
Abstract
The Red Sea Atlantis II brine pool is an extreme environment that displays multiple harsh conditions such as high temperature, high salinity and high concentrations of multiple, toxic heavy metals. The survival of microbes in such an environment by utilizing resistant enzymes makes them an excellent source of extremophilic enzymes. We constructed a fosmid metagenomic library using DNA isolated from the deepest and most secluded layer of this pool. We report the isolation and biochemical characterization of an unusual esterase: EstATII. EstATII is thermophilic (optimum temperature, 65°C), halotolerant (maintains its activity in up to 4.5 M NaCl) and maintains at least 60% of its activity in the presence of a wide spectrum of heavy metals. The combination of biochemical characteristics of the Red Sea Atlantis II brine pool esterase, i.e., halotolerance, thermophilicity and resistance to heavy metals, makes it a potentially useful biocatalyst.
Similar content being viewed by others


Introduction
Until recently and despite its uniqueness, the Red Sea has received little attention compared with other marine environments. The Red Sea formed 3–5 million years ago when the Arabian and African plates began to split1 and it is characterized by high temperature and salinity due to the high rate of evaporation, lack of major river inflows and low rate of rainfall. The Red Sea is characterized by the presence of deep-sea, hypersaline anoxic basins called brine pools, which are large bodies of water at the bottom of the ocean that are characterized by high temperature and salinity. To date, 25 brine pools have been found in the Red Sea1,2. Atlantis II Deep (Figure 1) is the largest brine pool in the Red Sea and it has the highest temperature and is the most dynamic1,3. This brine pool has a maximum depth of 2,194 m and is stratified into several layers that increase in temperature and salinity with increasing depth: the brine-seawater interface, the upper convective layer, the middle convective layer and the lower convective layer (LCL)1,3. The lowest layer, LCL, is characterized by a temperature of 68.2°C, pH value of 5.3 and salinity of 270 psu, which is 7.5 times that of normal seawater1,3. Atlantis II Deep is nearly anoxic and contains high concentrations of iron, zinc, copper and other heavy metals1,3. Together, these extreme conditions make the Atlantis II brine pool an attractive site for mining for biocatalysts, such as lipolytic enzymes, which are predicted to possess desirable traits that include but are not limited to thermo-tolerance, halo-tolerance, pH plasticity and resistance to inhibition by heavy metals.

Atlantis II Brine Pool Sample Site.
Location of the Atlantis II Brine Pool (Latitude [N] 21° and Longitude [E] 38°) from which the samples were obtained during the KAUST Red Sea 2010 expedition. The figure was generated using Map data ©2012 Google, ORION-ME.
Industrialized societies are moving toward white (industrial) biotechnology, which has proven to be environmentally sound and commercially efficient4. Doing so poses a continuous demand for novel biocatalysts, preferably biocatalysts that demonstrate high activity over a wide range of conditions such as temperature, salinity, pH and metal concentrations. Biocatalysts of microbial origin represent the majority of biocatalysts that are used in industrial and biotechnological processes5, due to the ability of prokaryotes to populate and adapt to different environments. For example, we can derive a wide array of biocatalysts that are robust (within a flexible range of conditions) from regions such as hydrothermal vents and Antarctic desert soils. These characteristics make these enzymes desirable for industry6.
Metagenomics is a powerful tool for accessing the genomes of the unculturable majority of prokaryotes and to investigate their potential as sources of novel biocatalysts. This technology has led to the identification and characterization of a vast number of biocatalysts that are active under a wide range of conditions reflecting the environment from which they originate, thus making them desirable for industrial use7,8,9,10.
Microbial lipolytic enzymes possess a huge potential as industrial biocatalysts. These enzymes are characterized by substrate specificity and regio- and enantioselectivity that surpass that of any other enzyme, which makes their application potential boundless11. Using lipolytic enzymes in industrial and biotechnological applications is estimated to be a billion-dollar business12. The applications of these enzymes include but are not limited to leather manufacturing, flavor development in the dairy industry, oil biodegradation and the synthesis of pharmaceuticals and chemicals12,13,14,15.
Here, we report the isolation and biochemical characterization of an unusual esterase, EstATII, from the LCL of the Atlantis II brine pool.
Results
Screening the metagenomic library for lipolytic activity
The constructed fosmid library comprised 10,656 clones that were manually placed into 111 96-well plates for ease of handling. Functional screening of the fosmid library on tributyrin agar detected five recombinant clones that formed a clear halo zone indicative of putative lipolytic activity.
Sequencing, identification and sequence analysis
Fosmids of the five positive, recombinant clones were pyrosequenced to identify the genes responsible for the putative lipolytic activity. Following assembly, four large contigs (>10 kb) and seven smaller contigs (ranging from 1.4–4.9 kb) were obtained; the largest contig obtained was ~32 kb (1Table 2). A 945-bp ORF encoding a putative esterase/lipase (designated EstATII) was identified. The maximum identity to sequences in the database was 65%, which was with an alpha/beta hydrolase domain-containing protein from Pseudomonas mendocina. The highest identity to a lipolytic enzyme in the database was 56% to an esterase from Pseudomonas aeruginosa. EstATII was the only lipolytic enzyme that was detected; however, other sulfatase-encoding ORFs were detected that could be responsible for false-positive activity, as previously reported22. EstATII consists of 945 bp, which corresponds to 314 amino acids. A domain search was conducted using the CD-search tool and detected an alpha/beta hydrolase fold domain [Pfam ID: pfam07859], which is the catalytic domain that is found in members of the alpha/beta hydrolase family, between residues 85 and 286. An esterase/lipase domain (cd00312) was also detected. In addition, two prokaryotic Clusters of Orthologous Groups (COGs) were identified, namely, COG0657 and COG2272, which are involved in lipid metabolism.
The catalytic triad residues, Ser160, Asp204 and His282, were identified in EstATII and the catalytic, nucleophilic residue Ser160 was found in the consensus pentapeptide GDSAG, which is characteristic of the hormone-sensitive lipase (HSL) family. Another motif characteristic of the HSL family (HGG), which contributes to the formation of the oxyanion hole, was also identified in the sequence ( Figure 3). EstATII was predicted to be soluble because a signal peptide was not detected. A transmembrane domain was identified using the TMAP program23 and this domain consisted of a stretch of 24 amino acids at the N-terminal side of amino acids 29–52 (data not shown). The membrane-imbedded domain was repeatedly reported in other identified esterases24.

Phylogenetic Analysis and Classification of EstATII.
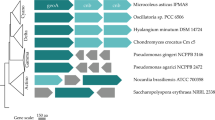
A multiple sequence alignment of EstAII with 41 lipolytic enzymes (representing the eight families of the bacterial lipolytic enzymes, as classified by Arpigny and Jaeger in 199917) was used to construct a phylogenetic tree. EstATII groups with members of family IV, also known as the HSL family. The confidence level of the tree was estimated by permutations (10,000 replicates) and the tree was constructed using MEGA 5. The scale represents the number of amino acid substitutions.

Conserved Motifs Found in Members of the Hormone-Sensitive Lipase (HSL) Family and in EstATII.
Multiple sequence alignment of EstATII with other members of the HSL family: Pseudomonas sp. B11-1 (AF034088), Archaeoglobus fulgidus (AE000985), Alcaligenes eutrophus (L36817), Escherichia coli (AE000153) and Moraxella sp. (X53868) was performed using ClustalW and visualized using the BoxShade server. The alignment displays the conserved motif HGG that is involved in the formation of the oxyanion hole and the nucleophilic catalytic serine residue in the pentapeptide GDSAG, which is conserved in the HSL family.
Bacterial lipolytic enzymes were classified into eight families by Arpigny and Jaeger in 199917. To determine whether EstATII is classified as a member of one of these families, a multiple sequence alignment of EstATII with 43 sequences of bacterial lipolytic enzymes, representing the eight families, was performed. A phylogenetic tree was then constructed and EstATII was grouped with members of family IV, which is also known as the HSL family (Figure 2).
Overexpression and purification of EstATII
To investigate the biochemical characteristics of EstATII, the gene was expressed as an N-terminal, His-tagged recombinant protein in the expression vector pET-SUMO in E. coli BL21 (DE3) cells. The overexpressed protein had the expected molecular weight of 46 kDa and western blot analysis of the purified protein revealed a single band of the expected size (Figure 4).

Overexpression, Purification and Western Blot Analysis of Recombinant EstATII.
Lane 1: molecular weight marker (size indicated in KDa), Lane 2: un-induced sample, Lane 3: sample induced with 0.5 mM IPTG, Lane 4: protein lysate, Lanes 5 and 6: flow-through, Lane 7: wash step, Lane 8: purified EstATII and Lane 9: western blot analysis of the purified EstATII.
Biochemical characterization of EstATII
Effect of temperature and pH on the EstATII activity
The effect of temperature on the activity of EstATII was assayed at temperatures ranging from 30°C to 80°C. The activity of the enzyme increased reproducibly with increasing temperature until 65°C, after which the activity began to decrease. High activity of the enzyme (>70%) was observed at temperatures ranging from 45°C to 75°C. The apparent optimum temperature of EstATII is 65°C; however, the enzyme remained active even after reaching 80°C (Table 3).
The effect of pH on the activity of EstATII was assayed within a range of 3–9. The enzyme exhibited significant activity (>50%) at pH 7–9 and the highest activity occurred at pH 8.5. No activity was observed at pHs lower than 5.5 (Table 3).
Effect of the NaCl concentration on the EstATII activity
The EstATII activity was assayed at different molar concentrations of NaCl ranging from 0 M to 4.5 M. The enzyme exhibited the highest activity in the presence of 2 M NaCl; however, the enzyme activity was maintained in up to 4.5 M NaCl (Table 3).
Substrate specificity of EstATII
To assess whether EstATII is a lipase or an esterase, its substrate specificity was investigated using an array of p-nitrophenol esters with various chain lengths. EstATII was active toward short-chain fatty acid esters (C2, C4 and C5); however, it showed no activity toward long-chain fatty acid esters (C10, C12, C14 and C16), which indicated that EstATII is an esterase and not a lipase25 (Table 3).
Effect of metal ions on the EstATII activity
The effect of metal ions was assessed with and without EDTA chelation. The enzyme activity was not affected by EDTA chelation, suggesting that EstATII is not a metalloenzyme10. The enzyme activity was promoted by Ba2+ (158%), Mg2+ (111%) and Co2+ (104%) and the enzyme was resistant to inhibition by the remainder of the tested metal ions, i.e., the activity remained above 60% (Table 4). Upon investigating the effect of heavy metal ions on the activity of EstATII compared with other esterases (thermophilic and mesophilic), it was noticed that Cu2+, Zn2+ and Hg2+ exhibited a strong inhibitory effect (>50%) on the activity of most esterases that were included in the comparison (Table 4). Although EstATII displayed significant resistance to all heavy metal ions that were tested, resistance to these three heavy metals is of particular interest due to their strong inhibitory effect on most of the identified esterases.
Effect of detergents, reducing agents and modifying agents on the EstATII activity
The effect of detergents (ionic and non-ionic) was tested at two concentrations: 0.1% and 1%. At 0.1%, Tween 80 exhibited no effect on the enzyme activity; Triton X-100 and Tween 20 were tolerated by the enzyme (42.3% and 87.5% activity, respectively), while SDS dramatically reduced the enzyme activity to 6%. At 1%, EstATII was tolerant to the effects of Tween 20 and 80 (40.1% and 54.5% activity, respectively), while SDS and Triton X-100 abolished the enzyme activity. The reducing agent β-mercaptoethanol (at a final concentration of 1 mM) increased the enzyme activity to 117.9%, while 1-mM DTT slightly reduced the enzyme activity to 78.9%. DEPC, the histidine-residue modifier, reduced the activity by one-half and this result was in agreement with the involvement of a histidine residue in the catalytic triad (Table 3).
Discussion
We have identified an unusual esterase (EstATII) from the LCL of the Red Sea Atlantis II brine pool in the Red Sea, using a function-based approach. Sequence and phylogenetic analyses of EstATII revealed that it is a new member of the HSL family (Family IV). EstATII was grouped with members of the HSL family and was found to harbor conserved motifs that are characteristic of this family. A wide range of p-nitrophenol esters with various chain lengths (Table 3) was used to identify EstATII as an esterase.
To address the biotechnological potential of EstATII as a biocatalyst, we compared it to 23 recently studied, thermophilic and mesophilic esterases (Table 4). The selected esterases represent different families and were isolated from either a single isolate or a metagenomic library. These enzymes included selected thermophilic esterases from G. obscurus26, Rhodococcus sp. LKE-02827 and Bacillus subtilis DR880628, as well as EstCS2 from a compost-soil metagenomic library29 and EstY from a metagenomic library of the Yangtze River30. The mesophilic esterases that were selected for this comparison included EstPc, a cold-adapted esterase from Psychrobacter cryohalolentis K5T31; EstC, a cold-active esterase from Streptomyces coelicolor A3(2)32; EstIM1 from a metagenomic library of mountain soil33; and Est_p1 from a metagenomic library of neritic sediments of the South China Sea34.
The characteristics of the Red Sea Atlantis II (EstATII) enzyme reflect the environment from which it was isolated. The enzyme is thermophilic (optimum temperature, 65°C). Although some previously studied thermophilic esterases exhibit higher optimal temperatures (e.g., esterases that were isolated from G. obscures26 and Anoxybacillus gonesis A435), these esterases were less active in the presence of Cu2+, Zn2+, Mn2+ and Hg2+ (Table 3). It is noteworthy that EstATII maintains significant activity at a wide range of temperatures, from 30–80°C (Table 3), is halotolerant (optimum activity at 2 M NaCl) and maintains activity in up to 4.5 M NaCl.
In addition and of particular interest, EstATII displays significant resistance to inhibition by all heavy metal ions that were tested in this work (Ca2+, Mg2+, Cu2+, Zn2+, Co2+, Mn2+, Hg2+, Fe3+and Ba2+). In particular, it was found that most of the compared esterases were particularly inhibited by three of the tested metal ions: Cu2+, Zn2+ and Hg2+. In our comparison, an enzyme was considered significantly inhibited by a given heavy metal ion if its relative activity dropped below 50%. Copper significantly inhibited 37.5% of the thermophilic esterases and 46% of the mesophilic esterases (~43% of all esterases included for comparison). Zinc inhibited 60% of the thermophilic esterases and 75% of the mesophilic esterases (~68% of all esterases included for comparison). Mercury significantly inhibited ~71.5% of the thermophilic esterases and 100% of the mesophilic esterases (~78% of all esterases included for comparison). Nineteen of the 23 esterases that were presented in our analysis exhibited significant inhibition by at least one of these three heavy metal ions. Only 4 esterases displayed resistance to inhibition by all the tested metal ions: EstATII (this study), EstR (isolated from Ralstonia sp. M1, thermophilic, optimum temperature of 60°C)36, EstA (isolated from a metagenomic library from the South China Sea, mesophilic, optimum temperature of 45°C)37 and EstH112 (isolated from the metagenome of a Korean intertidal flat sediment, mesophilic, optimum temperature of 35°C)38. However, EstATII has higher activity than EstR in the presence of 1 mM Ca2+, Mg2+, Cu2+, Mn2+, Hg2+ and Fe3+, and, as stated, EstA and EstH112 are mesophilic enzymes.
Therefore, the Red Sea Atlantis II esterase EstATII displays a combination of extremophilic properties. It is thermophilic, halotolerant and maintains significant activity in the presence of heavy metals. These characteristics make it a potentially useful biocatalyst. Additionally, the present study provides a promising example of utilizing sequence-based and activity-based metagenomics in mining for potential industrial biocatalysts.
Methods
Sample collection, DNA isolation and fosmid library construction
Water samples were collected from the Atlantis II brine pool's LCL during the KAUST Red Sea 2010 expedition (Latitude [N] 21° and Longitude [E] 38°). Collected water samples were immediately processed by serial filtration using mixed cellulose ester filters (nitrocellulose/cellulose acetate, Millipore, Ireland) with pore sizes of 3, 0.8 and 0.1 μm. Filters were stored in sucrose buffer until DNA extraction. DNA extraction from the 0.1-μm filters was performed as previously described by Rusch et al16 and the fosmid library was constructed using the Copy Control Fosmid Library Production Kit (Epicentre). The metagenomic DNA was sheared and ~40-kb DNA fragments were selected and subsequently cloned into fosmids and transformed into Escherichia coli host cells. The constructed library (fosmid vector pCC2FOS) was spread over 11 large agar plates. Next, individual colonies were picked and each colony was transferred to one well of 96-well ELISA plates, such that each well contained only one fosmid—resulting in 111 96-well ELISA plates with 10,656 clones.
Functional screening for lipolytic activity, sequencing and identification of lipolytic genes
Transformants were grown on LB agar plates supplemented with 12.5 μg chloramphenicol/mL and 1% Tributyrin (Sigma-Aldrich). The plates were incubated at 37°C for 3 days and the appearance of a clear halo zone around a transformant was indicative of candidate lipolytic activity. Candidate transformants were selected for fosmid isolation using the Wizard Plus SV Minipreps DNA Purification System (Promega).
Fosmids were digested using BamHI to assess their diversity (data not shown) and were subjected to pyrosequencing using the GS FLX Titanium pyrosequencer (454 Life Sciences). Table 1 summarizes the pyrosequencing data. The obtained sequences were assembled using a GS FLX de novo assembler and open reading frames (ORFs) were identified using the ORF Finder tool (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) that was provided by the National Center for Biotechnology Information (NCBI). The putative function of each ORF was annotated by comparing the amino acid sequences to the non-redundant protein database using BLASTP.
Sequence analysis and phylogenetic tree construction
A domain search was conducted using the Conserved Domain (CD)-search tool that was provided by NCBI (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and prediction of signal peptide sequences was performed using SignalP 3.0 servers. For the phylogenetic analysis, sequences of 43 bacterial lipolytic enzymes (representing the eight families of bacterial lipolytic enzymes as classified by Arpigny and Jaeger in 199917) were retrieved from the NCBI protein database. The 43 selected enzymes are displayed in Figure 2. Multiple sequence alignment of the retrieved sequences with EstATII was performed using the ClustalW version 1.83 program18 and a phylogenetic tree was constructed using the neighbor-joining method and the MEGA version 5.0519 software. Bootstrapping (10,000 replicates) was used to estimate the confidence of the tree.
Cloning of the EstATII gene
The EstATII gene was amplified using the forward primer (EstF) 5′-ATG TCC AGG TAC GTT GAT GAG C-3′ and the reverse primer (EstR) 5′-TCA GCT TAC CGA GTC GGT CT-3′ using DreamTaq polymerase (Fermentas). The primers were designed based on the 945-bp ORF of Contig 1 (Table 2), which was annotated as a putative lipolytic sequence by BlastP. The amplified fragment was cloned into the pET-SUMO vector (Champion™ pET SUMO Protein Expression System kit, Invitrogen) according to the manufacturer's instructions. Recombinant plasmids were transformed into E. coli BL21 (DE3) chemically competent cells and colony PCR using the gene primers was performed to verify the presence of the insert, while colony PCR using the gene's forward primer and the vector's reverse primer was performed to verify the orientation of the gene.
Overexpression and purification of the recombinant EstATII enzyme
E. coli BL21(DE3) cells (200 mL) harboring the pET-SUMO/EstATII plasmid were grown in LB at 37°C until the culture reached an OD600 = 0.4–0.6. The culture was then induced by adding isopropyl-b-D thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM and further incubated for 3 hours at 37°C. The cells were harvested by centrifugation at 10,000 × g for 15 min at 4°C. The purification procedure was performed using the His SpinTrap™ (GE Healthcare) according to manufacturer's instructions and the purified protein was dialyzed using 50 mM NaH2PO4 buffer, pH 8.0, analyzed by SDS-PAGE and stored at 4°C until further use. Protein isolation was confirmed by western blot analysis using an anti-His-G antibody (Invitrogen).
Characterization of EstATII
Enzyme activity was determined by measuring the formation of p-nitrophenol (pNP) from the enzymatic hydrolysis of the p-nitrophenyl ester, p-nitrophenyl butyrate (Sigma-Aldrich). The reaction mixture contained p-nitrophenyl butyrate (pNPB) at a final concentration of 0.1 mM and 50 mM Tris-HCl, pH 8. In this study, the standard reaction was conducted at 65°C, initiated by the addition of EstATII and terminated by the addition of 10% SDS. Measurements were performed at 410 nm using an UV-spectrophotometer (Ultrospec 3100 pro, Amersham Biosciences) unless stated otherwise20. All experiments were performed in triplicate. The substrate specificity of EstATII was determined using p-nitrophenol esters with varying side chain lengths. The short-chain fatty acid esters that were used included PNP-acetate (C2), PNP-butyrate (C4) and PNP-valerate (C5); the long-chain fatty acid esters that were used included PNP-decanoate (C10), PNP-dodecanoate C12, PNP-myristate (C14) and PNP-palmitate (C16). The optimum temperature for the activity of EstATII was determined to be within a temperature range of 30–80°C. The optimum pH for the activity of EstATII was determined to be within a pH range of 3–9.5 using the following buffers: 50 mM sodium acetate (pH 3–5.5), 50 mM sodium phosphate (pH 6 and 7.5) and 50 mM Tris-HCL (pH 7.5–9.5). Formation of pNP was measured at 348 nm (the pH-independent, isosbestic wavelength of pNP)21. The effect of the NaCl concentration on the enzyme activity was measured at different NaCl concentrations (0–4.5 M) under standard assay conditions. To assess the effect of metal ions on the enzyme activity, cations were added at a final concentration of 1 mM and the relative activity was measured using the above-described, standard conditions. The effects of detergents (at final concentrations of 0.1 and 1%) and inhibitors (final concentration, 1 mM) on the enzyme activity were tested using the above-described standard assay conditions.
GenBank accession number for the EstATII gene
KC958722
References
Qian, P. Y. et al. Vertical stratification of microbial communities in the Red Sea revealed by 16S rDNA pyrosequencing. ISME J. 5, 507–18 (2010).
Antunes, A., Ngugi, D. K. & Stingl, U. Microbiology of the Red Sea (and other) deep-sea anoxic brine lakes. Environ. Microbiol. Rep. 3, 416–433 (2011).
Swift, S. A., Bower, A. S. & Schmitt, R. W. Vertical, horizontal and temporal changes in temperature in the Atlantis II and Discovery hot brine pools, Red Sea. Deep-Sea Res. I 64, 118–128 (2012).
Lorenz, P. & Eck, J. Metagenomics and industrial applications. Nat. Rev. Microbiol. 3, 510–516 (2005).
Uchiyama, T. & Miyazaki, K. Functional metagenomics for enzyme discovery: challenges to efficient screening. Curr. Opin. Biotechnol. 20, 616–622 (2009).
Schmeisser, C., Steele, H. & Streit, W. R. Metagenomics, biotechnology with non-culturable microbes. App. Microbiol. Biotechnol. 75, 955–962 (2007).
Glogauer, A. et al. Identification and characterization of a new true lipase isolated through metagenomic approach. Microb. Cell Fact. 10, 1–15 (2011).
Jeon, J. H. et al. Novel lipolytic enzymes identified from metagenomic library of deep-sea sediment. Evid. Based Complement. Alternat. Med. 2011, 1–9 (2011).
Steele, H. L., Jaeger, K. E., Daniel, R. & Streit, W. R. Advances in recovery of novel biocatalysts from metagenomes. J. Mol. Microbiol. Biotechnol. 16, 25–37 (2009).
Zhang, T. & Han, W. J. Gene cloning and characterization of a novel esterase from activated sludge metagenome. Microb. Cell Fact. 8, 1–8 (2009).
Salameh, M. & Wiegel, J. Lipases from extremophiles and potential for industrial applications. Adv. Appl. Microbiol. 61, 253–283 (2007).
Hasan, F., Shah, A. A. & Hameed, A. Industrial applications of microbial lipases. Enzyme Microb. Technol. 39, 235–251 (2006).
Aravindan, R., Anbumathi, P. & Viruthagiri, T. Lipase applications in food industry. Indian J. Biotechnol. 6, 141–158 (2007).
Gandhi, N. N. Applications of lipase. J. Am. Oil Chem. Soc. 74, 621–634 (1997).
Hata, K., Matsukura, M., Taneda, H. & Fujita, Y. Mill-Scale Application of Enzymatic Pitch Control During Paper Production (American Chemical Society, 1996).
Rusch, D. B. et al. The Sorcerer II global ocean sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol. 5, e77 (2007).
Arpigny, J. L. & Jaeger, K. E. Bacterial lipolytic enzymes: classification and properties. J. Biochem. 343, 177–183 (1999).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–80 (1994).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–9 (2011).
Henne, A., Schmitz, R. A., Bomeke, M., Gottschalk, G. & Daniel, R. Screening of environmental DNA libraries for the presence of genes conferring lipolytic activity on Escherichia coli. Appl. Environ. Microbiol. 66, 3113–6 (2000).
Rhee, J. K., Ahn, D. G., Kim, Y. G. & Oh, J. W. New thermophilic and thermostable esterase with sequence similarity to the hormone-sensitive lipase family, cloned from a metagenomic library. Appl. Environ. Microbiol. 71, 817–25 (2005).
Litthauer, D., Abbai, N. S., Piater, L. A. & van Heerden, E. Pitfalls using tributyrin agar screening to detect lipolytic activity in metagenomic studies. Afr. J. Biotechnol. 9, 4282–4285 (2010).
Persson, B. & Argos, P. Prediction of transmembrane segments in proteins utilising multiple sequence alignments. J. Mol. Biol. 237, 182–92 (1994).
Wijeyesakere, S. J., Richardson, R. J. & Stuckey, J. A. Modeling the tertiary structure of the patatin domain of neuropathy target esterase. The Protein J. 26, 165–72 (2007).
Hausmann, S. & Jaeger, K. E. Lipolytic Enzymes from Bacteria (Springer-Verlag Berlin Heidelberg, 2010).
Jaouani, A. et al. Purification and characterization of a highly thermostable esterase from the actinobacterium Geodermatophilus obscurus strain G20. J. Basic Microbiol. 52, 653–60 (2012).
Kumara, L., Singhb, B., Adhikaric, D. K., Mukherjeed, J. & Ghoshc, D. A thermoalkaliphilic halotolerant esterase from Rhodococcus sp. LKE-028 (MTCC 5562): enzyme purification and characterization. Process Biochem. 47, 983–991 (2012).
Asoodeha, A. & Ghanbarib, T. Characterization of an extracellular thermophilic alkaline esterase produced by Bacillus subtilis DR8806. J. Mol. Catal. B: Enzym. 85, 49–55 (2013).
Kang, C.-H. et al. A novel family VII esterase with industrial potential from compost metagenomic library. Microb. Cell Fact. 10, 1–8 (2011).
Wu, C. & Sun, B. Identification of novel esterase from metagenomic library of Yangtze River. J. Microbiol. Biotechnol. 19, 187–193 (2009).
Novototskaya-Vlasova, K., Petrovskaya, L., Yakimov, S. & Gilichinsky, D. Cloning, purification and characterization of a cold-adapted esterase produced by Psychrobacter cryohalolentis K5T from Siberian cryopeg. FEMS Microbiol. Ecol. 82, 367–75 (2012).
Brault, G., Shareck, F., Hurtubise, Y., Le'pine, F. & Doucet, N. Isolation and characterization of EstC, a new cold-active esterase from Streptomyces coelicolor A3(2). PLoS One 7, e32041 (2012).
Ko, K. C. et al. Identification and characterization of a novel cold-adapted esterase from a metagenomic library of mountain soil. J. Ind. Microbiol. Biotechnol. 39, 681–9 (2012).
Peng, Q. et al. A novel esterase gene cloned from a metagenomic library from neritic sediments of the South China Sea. Microb. Cell Fact. 10, 1–11 (2011).
Faiz, Ö., Colak, A., Saglam, N., Çanakçi, S. & Beldüz, A. O. Determination and characterization of thermostable esterolytic activity from a novel thermophilic bacterium Anoxybacillus gonensis A4. J. Biochem. Mol. Biol. 40, 588–594 (2007).
Quyen, D. T., Dao, T. T. & Thanh Nguyen, S. L. A novel esterase from Ralstonia sp. M1: gene cloning, sequencing, high-level expression and characterization. Protein Expr. Purification 51, 133–140 (2007).
Chu, X., He, H., Guo, C. & Sun, B. Identification of two novel esterases from a marine metagenomic library derived from South China Sea. Appl. Microbiol. Biotechnol. 80, 615–25 (2008).
Oh, K. H. et al. Characterization of a novel esterase isolated from intertidal flat metagenome and its tertiary alcohols synthesis. J. Mol. Catal. B: Enzym. 80, 67–73 (2012).
Rao, L. et al. A thermostable esterase from Thermoanaerobacter tengcongensis opening up a new family of bacterial lipolytic enzymes. Biochimica et Biophysica Acta 1814, 1695–1702 (2011).
Soliman, N. A., Knoll, M., Abdel-Fattah, Y. R., Schmid, R. D. & Lange, S. Molecular cloning and characterization of thermostable esterase and lipase from Geobacillus thermoleovorans YN isolated from desert soil in Egypt. Process Biochem. 42, 1090–1100 (2007).
Luo, Z. H. et al. Purification and characterization of an intracellular esterase from a Fusarium species capable of degrading dimethyl terephthalate. Process Biochem. 47, 687–693 (2012).
Fu, J. et al. Functional and structural studies of a novel cold-adapted esterase from an Arctic intertidal metagenomic library. App. Microbiol. Biotechnol. 97, 3965–78 (2012).
Metin, K., Ateslier, Z. B. B., Basbulbul, G. & Biyik, H. H. Characterization of esterase activity in Geobacillus sp. HBB-4. J. Basic Microbiol. 46, 400–409 (2006).
Jeon, J. H. et al. Identification of a new subfamily of salt-tolerant esterases from a metagenomic library of tidal flat sediment. App. Microbiol. Biotechnol. 93, 623–31 (2012).
Acknowledgements
This work was supported by King Abdullah University for Science and Technology Global Collaborative Partners (GCR) program. YMM was partly funded by an Al-Alfi Biotechnology Fellowship.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: R.S., H.D., Y.M.M. Performed the experiments: Y.M.M., M.G., A.S., A.O. Analyzed the data: R.S., Y.M.M., H.D. Wrote the paper: Y.M.M., R.S.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Mohamed, Y., Ghazy, M., Sayed, A. et al. Isolation and characterization of a heavy metal-resistant, thermophilic esterase from a Red Sea Brine Pool. Sci Rep 3, 3358 (2013). https://doi.org/10.1038/srep03358
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03358
This article is cited by
-
Metagenomic profiling of antibiotic resistance genes in Red Sea brine pools
Archives of Microbiology (2023)
-
Molecular study on recombinant cold-adapted, detergent- and alkali stable esterase (EstRag) from Lysinibacillus sp.: a member of family VI
World Journal of Microbiology and Biotechnology (2022)
-
Magnetic Molecularly Imprinted Polymer (MMIP) Mediated Bacterial Esterase-Based Assay for Captan Detection in Milk
Food Analytical Methods (2022)
-
Structure-guided protein engineering increases enzymatic activities of the SGNH family esterases
Biotechnology for Biofuels (2020)
-
Anthropogenic Pb contribution in soils of Southeast China estimated by Pb isotopic ratios
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
