
Abstract
Neural stem cells (NSCs) generate new neurons in vivo and in vitro throughout adulthood and therefore are physiologically and clinically relevant. Unveiling the mechanisms regulating the lineage progression from NSCs to newborn neurons is critical for the transition from basic research to clinical application. However, the direct analysis of NSCs and their progeny is still elusive due to the problematic identification of the cells. We here describe the isolation of highly purified genetically unaltered NSCs and transit-amplifying precursors (TAPs) from the adult subependymal zone (SEZ). Using this approach we show that a primary cilium and high levels of epidermal growth factor receptor (EGFR) at the cell membrane characterize quiescent and cycling NSCs, respectively. However, we also observed non-ciliated quiescent NSCs and NSCs progressing into the cell cycle without up-regulating EGFR expression. Thus, the existence of NSCs displaying distinct molecular and structural conformations provides more flexibility to the regulation of quiescence and cell cycle progression.
Similar content being viewed by others


Introduction
The lateral subependymal zone (SEZ) is the largest germinal niche in the adult rodent brain giving rise thousands of new olfactory bulb interneurons every day1. Nevertheless, neural stem cells (NSCs) in the adult SEZ are largely quiescent2,3,4. Upon activation and cell division, NSCs generate rapidly proliferating transit amplifying precursors (TAPs) that progressively differentiate into neuroblasts5,6. The expression of epidermal growth factor receptor (EGFR) at the cell membrane increases during the transition from NSCs to TAPs and rapidly decreases during neuronal differentiation and cell cycle exit5.
Previous studies have suggested that adult NSCs derive from a subset of embryonic radial glia (RG) precursors7,8 and maintain RG characteristics such as the expression of Prominin-19, apical/basal polarity and a primary cilium contacting the lateral ventricle10,11,12. Primary cilia are organelles essential for the transduction of key developmental signals13. In RG their presence and length is negatively correlated with cell cycle progression14,15. Ablation of primary cilia during development is also associated with an impairment of the transition from RG to adult NSCs8,16 whereas, cilia deletion in the postnatal niche affects rapid proliferation and quiescence in the hippocampus17 and in the SEZ18, respectively. However, the relationship between cilia, proliferation and lineage progression in the intact adult niche has not been directly investigated.
Understanding the mechanisms underlying the progression through the neural lineage is a prerequisite for the manipulation of adult neurogenesis. An important step towards this goal is the purification of NSCs and more differentiated progenitors. We have previously shown that from late development onwards cells expressing high levels of EGFR at the cell membrane (Eh) are enriched in clone forming NSCs and especially in TAPs5,19. Indeed, analysis of EGFR and Prominin-1 expression has been used for the purification of NSCs from the SEZ of adult transgenic mice expressing GFP under the control of the GFAP promoter18,20. However, it remains still unclear whether all adult NSCs are Eh cells or if these cells represent the subset of cycling NSCs. Moreover, the use of reporter genes, which has been instrumental for NSC isolation, involves genetic manipulation, possible leaky reporter expression and it does not allow the analysis of gene transcription in genetically unmodified cells.
We here describe a flow cytometry-based approach to obtain highly purified populations of naïve NSCs and TAPs. Moreover, we have investigated the relationship between proliferation and cilia in the isolated populations. We found that Eh NSCs represent a homogenous population of cycling cells. Instead quiescent NSCs only express low levels of membrane EGFR (El), although some El NSCs progressing into the cell cycle were also observed. In contrast to actively cycling NSCs, most long-term quiescent NSCs display a primary cilium, although some El NSCs progressing into the cell cycle were also observed. However, the extension of a primary cilium is not a prerequisite for the entry into quiescence, but rather a distinctive characteristic, of a pool of quiescent NSCs.
Results
Transcriptome analysis of Eh cells reveals an up-regulation of cell cycle genes
We have previously reported extensive characterization of neonatal Eh and El cells showing that the two cell populations are highly enriched in precursors and neuroblasts, respectively5,21.To gain further insight into the functional characteristics of Eh and El cells in the neonatal SEZ, we have analysed their trascriptome using the Affymetrix mouse 430_2.0 microarray. Out of 45.101 probe sets represented on the microarray, 1.718 in El and 597 in Eh cells transcripts were up-regulated at least two-fold. Confirming the reliability of the approach used for their isolation, in the latter population Egfr transcripts displayed the strongest up-regulation and 10.4% of the up-regulated genes (i.e. 62 genes corresponding to 0.138% of all genes analysed) were virtually not expressed in El cells. On the other hand, 26.05% of the genes up-regulated in El cells (i.e. 453 genes corresponding to 1.0% of all genes analysed) were solely expressed in this population.
More than 20% of the transcripts up-regulated in Eh cells were involved in cell cycle regulation whereas genes upregulated in El cells influenced electrical membrane properties (Fig. 1A and Fig. S1A). Transcripts up-regulated in Eh cells were further analysed according to predefined cell cycle pathway annotated by KEGG22 using the DAVID bioinformatics resource. Compared to the El counterpart, transcripts for the ataxia telangiectasia mutated homolog (Atm), the retinoblastoma-like protein 1 (Rbl1), the growth arrest and DNA-damage-inducible 45 gamma (Gadd45g) and Geminin (Gmnn) were all expressed more than two-fold in Eh cells (data not shown). To validate differences in gene expression and precisely quantify them we performed quantitative real time PCR (qPCR) from lysates of Eh and El cells sorted from neonatal and adult mice (Fig. 1B). Transcript levels of Rbl1, Gadd45g and Gmnn were all significantly higher in neonatal Eh cells than in the El counterpart (Fig. 1B). On the other hand, Atm mRNA was significantly up-regulated in adult but not in neonatal Eh cells (Fig. 1B). In adult Eh cells, levels of all transcripts analysed were up-regulated compared to the age-matched El counterpart with the exception of Rbl1 which showed a non-significant increase (Fig. 1B). However, this lack of significance is likely due to the low levels of Rbl1 expression in adult El cells, as transcripts were not detected in three out of five samples, thereby limiting the possibility of directly comparing transcript levels in the two populations. We have previously shown that albeit most neonatal Eh cells are TAPs, a subset of this population consists of NSCs as indicated by the expression of GFAP and Prominin-123. This co-selection approach allows the direct comparison of clone forming cells within the Prominin-1+/Eh (P+/Eh) and Prominin-1−/Eh (P−/Eh) populations, as they are equally enriched in clonogenic precursors. As P+/Eh cells represent approximately one tenth of the total Eh population, we next investigated whether genes associated with cell cycle regulation were also up-regulated in this cellular subset. Upon comparison with P−/El cells, which within the neonatal SEZ mainly represent neuroblasts21, the lowest transcript levels were observed in the P+/El population (Fig. 1B). Instead, all genes analysed were up-regulated in P+/Eh and especially in P−/Eh cells (Fig. 1B), indicating that cells in both populations are actively cycling. Thus, neonatal and adult Eh cells display increased expression of genes involved in cell cycle regulation. Moreover, a similar transcription profile is observed also in the P+/Eh subset, indicating that they represent cycling NSCs.

Gene transcription analysis of precursors isolated from the neonatal and adult SEZ.
(A) Hierarchical clustering heatmaps illustrating differential expression of transcripts affecting cell cycle in Eh cells and electrical membrane properties in El cells isolated from the neonatal SEZ. (B) Quantitative analysis by qPCR of the indicated transcripts in the given populations sorted from the dissociated SEZ at postnatal day (P) 7 and 56. Values are the means of RQ from ddCT ± SEM normalized to El cells (upper row panels) and P−/El cells (lower row panels). n ≥ 3. Statistical differences between El and the age corresponding Eh cells were calculated by t-test whereas populations sorted on the basis of both P and E expression were analysed by ANOVA. * and # indicate significantly different from the population used for normalization and from the preceding population on the X axis, respectively.
Isolation of P+/Eh and P−/Eh cells from the adult SEZ
We next investigated whether P+/Eh and P−/Eh isolated from the SEZ of adult mice also exhibit characteristics of cycling NSCs and TAPs, respectively. Since samples derived from dissociated adult tissue contain more contaminating debris and fewer immunolabelled cells than their neonatal counterpart, we optimized the gate setting procedure for the isolation of adult cells. We firstly defined the Prominin-1 gates using dissociated unstained SEZ cells as negative control (Fig. 2Aa). This procedure, hereafter referred to as gate setting I, was previously used for the isolation of neonatal cells23,24. Since adult SEZ cells displayed high levels of autofluorescence (Fig. S2A), coinciding with the levels of Prominin-1 immunoreactivity detected in most P+/Eh cells, (Fig. S2B), this procedure resulted in a poor yield of double positive cells (Fig. S3Aa). Moreover, the increase in autofluorescence was mostly observed in adult El but not Eh cells (Fig. S2A). Therefore, we next identified P+ and P− cells separately in Eh (Fig. 2Ab) and El (Fig. 2Ac) cells. This approach, hereafter referred to as gate setting II, resulted in a greater than threefold increase in the number of P+/Eh cells (Fig. S3Aa, P ≤ 0.001) without affecting their clonal enrichment (Fig. S3Ab). However, P+/Eh cells sorted with gate setting II included cells displaying low light scattering levels (Fig. 2Ad) like P−/El (Fig. 2Ae) and especially P−/Eh cells (Fig. 2Af), suggesting that they represent contaminating cells. Therefore, we next used an additional gate to sort only those P+/Eh population displaying high light scattering values (Fig. 2Ad). Compared to gate setting I, this sorting procedure, hereafter referred to as gate setting III, delivered a higher number of P+/Eh cells (Fig. S3Aa) containing more clone-forming cells (Fig. S3Ab). Importantly, the different gate setting procedures did not affect the yield and clonogenic activity of the remaining populations (data not shown). Consistent with the hypothesis that P+/Eh cells represent cycling NSCs and P−/Eh cells are TAPs, the latter gave rise to smaller clones (Fig. 2Ah), which were less capable of generating secondary clones than those obtained from the P+/Eh subset (Fig. 2Ai). Moreover, whereas the remaining populations mainly generated spherical clones (Fig. S3Ba–b and data not shown), P−/Eh cells generated either round (Fig. S3Bc–d) or flat clones (Fig. S3Be–f). The latter contained fewer cells (Fig. S3Ca) and gave rise to less secondary clones (Fig. S3Cb) than their spherical counterpart. These observations suggest that P−/Eh cells include clonogenic precursors at different stages of lineage progression. Finally, all four cellular subsets were capable of multilineage differentiation (Fig. S3D) producing mostly GABAergic neurons (Fig. S3E).

Prospective isolation and antigenic characterization of cells sorted from the adult SEZ according to EGFR and Prominin-1 expression.
(A) Representative examples of FACS plots illustrating the distribution of all the dissociated SEZ (a, g) and the named populations (b–f) according to forward (FSC), side (SSC) scatter and fluorescence levels as indicated. For primary and secondary clones the quantification of clone size is shown in (h) and of the percentage of plated cells forming clones is given in (i). Secondary clones were analysed only in P+/Eh and P−/Eh cells. Values are the percentage means ± SEM. n = 3. * indicates significantly different from the P+/Eh counterpart, t-test. (B) Quantitative analysis by qPCR of the given transcripts in the mentioned populations sorted according the indicated gate-sorting procedures. Values are the means of relative expression levels from ddCT ± SEM normalized to P−/El cells. n = 3, ANOVA.
We next analysed gene transcription signatures to further evaluate the purity achieved upon isolation with each gate setting. Transcript levels of the intermediate filament protein Gfap and of the transcription factors Dlx2 and Mash1 are characteristically distinct in NSCs and TAPs. Indeed, we have previously shown that upon normalization to the P−/El population P−/Eh cells express higher levels of Dlx2 and Mash1 than the P+/Eh counterpart, whereas the opposite is observed for Gfap transcripts23,24. A similar analysis in adult populations isolated with the various gating procedures revealed that only gate setting III delivered P+/Eh and P−/Eh cells exhibiting relative transcription signatures similar to those observed in the neonatal populations (Fig. 2B). This confirms the high degree of purity obtained by cell sorting according to gate setting III and it underscores the importance of gene transcription analysis as a tool to verify the purity of the sorted populations.
In vivo analysis of proliferation and cell cycle profiling
We next investigated the proliferation dynamics of the four populations in the adult SEZ. Proliferating cells were labelled by means of intraperitoneal injection of DNA base analogues before sorting with gate setting III. To evaluate the potential toxicity of this approach, we first measured the effect of bromodeoxyuridine (BrdU) and iododeoxyuridine (IdU) on cell viability. Analysis of propidium iodide (PI) incorporation into dissociated SEZ cells 12 hours after intraperitoneal injection revealed that IdU caused a milder impairment of cell viability than BrdU (Fig. S4Aa). Moreover, most PI+ cells represented El cells (Fig. S4Ab–c). This likely reflects the fact that the latter are the vast majority of the dissociated SEZ cells and it proves that IdU injection does not selectively compromise the recovery of Eh cells. Therefore, we gave the mice one intraperitoneal injection of IdU and sacrificed them after 12 hours or 30 days to quantify in each sorted population the proliferation rate and the label retention, respectively (Fig. 3A). In these and the following experiments, only those cells displaying homogenous nuclear labelling were considered as IdU+ (Fig. 3Aa). When analysed 12 hours after IdU injection, P+/Eh cells displayed the highest rate of IdU incorporation (83.6% ± 1.6), which is consistent with the notion that they represent activated NSCs (Fig. 3Ab). Most P−/Eh (70.3% ± 2.2) and P−/El (73.2% ± 1.5) cells were also IdU+, whereas only 21.9% ± 3.1 of the P+/El cells displayed IdU immunoreactivity. Interestingly, a similar percentage (23.9% ± 0.8) of P+/El cells still retained IdU after 30 days, indicating that in this cell population most cells dividing at any given time are label-retaining cells (LRCs), in contrast after 30 days the number of IdU+ cells was dramatically reduced in all the remaining populations, indicating that most proliferating cells in these populations either undergo consecutive rounds of replication or leave the SEZ.

Proliferation and cell cycle profiling of precursors sorted from the adult SEZ according to EGFR and Prominin-1 expression.
(A) Collage of representative images illustrating IdU immunoreactivity and DAPI counterstaining of the nuclei (a) and relative quantification (b) of SEZ cells sorted 12 hrs and 30 days after IdU injection. Arrowheads point to immunonegative and arrows to immunopositive cells. Scale bar: 10 μm. Values are the percentage means ± SEM. n = 3. * and # indicate significantly different from Ph/El and both P−/Eh and P−/El respectively, ANOVA. (B) Representative FACS plots (a) and quantification (b) of the indicated populations illustrating the cell cycle phase distribution. n = 5. * indicates significantly different from the counterpart in all other populations, ANOVA.
It has been previously shown that the quiescent state (G0) of cells is characterized by a diploid (2N) DNA content and a low RNA content, which increases upon progression into the G1 phase of the cell cycle25. Therefore, we next used nucleic acid staining to obtain a cell cycle profile of the sorted populations. We first analysed SEZ cells sorted on the basis of Prominin-1 expression without additional analysis of EGFR expression by staining with EGF-Alexa647, as the latter retains mitogenic activity19 and might promote cell cycle entry, during the plating of the cells before the immunostaining (see below). Upon staining the DNA with Hoechst 33342 (Ho) and the RNA with Pyronin Y (Py), P+ (Fig. S4Ba) and P− (Fig. S4Ca) cells could be gated into five groups corresponding to the cell cycle phases G0 (Ho−/Py−), G0/G1 (Ho−/Py+/−), G1 (Ho−/Py+), S (Ho+/−/Py+) and G2/M (Ho+/Py+). Interestingly, the analysis of the distribution in forward (FSC) and side (SSC) scatter of each gated population revealed that cell cycle progression is associated with increasing values of light scattering in P+ cells (Fig. S4Bb), but not within the P− population (Fig. S4Cb), which likely explains the increased purity of P+/Eh NSCs sorted with gate sorting III procedure. We next sorted and plated each group of P+ (Fig. S4Bc) and P− (Fig. S4Cc) cells to detect cycling and mitotic cells after immunostaining with Ki67 and PHH3 antibodies, respectively. Independent of Prominin-1 expression, virtually all Ho−/Py+ and Ho+/−/Py+ cells displayed strong immunoreactivity to Ki67 but not to PHH3, which was detected only within the Ho+/Py+ population. Instead, some Ho−/Py− and Ho−/Py+/− exhibited a faint Ki67 immunostaining, indicating that they were in mid-G1 phase26,27. In isolated P+ cells, mid-G1 cells were observed only within the Ho−/Py+/− population, whereas Ki67 immunoreactivity was not detected in the Ho−/Py− group, confirming that these cells are indeed in G0 (Fig. S4Bd and Table S1). In contrast, within the P− population both Ho−/Py− and Ho−/Py+/− subsets contained mid-G1 cells, representing around 27% and 84% of the total cells, respectively (Fig. S4Cd and Table S2). We then added the content of Ki67− and Ki67+ cells within the Ho−/Py−, Ho−/Py+/− and Ho−/Py+ subsets to calculate the distribution of P+ (Fig. S4Be) and P− (Fig. S4Ce) cells in the various phases of the cell cycle. This analysis highlighted a different cell cycle profile between P+ and P− cells with the first displaying more cells in G0 and fewer cells in the G1 and S phase of the cell cycle than the latter population. This suggests that the majority of P+ cells are quiescent, which is in agreement with the analysis of IdU incorporation. After having proved the reliability of our approach to cell cycle analysis, we next analysed the cell cycle profile of the four cellular subsets isolated based on both Prominin-1 and EGFR expression (Fig. 3Ba). We observed cycling and dividing cells also within the P+/El population, showing that P+ cells can progress into the cell cycle without up-regulating EGFR expression. However, in contrast to the remaining populations, most P+/Eh cells were in G1 and even more in the S and G2/M phase of the cell cycle (Fig. 3Bb). Thus, although the majority of both P+/Eh and P−/Eh cells undergoes DNA replication within 12 hours, the first population unlike the latter consists almost exclusively of cycling cells.
Analysis of apical-basal polarity in the sorted populations
Previous studies have indicated that NSCs in the SEZ contact the lateral ventricle extending an apical primary cilium10,11,12. Besides NSCs, also E1 and E2 ependymal cells contact the lateral vetricle with multiple or two motile cilia, respectively10. We used immunostaining with acetylated tubulin and with Ki67 antibodies to analyse the presence of cilia and the correlation with cell cycle in the four cellular subsets isolated from the adult SEZ based on Prominin-1 and EGFR expression (Fig. 4Aa). Ependymal multiciliated (mc) and biciliated (bc) cells were observed only within the P+/El population. The remaining P+/El cells displayed a single short cilium (sc), a single long cilium (lc) or were non-ciliated (nc) (Fig. 4Ab). Cilia were not detected in the remaining populations, except in a small subset of P−/Eh cells (1.5% ± 0.5), which displayed a short cilium. No Ki67 immunoreactivity was detected in mc P+/El and bc P+/El cells (Fig. 4Ac). Only few lc P+/El (5.4% ± 2.7) were Ki67+, whereas approximately half of sc P+/El (50% ± 2) and nc P+/El (50 ± 5.8) displayed Ki67 immunoreactivity (Fig. 4Ac). Most of the cells in the remaining populations were cycling, especially within the P+Eh population, where virtually all cells were Ki67+ (Fig. 4Ad). In contrast, the small subset of ciliated P−/Eh cells was largely Ki67−. Similar cell types, including uniciliated cells with a cilium of variable length were also observed upon immunostaining of the whole SEZ (Fig. 4Ba), indicating that the heterogeneity in terms of cilia presence/length is not a consequence of the sorting procedure. As previously reported10, Prominin-1 immunoreactivity was observed at the tip of the cilia (Fig. 4Bb, C).

Analysis of cilia in precursors of the adult SEZ.
(A) (a) Representative photomicrographs illustrating the given populations upon immunostaining and DAPI nuclear counterstaining as indicated. Scale bar: 10 μm. (b–c) Quantitative analyses of the percentage of sorted P+/El cells which are multiciliated (mc), biciliated (bc), uniciliated with long (lc) or short (sc) cilium and non-ciliated (nc) (b) and Ki67 immunopositive (Ki67+) and immunonegative (Ki67−) (c). * and # indicate significantly different from the remaining populations and mc, bc, lc respectively, ANOVA. (d) Quantification of the percentage of Ki67+in the indicated cell populations. n = 3. * indicates significantly different from the remaining populations, ANOVA. (B–C) Confocal photomicrographs illustrating a representative example of the apical side of the SEZ in whole-mount (B) and in coronal section (C) upon immunostaining as indicated. Higher magnification of multiciliated ependymal cells (a′) and cells with a single long or short cilium (a″). Double immunostaining (Bb and C) illustrates Prominin-1 expression at the tip of the cilia. Arrow points to a non-ciliated cell as demonstrated by analysis of sequential sections. Scale bar: 100 μm.
The presence of cilia within the four populations isolated from the SEZ of adult (Fig. 5A) and neonatal (Fig. S5) mice was also investigated by scanning electron microscopy (SEM), although this analysis provides qualitative but not quantitative information. Independent of age, cells with more than one cilium were uniquely observed within isolated P+/El cells, which also contained lc P+/El, sc P+/El as well as nc cells. This analysis also revealed that many P+/El cells displayed a rough cell surface with multiple villi and bleb-like structures. Blebs and villi were also observed in neonatal (Fig. S5) but not in adult (Fig. 5A) P+/Eh cells. Independent of EGFR expression and age, all P− cells displayed a relatively smooth cell surface. Although most P−/Eh cells analysed were non-ciliated, few cells in this population displayed a stumpy cilium, which is consistent with our previous observations. Similar cell surface structures were also observed in the intact adult SEZ (Fig. 5B). Interestingly, at higher magnification the tip of some cilia showed a cup shaped structure (Fig. 5Bc) reminiscent of Prominin-1 enriched structures previously observed in neuroepithelial cells28. Taken together, these analyses demonstrate that P+/El cells contain the main cell types previously identified at the apical surface of the lateral SEZ. They also show that virtually all P+/Eh cells lack cilia, in keeping with the previous observation that the cilium is retracted upon entering the cell cycle29.

Analysis by SEM of precursors in the adult SEZ.
Representative SEM images of the indicated sorted populations (A) and of the apical surface of the SEZ in whole-mount (B). Arrows and arrowhead point to uniciliated cells with seemingly a long (arrows) and a short (arrowhead) cilium. Asterisk points to two closely localized long cilia, which may belong to a biciliated E2 cell. Higher magnification view of the inset in (a) and a detail of the tips of cilia in ependymal multiciliated E1 cells are shown in (a′) and (c), respectively. Scale bars: 1 um and 200 nm in higher magnification views. (C) Quantitative analysis of the percentage of DiI+ labeled cells (a) and relative clonogenic activity (b) in sorted P+/Eh and P−/Eh populations. n = 3. * indicates significantly different from the P−/Eh counterpart, ANOVA.
We next labelled the apical side of the SEZ with DiI before dissociation and sorting to investigate whether P+/Eh and P−/Eh cells directly contact the ventricle. Using this approach we were able to label 90% ± 5.8 of the P+/Eh cells (Fig. 5Ca) as compared to only 2% ± 1.45 of the P−/Eh cells. Moreover, DiI labeled P+/Eh cells showed the same clonogenic capability as the DiI unlabelled counterpart (Fig. 5Cb). Thus, in contrast to P+/Eh NSCs, most P−/Eh cells are not in contact with the ventricle, which is consistent with the notion that TAPs are localized beneath the ependymal layer10.
Combined analysis of proliferation and cilia dynamics
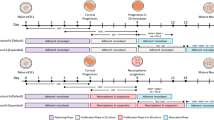
We next investigated cilia dynamics in rapidly proliferating cells (Fig. 6A) and LRCs (Fig. 6B) in the adult SEZ. To this end mice were sacrificed 12 hours (Fig. 6Aa) or 30 days (Fig. 6Ab) after IdU injection and upon dissection and sorting the four populations were processed for triple immunostaining with antibodies to acetylated tubulin, IdU and Ki67. Within the P+/El population, 12 hours after injection, nc cells represented the vast majority (86.5% ± 0.8) of IdU+ cells (Fig. 6Ab). Moreover, more than half (57.6% ± 0.4) of the total subset of nc P+/El cells had incorporated IdU (Fig. 6Ac). The remaining IdU+ cells were uniciliated cells (sc: 10.83% ± 0.9; lc: 1.9% ± 0.8), whereas no IdU incorporation was detected within mc and bc cells (Fig. 6Ab, c). At this time point, independent of the presence of a cilium, virtually all IdU+ cells within the P+/El population also displayed Ki67 immunoreactivity (Fig. 6Ad). In fact, out of 1414 P+/El cells examined, we could only find one lc P+/El cell, which was IdU+/Ki67− (Fig. 6Aa see the cell highlighted by the asterisk). Also the IdU+ cells present within the remaining sorted populations were largely Ki67+ (Fig. 6Ad). However, in these populations a few Ki67+/IdU− cells were also observed, which likely represent cells progressing through G1 (Fig. 6Ad). Finally, within the P−/Eh population, most of the cells with a single short cilium were IdU− and Ki67− with the exception of a few cases where a foci Ki67staining was observed (Fig. 6Aa).

Relationship between cilia and proliferation in the adult SEZ.
Analysis of the indicated populations sorted 12 hours (A) or 30 days (B) after IdU injection by triple immunostaining. (a) Collage of representative confocal images upon immunostaining and DAPI counterstaining. Arrows point to double immunopostive and arrowheads to single immunopositive cells. * points to the only lc P+/El cell observed which was IdU+/Ki67− (A upper row panels), a rare example of a P−/Eh cell with a short cilium and foci Ki67 immunostaining (A third row panels), a rare biciliated LRC (B). Scale bar: 10 μm. Quantitative analysis of the percentage of P+/El cells which display IdU immunoreactivity and are multiciliated (mc), biciliated (bc), unciliated with a long (lc) or short (sc) cilium and non-ciliated (nc). *indicates significantly different from the IdU− counterpart, t-test. (b). Quantitative analyses of the percentage of the indicated cellular subset displaying IdU (c) or Ki67/IdU (d) immunoreactivity as indicated. # indicates significantly different from IdU+ (c) and Ki67+ (d) cells in all other populations, ANOVA.
Thirty days after IdU injection, the IdU+ cells were more evenly distributed within the non-ependymal P+/El cells (Fig. 6Ba). In descending order, most LRCs were represented by nc (42% ± 0.1), sc (29.1% ± 0.6) and lc (25.5% ± 1.0) cells (Fig. 6Bb). Also at this time point IdU was not detected in mc P+/El cells (Fig. 6Bb). However, 30 days after IdU injection, we detected a few bc LRCs. These cells represented 41.9% ± 8.1 of the total bc cells (Fig. 6Bc), indicating that almost half of these cells are generated during a 30 day period. Likewise, although IdU immunoreactivity was never observed in ciliated P−/Eh cells analysed 12 hours after IdU injection, rare ciliated LRCs were detected after 30 days. As previously observed (see Fig. 4Ab), at this time point a similar small percentage of LRCs (Fig. 6Ba), which did not display Ki67 immunoreactivity (Fig. 6Bd), was present also in populations other than P+/El cells, including in P+/Eh cells. Since in this population quiescent cells were consistently observed only in tissue derived from mice that had been injected with IdU 30 days before, it is possible that they represented cells that had stopped dividing due to excessive incorporation of IdU. Supporting this conclusion, we observed at this time point a similar slight decrease in the percentage of Ki67+ cells across all sorted populations (Fig. 6 compare panel Ad with panel Bd). Moreover, these variations were not due to an effect of IdU injections on the number of cells in the four populations (data not shown). To further investigate the effect of IdU we quantified the percentage of the various types of P+/El cells found in the dissociated SEZ of control non-injected mice and of animals injected with IdU 12 hours and 30 days before (Table S3). We did not include in this analysis mc cells as they never incorporated IdU and their number was not significantly affected by IdU injection (see Figs 4Ab, 6Ab and 6Bb for comparison). Thirty days after IdU injection a significantly higher proportion of sc and lc cells and fewer nc cells were present in the SEZ of injected mice than in the non-injected counterpart (Table S3). The effect of IdU on sc was also observed at 12 hours, however at this time the number of lc cells was not affected and the proportion of nc cells displayed only a non-significant trend decrease. Since the increase in the proportion of ciliated cells was only observed within the population of P+/El cells, it likely reflects the effect of IdU in inducing quiescence rather than in promoting cilia extension.
Thus, cell cycle profiling along with IdU labeling demonstrate that the P+/El cellular subset is composed of mainly quiescent NSCs. In contrast, the P+/Eh population largely contains actively cycling cells. Moreover, our analyses also show that the extension of a primary cilium is not a prerequisite to enter quiescence, although there is a positive correlation between the presence/length of a primary cilium and quiescence.
Discussion
We here describe a method for the simultaneous isolation of cycling NSCs and TAPs at a high degree of purity, which can be easily standardized across different laboratories. Our data also underscore the importance of transcriptional signature analysis for the confirmation of the identity of the isolated cell populations.
We found that, despite its limitations30, clonal analysis represents a very powerful tool to investigate the stemness of neural precursors as it allows to detect intrinsic differences in self-renewal and differentiation potential between cycling NSCs and TAPs. However, this approach is not suitable for the analysis and the detection of NSCs within the P+/El compartment, as these cells proliferate in vitro very slowly and generate very small clones. The limited ability of adult P+/El NSCs to proliferate and form clones was not observed in similar cell types isolated from the neonatal SEZ23,24. The scarce proliferation in the compartment of adult P+/El NSCs may reflect the inability of these cells to up-regulate EGFR expression at the cell surface, which is a characteristic of highly proliferative NSCs and intermediate precursors. An association between EGFR expression and proliferation was also observed in several niches of the developing telencephalon24, although it is still not clear whether neural precursor proliferation in vivo depends on EGFR activation.
Due to their scarce ability to form clones, we here determined the presence of NSCs within the P+/El population by analysing the presence of primary cilia. The presence of primary cilia in NSCs likely reflects their origin from RG. However, whereas the primary cilium of embryonic cortical precursors is extended during G0/G114, our data show that in adult NSCs quiescence and extension of a primary cilium are not strictly associated. If NSCs represent a homogenous pool of RG descendants, one possible interpretation of our data could be that the observed heterogeneity in terms of cytoarchitecture, i.e. presence/absence of a primary cilium, may reflect different statuses of quiescence. For example, ciliated LRCs may represent long-term quiescent NSCs whereas non-ciliated LRCs may be more likely to re-enter the cell cycle. Consistent with this hypothesis, in embryonic RG the presence of a primary cilium not only inversely correlates with cell cycle progression but it also prevents cell division14,15. Indeed, our data indicate that the quiescence-inducing effect of IdU is associated with the transition of nc cells to ciliated LRCs. An alternative explanation may be that despite the similar antigenic profile, uniciliated and non-ciliated cells may represent functionally distinct NSC types: a ciliated RG like cell and a non-ciliated precursor type. Indeed, lineage analysis of isolated SEZ NSCs have proposed the existence of independent pools of NSCs which are differentially affected by regulatory signals31.
Since primary cilia mediate the response to key regulatory signalling pathways8,32,33 our findings indicate that different cellular conformations may affect the ability of NSCs to sense key environmental signals regulating quiescence and cell cycle progression.
Experimental procedures
Tissue dissection and FACS analysis
All animal experiments were approved by the Regierungspräsidium Karlsruhe and the local authorities at the University of Heidelberg. Eight week-old and neonatal C57/BL6 mice were killed by CO2 inhalation followed by cervical dislocation and decapitation, respectively. For single staining, after dissociation, cells were incubated with 20 ng/ml EGF-Alexa 488 (Molecular Probes) at 4°C for 30 minutes, as previously described19. For Prominin-1 immunolabelling and EGFR staining dissociated cells were incubated at 4°C with anti-Prominin-1 antibody conjugated to phycoerythrin (PE) (1:400, Milteny). After 1 hour the excess of antibody was removed and cells were stained with EGF-Alexa 647, as previously described24. Cells were sorted with a FACSAria cytometer (Becton-Dickinson) at single cell precision. Gates were set based on the following controls: 1) Unstained cells as control for autofluorescence and cells stained with 1 μg/ml propidium iodide (PI) (Sigma) to exclude dying cells, PI− live cells were gated in P2; 2) Cells that had been incubated with 20 ng/ml EGF for 1 hour before staining with EGF-Alexa; 3) Cells stained only with PE-conjugated anti-Prominin-1 antibodies. Interference of PI into the PE channel was compensated with this control.
Clonal assays and differentiation
For clonal analysis, independent of Prominin-1 expression, 1 Eh cell and 20 El cells per well were plated in 96 well plates (Nunc) in 50 μl NS-A medium (Euroclone) supplemented with 2% B27 (Gibco), 10 ng/ml FGF-2 and 20 ng/ml EGF (all from Peprotech)19,24 using the FACSAria-automated cell deposition and grown at 37°C with 5% CO2. After 7 days clones were counted and mechanically dissociated for cell counting. For secondary clone formation 50 P+/Eh or P−/Eh cells per well were plated in 96 well plates and grown for further 7 days. To examine the differentiation capacity of individual cellular subsets 10–25,000 cells were plated in μ-slide wells (Ibidi) coated with matrigel (Becton-Dickinson) in NS-A medium supplemented with 2% B27, 1% FCS (GIBCO) and 2 ng/ml FGF-2 and grown at 37°C in 5% CO2 for 7 days.
Whole-mount dissection and immunostaining
The lateral ventricular whole mount was dissected as previously described34. The dissected lateral wall was fixed in 4% PFA/0.1% Triton X-100 overnight at 4°C. For immunostaining, whole-mounts were washed three times in PBS and incubated for 48 h at 4°C with primary antibodies, further washed three times with PBS and incubated for 48 h at 4°C with the secondary antibodies. Before mounting on glass coverslips, the ventricular walls were further dissected from the underlying parenchyma to reach a total thickness of about 100–200 μm.
Immunofluorescence
Coronal sections and isolated cells were fixed in 4% PFA and processed for immunostaining as previously described24. For IdU detection mice were given a single intraperitoneal injection of IdU (100 mg/kg body weight, dissolved in water) and were sacrificed after 12 hrs or 30 days. After fixation cells were permeablized with 0.5% NP-40 followed by treatment with 300 ug/ml DNAase A (all from Sigma) and incubation with a primary antibody to IdU and/or other antigens, as indicated in the text, in presence of DNAase A. After washing twice cells were stained with appropriate secondary antibody and DAPI for nuclear counterstain. Immunopositive cells were quantified using a SP2 confocal microscope (Leica). For each antigen, cells were counted in one entire well. Immunopositive cells were counted as percentage of total cell numbers revealed by DAPI nuclear counterstain. See supplementary experimental procedures for a complete list of the antibodies used.
Cell cycle analysis
Each sorted population was incubated with 0.6 μg/ml Hoechst 33342 (Sigma) at 4°C for 10 minutes followed by further 10 minute incubation with 0.6 μg/ml Pyronin Y (Sigma) at 4°C. Cells were analysed by FACS by using violet and red lasers to detect Hoechst and Pyronin Y, respectively. Cells stained with either dye were used to check the inference of each channel with PI.
Sample preparation for Affymetrix analysis
To avoid a deterioration of the cellular sample, which could negatively affect the RNA quality, we separately processed the tissue derived from each animal. After sorting the Eh and El cell lysates collected from one mouse litter were pooled together. Using this procedure we collected for each cell population two independent RNA samples. Total RNA was extracted from sorted cells (500–20.000 cells, depending on the cellular subset) using the RNeasy total RNA isolation kit (Qiagen) according to the manufacturer's instructions. RNA was eluted with 30 μl H2O. RNA quality was examined by agarose gel electrophoresis and concentration was determined by UV absorbance.
Affymetrix gene-chip hybridization
Gene expression profiling was performed using Affymetrix GeneChipR Mouse Genom 420 2.0 according to the manufacturer's protocol. See supplementary experimental procedures for details.
Statistical analysis
For each quantitative analysis, the mean standard errors of at least three independent experiments and statistical significance tests (Student's t-test and ANOVA, Bonferroni post hoc test) were calculated using a statistical package (Graphpad Prism). Calculated p-values are indicated in the figure as follows: *,#: p ≤ 0.05, **, ##: p ≤ 0.01; ***, ###: p ≤ 0.001.
References
Petreanu, L. & Alvarez-Buylla, A. Maturation and death of adult-born olfactory bulb granule neurons: role of olfaction. J Neurosci 22, 6106–6113 (2002).
Morshead, C. M., Craig, C. G. & van der Kooy, D. In vivo clonal analyses reveal the properties of endogenous neural stem cell proliferation in the adult mammalian forebrain. Development 125, 2251–2261 (1998).
Morshead, C. M. et al. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron 13, 1071–1082 (1994).
Doetsch, F., Garcia-Verdugo, J. M. & Alvarez-Buylla, A. Regeneration of a germinal layer in the adult mammalian brain. Proc Natl Acad Sci U S A 96, 11619–11624 (1999).
Cesetti, T. et al. Analysis of stem cell lineage progression in the neonatal subventricular zone identifies EGFR+/NG2− cells as transit-amplifying precursors. Stem Cells 27, 1443–1454 (2009).
Kuhn, G. H. & Blomgren, K. Developmental dysregulation of adult neurogenesis. Eur J Neurosci 33, 1115–1122 (2011).
Spassky, N. et al. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J Neurosci 25, 10–18 (2005).
Han, Y. G. et al. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci 11, 277–284 (2008).
Weigmann, A., Corbeil, D., Hellwig, A. & Huttner, W. B. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci U S A 94, 12425–12430 (1997).
Mirzadeh, Z., Merkle, F. T., Soriano-Navarro, M., Garcia-Verdugo, J. M. & Alvarez-Buylla, A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell 3, 265–278 (2008).
Shen, Q. et al. Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell-cell interactions. Cell Stem Cell 3, 289–300 (2008).
Tavazoie, M. et al. A specialized vascular niche for adult neural stem cells. Cell Stem Cell 3, 279–288 (2008).
Louvi, A. & Grove, E. A. Cilia in the CNS: the quiet organelle claims center stage. Neuron 69, 1046–1060 (2011).
Li, A. et al. Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat Cell Biol 13, 402–411 (2011).
Wilson, S. L., Wilson, J. P., Wang, C., Wang, B. & McConnell, S. K. Primary cilia and Gli3 activity regulate cerebral cortical size. Dev Neurobiol 72, 1196–1212 (2012).
Breunig, J. J. et al. Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc Natl Acad Sci U S A 105, 13127–13132 (2008).
Amador-Arjona, A. et al. Primary cilia regulate proliferation of amplifying progenitors in adult hippocampus: implications for learning and memory. J Neurosci 31, 9933–9944 (2011).
Beckervordersandforth, R., Tripathi, P., Ninkovic, J. & al, e. In vivo fate mapping and expression analysis reveals molecular hallmarks of prospectively isolated adult neural stem cells. Cell Stem Cell 7, 744–758 (2010).
Ciccolini, F., Mandl, C., Holzl-Wenig, G., Kehlenbach, A. & Hellwig, A. Prospective isolation of late development multipotent precursors whose migration is promoted by EGFR. Dev Biol 284, 112–125 (2005).
Fischer, J. et al. Prospective isolation of adult neural stem cells from the mouse subependymal zone. Nat Protoc 6, 1981–1989 (2011).
Cesetti, T., Fila, T., Obernier, K., Bengtson, C. P., Li, Y., Mandl, C., Holzl-Wenig, G. & Ciccolini, F. GABAA receptor signaling induces osmotic swelling and cell cycle activation of neonatal prominin+ precursors. Stem Cells 29, 307–319 (2011).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Obernier, K. et al. Expression of Tlx in both stem cells and transit amplifying progenitors regulates stem cell activation and differentiation in the neonatal lateral subependymal zone. Stem Cells 29, 1415–1426 (2011).
Carrillo-Garcia, C. et al. Multipotent precursors in the anterior and hippocampal subventricular zone display similar transcription factor signatures but their proliferation and maintenance are differentially regulated. Mol Cell Neurosci 44, 318–329 (2010).
Ross, J. S. DNA ploidy and cell cycle analysis in cancer diagnosis and prognosis. Oncology (Williston Park) 10, 867–882, 887; discussion 887–890 (1996).
Bruno, S. & Darzynkiewicz, Z. Cell cycle dependent expression and stability of the nuclear protein detected by Ki-67 antibody in HL-60 cells. Cell Prolif 25, 31–40 (1992).
Gerdes, J. et al. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 133, 1710–1715 (1984).
Dubreuil, V., Marzesco, A. M., Corbeil, D., Huttner, W. B. & Wilsch-Bräuninger, M. Midbody and primary cilium of neural progenitors release extracellular membrane particles enriched in the stemcell marker prominin-1. J Cell Biol 176, 483–495 (2007).
Wilsch-Bräuninger, M., Peters, J., Paridaen, J. T. & Huttner, W. B. Basolateral rather than apical primary cilia on neuroepithelial cells committed to delamination. Development 139, 95–105 (2012).
Pastrana, E., Silva-Vargas, V. & Doetsch, F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 8, 486–498 (2011).
Ortega, F. et al. Oligodendrogliogenic and neurogenic adult subependymal zone neural stem cells constitute distinct lineages and exhibit differential responsiveness to Wnt signalling. Nat Cell Biol 15, 602–613 (2013).
Jackson, E. L. et al. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron 51, 187–199 (2006).
Zhang, L., Yang, X., Yang, S. & Zhang, J. The Wnt/beta-catenin signaling pathway in the adult neurogenesis. Eur J Neurosci 33, 1–8 (2011).
Mirzadeh, Z., Han, Y. G., Soriano-Navarro, M., García-Verdugo, J. M. & Alvarez-Buylla, A. Cilia organize ependymal planar polarity. J Neurosci 30, 2600–2610 (2010).
Acknowledgements
This work and P.K. have been supported by research program “Adulte Stammzellen II” of the Baden-Württemberg Stiftung. We thank Denis Corbeil (Dresden) and J. Trotter (Mainz) for kindly providing the Prominin-1 and O4 antibodies, respectively.
Author information
Authors and Affiliations
Contributions
F.C. designed the research. P.K., C.M., K.O., I.S. and C.S. performed the experiments, S.W., J.W., J.A.G. and A.S. microarray analysis, G.H.-W. operated the FACS, A.H. performed SEM, K.O. and I.S. prepared figure 1, P.K. prepared all the remaining figures. F.C. and P.K. wrote the main manuscript. All of the authors reviewed and approved the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Khatri, P., Obernier, K., Simeonova, I. et al. Proliferation and cilia dynamics in neural stem cells prospectively isolated from the SEZ. Sci Rep 4, 3803 (2014). https://doi.org/10.1038/srep03803
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03803
This article is cited by
-
Mucociliary Wnt signaling promotes cilia biogenesis and beating
Nature Communications (2023)
-
Prominin-1 Modulates Rho/ROCK-Mediated Membrane Morphology and Calcium-Dependent Intracellular Chloride Flux
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
