemokine levels in the supernatant after MSCs (1 × 105 cells/plate, n = 3) were activated for 18 h in 10 cm2 plates. Values are calculated based on relative densitometry following manufacturer's protocol. (b) TNF-α accumulation in the supernatant after N-, L- or P-MSCs (104 cells/well, n = 3) were co-cultured with murine splenocytes (2 × 105 cells/well) with LPS for 18 h in 96 wells as quantified by ELISA following manufacturer's protocol. In some experiments, splenocytes were allowed to adhere to MSCs for 1 h, or indomethacin was added to the co-culture prior to activation of splenocytes. Values are mean ± SD. *p < 0.05, **p < 0.01, n.s. not significant, by ANOVA followed by Tukey's test for multiple comparisons. (c) TNF-α accumulation in the supernatant after N-, L- or P-MSCs (104 cells/well, n = 3) were co-cultured with murine splenocytes (2 × 105 cells/well) with LPS for 18 h in 96 wells in the presence of hyaluronidase.
Finally, we compared the immunosuppressive properties of MSC among our activation regimens. Both MSC groups (L- and P-MSCs) inhibited inflammatory responses of splenocytes exposed to LPS or ConcavalinA in vitro comparable to the effects seen with N-MSCs (Fig. 5b and Supplemental Fig. 2). The anti-inflammatory effects were abrogated by the addition of Indomethacin (Fig. 5b and Supplemental Fig. 2), suggesting the involvement of PGE2 on the immunosuppressive effects of MSCs as previously described10,27, but not exclusively linked to an activation pathway tested here. Interestingly, when splenocytes were allowed to adhere to activated MSCs for 1 h at 4°C prior to activation, there was an enhanced anti-inflammatory effect (Fig. 5b). However, no difference was observed when N-, L- or P-MSC were treated with hyaluronidase (Fig. 5c), suggesting HA does not directly suppress inflammatory responses in this in vitro model. These data indicate that activated MSCs are similarly capable of modulating inflammatory responses despite differences in cytokine secretion profile through prostaglandin-related mechanisms and that HA does not play a role in this process.
Discussion
Our results demonstrate that MSCs respond differently to TLR3 and TLR4 activation. TLR3 pre-activation significantly increases the number of leukocytes that bind to MSCs, predominantly through interacting with HA that accumulates into extended structures around P-MSCs. Conversely, MSCs activated by TLR4 displayed punctate HA accumulation that did not associate with the smaller increase in leukocyte binding, which was primarily caused by surface expression of VCAM-1 and ICAM-1.
HA is a linear, uniformly repetitive polysaccharide that, unlike other glycosaminoglycans (GAGs) whose size is relatively smaller (15–20 KDa), is usually found as a megadalton molecule and extended lengths of 2–25 μm. The specific size of HA is determined during synthesis by which of the three transmembrane glycosyltransferase (HAS 1, 2 or 3) is expressed. HAS1 and HAS2 are reported to express high molecular mass HA (>500 KDa), whereas HAS3 is associated with the synthesis of short molecular weight forms of HA (<300 KDa)28. In our study, the deposit of HA structures observed here did not correlate with changes in the gene expression of HAS1, HAS2. Moreover, HAS3 expression levels could not be detected on MSCs by PCR in our study. A previous study reported expression of HAS1, HAS2 and HAS3 decreased as MSC passage number increased, vanishing when MSCs reached senescence29. This decrease in HAS expression correlated with a decreased in VCAM-1 expression, which was rescued by exogenous HA29. Unlike this observation, in our study low passage MSCs displayed very low levels of VCAM-1 expression. Upon activation, our MSCs had increased VCAM-1 and ICAM-1 expression, but no changes corresponding to HAS expression as described above. Future studies, such as knockdown and overexpression experiments, are needed to determine the specific mechanisms by which Poly (I:C) and LPS directly change MSC cell surface molecules and the surrounding ECM.
In addition, the presence of Inter-alpha inhibitor (IαI), a protease inhibitor present in the serum, has been implicated with the assembly of HA cables in smooth muscle cells16. In our study, MSCs activated with Poly (I:C) were able to assemble HA cable structures in the absence of serum. However, serum-free HA cables looked thinner (data not shown) and serum was an essential component for the binding of U937 to the P-MSCs. The variations between other reports and the results described here may reflect sampling problems in obtaining MSCs with bone marrow aspirates, intrinsic donor differences in MSCs, usage of different passage cells, or different growth conditions30,31.
The different activation responses by MSCs are also reflected in changes to their secretome, where TLR4 activation induce the expression of several “proinflammatory” cytokines while TLR3 activation results in a mild activation state similar to normal MSCs. Despite this difference, all the MSCs were capable of strongly inhibiting the expression of TNF-α by activated splenocytes through mechanisms related to prostaglandin E2 (PGE2). PGE2 is a metabolite of arachidonic acid metabolism created by COX2 in response to inflammation and has been reported to be integral to the immunosuppression exerted by MSCs10,11,32,33. These results are partially in accordance with a previous study suggesting an immunosuppressive phenotype is generated in MSCs activated with TLR3 versus a pro-inflammatory phenotype from TLR4 activation9. Our study differs in that activation did not cause any functional change to MSC's ability to inhibit inflammation in LPS-stimulated splenocytes. This observation is supported by studies exploring the use of MSC in sepsis10,34, where MSCs exposed to TLR4 agonists were still capable of decreasing systemic inflammation.
Recent evidence indicates that MSC can use hyaluronic acid (HA) as a mechanism of immune modulation. MSCs exposed to inflammation are activated to express the anti-inflammatory protein TSG-6, whose interactions with HA have been associated with the immunomodulatory functions exerted by MSCs in animal models of peritonitis35 and diabetes36. The involvement of HA in many fundamental physiological and pathological processes including embryogenesis, cell adhesion, inflammation, multi-drug resistance and even malignant transformation37 suggests modulation of HA could be targeted as a potential therapeutic mechanism. Noteworthy in our study, the immunosuppressive effect exerted by MSCs was increased when leukocytes were allowed to adhere to activated MSCs prior to the addition of inflammatory stimuli, suggesting activated MSCs can ‘educate’ immune cells towards a non-inflammatory response. However, the fact that hyaluronidase treatment did not alter the immunosuppressive properties of MSCs suggests that HA expressed by MSCs does not directly inhibit inflammatory responses in our in vitro model.
Studies indicate that high molecular HA promotes immunotolerance by providing a co-stimulatory signal and increasing Foxp3 expression on T-cells38,39. Moreover, high molecular HA inhibits activation and proliferation of glial cells, decreasing scar formation in models of spinal cord and traumatic brain injury40,41. Scar formation represents a major obstacle for neuroregeneration, since it inhibits the regeneration of axons across the damaged area. In yet another study, topical Poly (I:C) administration was shown to accelerate skin wound healing by recruiting myeloid cells that were shown to acquire an anti-inflammatory phenotype42. Hence, it would be interesting to assess the role of MSC-derived HA on such models. Although studies have shown that TLR3 and TLR4 activation decreases MSC migration9, the ability to trap leukocytes itself, could increase MSC efficacy by creating the close proximity required by many of the therapeutic factors expressed by MSCs and modulate circulating leukocytes.
MSC pre-conditioning is being considered for the treatment of many conditions, including cardiovascular diseases43, diabetes44,45, multiple sclerosis46 and cancer26,30. The concept of pre-conditioning aims to enhance MSC responses and expression of therapeutic factors47. Once viewed only as a ‘filling material’ in the interstitial space, HA is now appreciated as a multifunctional molecule with important roles in various biological processes37. Hence, the observations here, combined with the realization that RNA from necrotic/apoptotic cells can trigger TLR3 responses48,49 and TNF-α seem to replicate the inflammatory and adhesive effects of LPS (data not shown), could significantly impact the usefulness of MSCs in many disease models. Future studies will better describe the role of HA in MSC biology and increase our understanding of alternate MSC activation states.
Methods
Cell culture
We obtained frozen vials of pre-characterized human MSCs isolated from the bone marrow of healthy donors from the Center for the Preparation and Distribution of Adult Stem Cells (NIH/NCRR grant P40 RR 17447-06) - http://medicine.tamhsc.edu/irm/msc-distribution.html. For Supplemental Figure 1, we also isolated MSCs from commercially available fresh bone marrow aspirates (AllCells, Emeryville, CA) according to the protocols previously established using density centrifugation and plastic adherence50. All cultures of MSCs were screened for typical MSC spindle-like morphology and growth kinetics. MSCs were expanded by thawing a frozen vial of 106 passage 2 or 3 cells and plating at 200 cells/cm2 in 225 cm2 flasks with 50 ml of complete culture medium (CCM) that consisted of α-minimal essential medium (α-MEM; Life Technologies, Grand Island, NY), 17% fetal bovine serum (FBS; lot-selected for rapid growth of MSCs; Atlanta Biologicals, Norcross, GA), 100 units/ml penicillin (Life Technologies), 100 μg/ml streptomycin (Life Technologies) and 2 mM L-glutamine (Life Technologies). MSCs were incubated with medium replaced every 2 days until about 70% confluent. The medium was discarded, the cultures were washed with phosphate-buffered saline (PBS, Life Technologies) and the adherent cells harvested with 0.25% trypsin/1 mM EDTA (Life Technologies) for 2–3 min at 37°C and resuspended with fresh media for subsequent experiments.
The THP-1 and U937 human lymphoma cell line were purchased from ATCC (Rockville, MD) and cultured in suspension culture in complete RPMI media, consisting of RPMI (Life Technologies) supplemented with 10% heat-inactivated serum (heated at 65°C for 30 min), 100 units/ml penicillin (Life Technologies), 100 μg/ml streptomycin (Life Technologies) and 2 mM L-glutamine (Life Technologies). Cells were grown in 225 cm2 flasks at 2 × 105 cells/ml and split at a 1:5 ratio two times a week.
We isolated peripheral blood mononuclear cells following an IRB approved protocol (HSC-MS-10-0190) with informed consent. Briefly, blood from healthy donors was diluted 1:2 with PBS and layered on top of Ficoll-Paque and centrifuged at 400 g for 30 min at room temperature. The mononuclear cell layer was carefully collected and rinsed twice with PBS and PBMCs were suspended in complete RPMI media.
MSC activation
Unless otherwise noted, MSCs were treated with Poly (I:C) (R&D Systems, Minneapolis, MN, 20 μg/ml) or LPS (Invivogen, San Diego, CA, 250 ng/ml) or media alone as a negative control, which consisted of CCM with FBS concentration reduced to 2% (2%CCM) and incubated for 24 h prior to analysis, unless otherwise specified.
VCAM-1 and ICAM-1 expression on MSCs
We plated MSCs in 6-well plates at 105 cells per well and allowed to adhere for 6 h. MSC were activated in 2% CCM, harvested and labeled with mouse anti-human VCAM-1 (1:100, clone 51-10C9, BD Biosciences, San Jose, CA) and mouse anti-human ICAM-1 antibodies (1:100, clone HA58, Biolegend, CA) for 15 min at 4°C. Data was acquired on an LSRII flow cytometer and analyzed with BD FACSDiva software (BD Biosciences, San Jose, CA). Unstained cells were used to set a CD106+ gate at the level of background fluorescence and approximately ten thousand singlet cells were analyzed per sample.
MSC proliferation
We activated MSCs (5 × 103 cells/well) in 96 well plates in 2% CCM for 30 h and 3-[4,5-dimethylthiazol-2yl]-2,5-diphenyl-tetrazolium bromide (MTT) assay (Life Technologies) was performed following manufacturer's protocol. Briefly, the MTT reagent was added (10 μl per well) and the plate incubated for 4 h to allow for intracellular reduction of the soluble yellow MTT to the insoluble purple formazan dye. Detergent reagent was added to each well to solubilize the formazan dye prior to measuring the absorbance of each sample in a microplate reader at 570 nm.
Adhesion assay
MSCs (2 × 104 cells/well) were plated in 24-well plates (Corning, Corning, NY) in 2% CCM and allowed to adhere for at least 6 h and then activated as above. U937 cells were labeled with CellTracker Red CMPTX (Life Technologies) following manufacturer's protocol. Briefly, U937 cells (2 × 106 cells/ml) were resuspended in pre-warmed CellTracker dye working solution (10 μM in serum–free RPMI) and incubated at 37°C. After 45 min, cells were resuspended in complete RPMI media. Following 30 min incubation at 37°C, cells were washed and resuspended at 106 cells/ml in complete RPMI media. Fluorescent-labeled U937 cells (5 × 105 cells/well) were allowed to bind to MSCs for 1 h at 4°C, as previously described16, to determine the extent to which activated MSCs could bind leukocytes without any significant physiological changes to the cells upon contact. Next, the plate was washed with cold PBS for removal of unbound cells and remaining cells were lysed with CyQUANT® Cell lysis buffer (Life Technologies) and 100 μl removed for fluorescence measurement. The number of bound cells/well was calculated based on a standard curve generated with a serial dilution of known numbers of fluorescent-labeled U937.
Adhesion inhibitor assays
To inhibit VCAM-1- and ICAM-1- dependent adhesion, we incubated U937 cells with selective, high affinity inhibitors of the VCAM-1 ligand VLA-4 (α4β1, 40 nM) and the ICAM-1 ligand LFA-1 (RWJ 50271, 50 μM) (both from Tocris Bioscience, MO) at 37°C for 15 min before the binding phase. To exclude the hyaluronic acid-dependent binding, we treated activated MSCs with hyaluronidase (100 μg/ml, MP Biomedicals, Santa Ana, CA) at 37°C for 10 min before adhesion, followed by washing of the cells and the binding phase.
Immunofluorescence
We grew MSCs on 4 or 8-well Lab-Tek slide chambers (Nalge Nunc Int, Roskilde, Denmark) in 2% CCM at 5 × 103 cells/well and allowed the cells to adhere for at least 6 h. MSCs were then activated with Poly (I:C) or LPS for 24 h and cells washed with PBS and fixed in an acid-formalin-ethanol solution (3.7% formaldehyde-PBS, 70% ethanol and 5% glacial acetic acid, all v/v)51. HA was detected using a biotin-conjugated HA binding protein (HABP) (4 μg/ml, Calbiochem, Billerica, MA) followed by FITC-conjugated streptavidin (1:1000, Biolegend, San Diego, CA). Coverslips were mounted with DAPI Fluoromount-G (Southern Biotech) to counterstain nuclei and slides were analyzed in a Nikon Eclipse TE 2000-U microscope (Nikon Instruments Inc., Melville, NY).
Real–Time RT-PCR
We plated MSCs (105 cells/well) in 6-well plates in 2% CCM and allowed the cells to adhere for at least 6 h. Following activation with Poly (I:C) or LPS for 8 h, RNA was isolated using RNAqueous (Life Technologies) following manufacturer's protocol. cDNA amplification and real-time PCR analysis was performed for 40 cycles on Step One Plus (Life Technologies). List of primers used: human HAS1 (Hs.PT.56a.965988), human HAS2 (Hs.PT.56a.40275566), human HAS3 (Hs.PT. 56a.2934680), human GAPDH (Hs.PT.39a.22214836), (all from IDT, Coralville, Iowa) and human TSG-6 (Hs01113602_m1) and RPL13A (Hs01926559_g1), both from Life Technologies.
Cytokine array
We plated MSCs (105 cells/plate) in 10 cm2 plates in 2% CCM and activated the cells as above for 18 h in the presence of Poly (I:C) (20 μg/ml), LPS (500 ng/ml) or TNF-α (20 ng/ml). Levels of cytokines, chemokines and acute-phase proteins from the supernatant were assessed using the Proteome Profiler Human Cytokine Array Kit, Panel (R&D Systems) following manufacturer's protocol.
Splenocyte activation
Mice and rats were purchased from Harlan (Indianapolis, IN, USA) for use in this study. The animals were housed on a 12 h light/dark cycle with ad libitum access to food and water. All protocols involving the use of animals were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (HSC-AWC-12-170 and HSC-AWC-13-065). After obtaining a fresh spleen from male C57BL/6 mice (20–30 g) or Sprague Dawley rats (250–300 g) under anaesthesia, the spleen was morselized by pushing it through a 70 μm mesh filter in order to exclude all connective tissue. The remaining material was suspended in ice cold PBS and centrifuged at 400 g for 8 minutes. Next the supernatant was removed and the sample suspended in 5 mL of red blood cell lysis buffer (Sigma-Aldrich, St. Louis, MO) and allowed to incubate on ice for 5 min. Subsequently, the sample was diluted with 5 ml of PBS and centrifuged at 400 g for 8 minutes. The supernatant was again removed and the pellet suspended in phenol free RPMI with 10% FBS and titrated 8–10 times after which the sample was run through a 40 μm mesh filter to remove any clumps. The splenocytes were counted and checked for viability via Trypan blue exclusion. Splenocytes (2 × 106 cells/ml) were activated with Poly (I:C) or LPS, alone or in the presence of MSCs or previously activated-MSCs in 96 well plates in the presence or absence of Indomethacin (1 μM, Sigma). In some experiments, splenocytes were allowed to adhere to MSCs 1 h at 4°C prior to LPS treatment. Culture supernatant was collected 18 h after LPS treatment and the samples analysed utilizing a TNF-α ELISA kit (Biolegend) following manufacturer's protocol.
References
Lee, R. H., Oh, J. Y., Choi, H. & Bazhanov, N. Therapeutic factors secreted by mesenchymal stromal cells and tissue repair. J Cell Biochem 112, 3073–3078, 10.1002/jcb.23250 (2011).
Prockop, D. J., Kota, D. J., Bazhanov, N. & Reger, R. L. Evolving paradigms for repair of tissues by adult stem/progenitor cells (MSCs). J Cell Mol Med 14, 2190–2199, 10.1111/j.1582-4934.2010.01151.x (2010).
Prockop, D. J. & Oh, J. Y. Mesenchymal stem/stromal cells (MSCs): role as guardians of inflammation. Mol Ther 20, 14–20, 10.1038/mt.2011.211 (2012).
Uccelli, A. & Prockop, D. J. Why should mesenchymal stem cells (MSCs) cure autoimmune diseases? Curr Opin Immunol 22, 768–774, 10.1016/j.coi.2010.10.012 (2010).
Menezes, G. B., Mansur, D. S., McDonald, B., Kubes, P. & Teixeira, M. M. Sensing sterile injury: opportunities for pharmacological control. Pharmacol Ther 132, 204–214, 10.1016/j.pharmthera.2011.07.002 (2011).
Hwa Cho, H., Bae, Y. C. & Jung, J. S. Role of toll-like receptors on human adipose-derived stromal cells. Stem Cells 24, 2744–2752, 10.1634/stemcells.2006-0189 (2006).
Pevsner-Fischer, M. et al. Toll-like receptors and their ligands control mesenchymal stem cell functions. Blood 109, 1422–1432, 10.1182/blood-2006-06-028704 (2007).
Tomchuck, S. L. et al. Toll-like receptors on human mesenchymal stem cells drive their migration and immunomodulating responses. Stem Cells 26, 99–107, 10.1634/stemcells.2007-0563 (2008).
Waterman, R. S., Tomchuck, S. L., Henkle, S. L. & Betancourt, A. M. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS One 5, e10088, 10.1371/journal.pone.0010088 (2010).
Nemeth, K. et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med 15, 42–49, 10.1038/nm.1905 (2009).
Ylostalo, J. H., Bartosh, T. J., Coble, K. & Prockop, D. J. Human mesenchymal stem/stromal cells cultured as spheroids are self-activated to produce prostaglandin E2 that directs stimulated macrophages into an anti-inflammatory phenotype. Stem Cells 30, 2283–2296, 10.1002/stem.1191 (2012).
Meisel, R. et al. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood 103, 4619–4621, 10.1182/blood-2003-11-3909 (2004).
Ren, G. et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2, 141–150, 10.1016/j.stem.2007.11.014 (2008).
Langer, H. F. & Chavakis, T. Leukocyte-endothelial interactions in inflammation. J Cell Mol Med 13, 1211–1220, 10.1111/j.1582-4934.2009.00811.x (2009).
de La Motte, C. A., Hascall, V. C., Calabro, A., Yen-Lieberman, B. & Strong, S. A. Mononuclear leukocytes preferentially bind via CD44 to hyaluronan on human intestinal mucosal smooth muscle cells after virus infection or treatment with poly(I.C). J Biol Chem 274, 30747–30755 (1999).
de la Motte, C. A., Hascall, V. C., Drazba, J., Bandyopadhyay, S. K. & Strong, S. A. Mononuclear leukocytes bind to specific hyaluronan structures on colon mucosal smooth muscle cells treated with polyinosinic acid:polycytidylic acid: inter-alpha-trypsin inhibitor is crucial to structure and function. Am J Pathol 163, 121–133 (2003).
Siegel, G. et al. Bone marrow-derived human mesenchymal stem cells express cardiomyogenic proteins but do not exhibit functional cardiomyogenic differentiation potential. Stem Cells Dev 21, 2457–2470, 10.1089/scd.2011.0626 (2012).
Ren, G., Roberts, A. I. & Shi, Y. Adhesion molecules: key players in Mesenchymal stem cell-mediated immunosuppression. Cell Adh Migr 5, 20–22, 10.4161/cam.5.1.13491 (2011).
Huo, Y., Hafezi-Moghadam, A. & Ley, K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res 87, 153–159 (2000).
Solito, E., Romero, I. A., Marullo, S., Russo-Marie, F. & Weksler, B. B. Annexin 1 binds to U937 monocytic cells and inhibits their adhesion to microvascular endothelium: involvement of the alpha 4 beta 1 integrin. J Immunol 165, 1573–1581 (2000).
Xiao, Q. et al. TNF-alpha increases bone marrow mesenchymal stem cell migration to ischemic tissues. Cell Biochem Biophys 62, 409–414, 10.1007/s12013-011-9317-y (2012).
Uchibori, R. et al. NF-kappaB Activity Regulates Mesenchymal Stem Cell Accumulation at Tumor Sites. Cancer Res 73, 364–372, 10.1158/0008-5472.CAN-12-0088 (2013).
Jiang, D., Liang, J. & Noble, P. W. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol 23, 435–461, 10.1146/annurev.cellbio.23.090506.123337 (2007).
Lee, R. H. et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 5, 54–63, 10.1016/j.stem.2009.05.003 (2009).
Quiroz, F. G. et al. Housekeeping gene stability influences the quantification of osteogenic markers during stem cell differentiation to the osteogenic lineage. Cytotechnology 62, 109–120, 10.1007/s10616-010-9265-1 (2010).
Waterman, R. S., Henkle, S. L. & Betancourt, A. M. Mesenchymal stem cell 1 (MSC1)-based therapy attenuates tumor growth whereas MSC2-treatment promotes tumor growth and metastasis. PLoS One 7, e45590, 10.1371/journal.pone.0045590 (2012).
Bartosh, T. J. et al. Aggregation of human mesenchymal stromal cells (MSCs) into 3D spheroids enhances their antiinflammatory properties. Proc Natl Acad Sci U S A 107, 13724–13729, 10.1073/pnas.1008117107 (2010).
Lee, J. Y. & Spicer, A. P. Hyaluronan: a multifunctional, megaDalton, stealth molecule. Curr Opin Cell Biol 12, 581–586 (2000).
Jung, E. M. et al. Evidences for correlation between the reduced VCAM-1 expression and hyaluronan synthesis during cellular senescence of human mesenchymal stem cells. Biochem Biophys Res Commun 404, 463–469, 10.1016/j.bbrc.2010.12.003 (2011).
Lee, R. H., Yoon, N., Reneau, J. C. & Prockop, D. J. Preactivation of human MSCs with TNF-alpha enhances tumor-suppressive activity. Cell Stem Cell 11, 825–835, 10.1016/j.stem.2012.10.001 (2012).
Phinney, D. G. et al. Donor variation in the growth properties and osteogenic potential of human marrow stromal cells. J Cell Biochem 75, 424–436 (1999).
Aggarwal, S. & Pittenger, M. F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 105, 1815–1822, 10.1182/blood-2004-04-1559 (2005).
Corcione, A. et al. Human mesenchymal stem cells modulate B-cell functions. Blood 107, 367–372, 10.1182/blood-2005-07-2657 (2006).
Krasnodembskaya, A. et al. Human mesenchymal stem cells reduce mortality and bacteremia in gram-negative sepsis in mice in part by enhancing the phagocytic activity of blood monocytes. Am J Physiol Lung Cell Mol Physiol 302, L1003–1013, 10.1152/ajplung.00180.2011 (2012).
Choi, H., Lee, R. H., Bazhanov, N., Oh, J. Y. & Prockop, D. J. Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis by decreasing TLR2/NF-kappaB signaling in resident macrophages. Blood 118, 330–338, 10.1182/blood-2010-12-327353 (2011).
Kota, D. J., Wiggins, L. L., Yoon, N. & Lee, R. TSG-6 produced by hMSCs delays the onset of autoimmune diabetes by suppressing Th1 development and enhancing tolerogenicity. Diabetes, 10.2337/db12-0931 (2013).
Girish, K. S. & Kemparaju, K. The magic glue hyaluronan and its eraser hyaluronidase: a biological overview. Life Sci 80, 1921–1943, 10.1016/j.lfs.2007.02.037 (2007).
Bollyky, P. L. et al. Cutting edge: high molecular weight hyaluronan promotes the suppressive effects of CD4+CD25+ regulatory T cells. J Immunol 179, 744–747 (2007).
Bollyky, P. L. et al. ECM components guide IL-10 producing regulatory T-cell (TR1) induction from effector memory T-cell precursors. Proc Natl Acad Sci U S A 108, 7938–7943, 10.1073/pnas.1017360108 (2011).
Khaing, Z. Z. et al. High molecular weight hyaluronic acid limits astrocyte activation and scar formation after spinal cord injury. J Neural Eng 8, 046033, 10.1088/1741-2560/8/4/046033 (2011).
Lin, C. M. et al. Hyaluronic acid inhibits the glial scar formation after brain damage with tissue loss in rats. Surg Neurol 72 Suppl 2, S50–54, 10.1016/j.wneu.2009.09.004 (2009).
Lin, Q. et al. Toll-like receptor 3 ligand polyinosinic:polycytidylic acid promotes wound healing in human and murine skin. J Invest Dermatol 132, 2085–2092, 10.1038/jid.2012.120 (2012).
Ranganath, S. H., Levy, O., Inamdar, M. S. & Karp, J. M. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell 10, 244–258, 10.1016/j.stem.2012.02.005 (2012).
Davis, N. E., Hamilton, D. & Fontaine, M. J. Harnessing the immunomodulatory and tissue repair properties of mesenchymal stem cells to restore beta cell function. Curr Diab Rep 12, 612–622, 10.1007/s11892-012-0305-4 (2012).
Khan, M., Akhtar, S., Mohsin, S., S, N. K. & Riazuddin, S. Growth factor preconditioning increases the function of diabetes-impaired mesenchymal stem cells. Stem Cells Dev 20, 67–75, 10.1089/scd.2009.0397 (2011).
Darlington, P. J., Boivin, M. N. & Bar-Or, A. Harnessing the therapeutic potential of mesenchymal stem cells in multiple sclerosis. Expert Rev Neurother 11, 1295–1303, 10.1586/ern.11.113 (2011).
Mastri, M. et al. Activation of Toll-like receptor 3 amplifies mesenchymal stem cell trophic factors and enhances therapeutic potency. Am J Physiol Cell Physiol 303, C1021–1033, 10.1152/ajpcell.00191.2012 (2012).
Cavassani, K. A. et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med 205, 2609–2621, 10.1084/jem.20081370 (2008).
Kariko, K., Ni, H., Capodici, J., Lamphier, M. & Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 279, 12542–12550, 10.1074/jbc.M310175200 (2004).
Sekiya, I. et al. Expansion of human adult stem cells from bone marrow stroma: conditions that maximize the yields of early progenitors and evaluate their quality. Stem Cells 20, 530–541, 10.1634/stemcells.20-6-530 (2002).
Lin, W., Shuster, S., Maibach, H. I. & Stern, R. Patterns of hyaluronan staining are modified by fixation techniques. J Histochem Cytochem 45, 1157–1163 (1997).
Acknowledgements
This study was performed with the generous support of the Clare A. Glassell Family Pediatric Stem Cell Research Fund.
Author information
Authors and Affiliations
Contributions
D.J.K. designed project, performed experiments, interpreted data and wrote manuscript; B.D.C. performed experiments; R.A.H. performed experiments, interpreted data; P.S. performed flow cytometry analysis, C.S.C. Jr.: helped to draft and critically revised the manuscript, S.D.O. designed and supervised project, interpreted data and wrote manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental Figures 1-3
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Kota, D., DiCarlo, B., Hetz, R. et al. Differential MSC activation leads to distinct mononuclear leukocyte binding mechanisms. Sci Rep 4, 4565 (2014). https://doi.org/10.1038/srep04565
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04565
This article is cited by
-
The effect of TLR3 priming conditions on MSC immunosuppressive properties
Stem Cell Research & Therapy (2023)
-
The critical role of apoptosis in mesenchymal stromal cell therapeutics and implications in homeostasis and normal tissue repair
Cellular & Molecular Immunology (2023)
-
The secretion profile of mesenchymal stem cells and potential applications in treating human diseases
Signal Transduction and Targeted Therapy (2022)
-
Human Fetal Cartilage-Derived Progenitor Cells Exhibit Anti-Inflammatory Effect on IL-1β-Mediated Osteoarthritis Phenotypes In Vitro
Tissue Engineering and Regenerative Medicine (2022)
-
Activation of TLR4 of Mesenchymal Stem Cells Enhances the Regenerative Properties of Their Secretomes
Bulletin of Experimental Biology and Medicine (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
