
Abstract
Targeting embryonic stem cells (ESCs) is important for ESC labeling, drug delivery and cell fate control. In this study, we identified twenty-two phage clones that bind specifically to the hESC cell line X-01, which was derived from human blastocysts of Chinese origin. One phage (H178), which displays the sequence VGGEAWSSPTDL, showed higher binding affinity to hESCs than to a monkey ES cell line (RS366.4) and two mouse ES cell lines (R1 and E14). Using quantum dots (QDs) conjugated to the H178 phage, we demonstrate that the phage can specifically bind to hESCs in vitro. Our results suggest a possible interaction between the selected peptide and the stem cell extracellular matrix (ECM). The selection method described here allows rapid and efficient screening of unique phage clones and targeting cells. The phages displaying peptides identified by this study have potential applications for cargo delivery and receptor studies.
Similar content being viewed by others


Introduction
Human embryonic stem cells (hESCs) have great potential for use in regenerative medicine, drug discovery and personalized therapy owing to their ability to differentiate into different cell lineages but also to maintain an undifferentiated state in vitro1,2,3. Specific interactions with the microenvironment are important for embryonic stem cell maintenance, survival and differentiation. Therefore, identifying ligands that interact specifically with unique sites on the ESC surface may provide a way to target receptors and modulate their activities, enhance the efficiency of cargo delivery (of drugs, nano-particles and fluorophores) and control the stem cell fate decision.
Recent progress has been made in using phage display technology4 to identify stem cell-specific biomarkers5,6,7,8,9,10. Phage-displayed peptides can be used to deliver cargo or to perform functions such as homing to bone marrow and binding to primitive hematopoietic stem cells7, supporting EC/ES cell cultures5, labeling mouse and rhesus monkey ES cells8,9 and discovering new membrane receptors10. These peptides can also be used as lead compounds in drug design and as alternatives to antibodies for target validation in genomics and drug discovery both in vitro and in vivo11,12. There are several advantages of this bacteriophage amplification system: 1) phage clones display sequences identical to specific peptides; 2) phage clones are simple to prepare and purify; 3) phage recombination can produce induced pluripotent stem cells (iPSCs) with intact endogenous gene function13; and 4) phages can tolerate drastic modifications to their coat proteins and serve as a solid support for organic synthesis14. Owing to the specificity, stability and safety of phages, some groups report that chemically modified or fluorophore-conjugated phages can be used specifically to detect bacteria15,16,17,18,19 and cancer cells19,20. However, using phages to deliver cargo to human ESCs has not been previously reported.
Quantum dots are resistant to photo bleaching, highly sensitive and non-invasive and are thus ideal candidates for ESC labeling21,22. In the current study, by combining the advantages of phages and QDs, we have developed a rapid and specific human ESC targeting method. Twenty-two phage clones that specifically bind to the hESC line X-01 were identified from 190 selected phage clones. The rate of binding of these phage clones to the hESC line X-01, monkey ESC line RS366.4 and mouse ESC lines R1 and E14 were compared to identify the most hESC-specific phage. The selected phage, H178, when chemically conjugated to water-soluble QDs, targeted hESCs in vitro. Cell labeling and homology analysis suggest a possible interaction between the phage and the stem cell extracellular matrix (ECM). The QD-phage conjugate described here provides a rapid and effective platform for cell targeting and has potential applications for drug delivery and receptor studies.
Results
Screening of specific ESC-binding phages
The human ESC X-01 cell line was cultured on CF-1 mouse embryo fibroblast (MEF) feeder cells. The cells displayed the compact colony morphology that is typical of ESCs, with high nuclear to cytoplasmic ratios and well-defined colony borders (Supplementary Fig. S1, A). ESCs express more alkaline phosphatase (AKP) than the MEF feeder cells and differentiated cells and therefore stain darker (Supplementary Fig. S1, A, C). Immunocytochemical staining revealed that the hESCs line expressed transcription factors such as Nanog and Oct4 and ES-specific cell surface markers, including SSEA-4 and TRA-1-60. RT-PCR analysis demonstrated that all of the ESC lines used in this study express high levels of hESC marker genes, including Oct4, Nanog and Sox2 (Supplementary Fig.S1,D). The differentiated hESCs did not display the compact morphology characteristic of undifferentiated ESCs (Supplementary Fig. S1, A, b) and did not express AKP or the ES-specific marker genes (Supplementary Fig.S1, A, D).
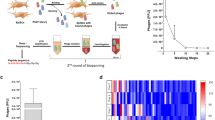
The phage pool was enriched in phages that specifically bound to hESCs by two rounds of bio-panning. To eliminate the possibility of selecting non-relevant phages, each round included two subtraction steps using d-ESCs (differentiated human ESCs), two subtractions steps using CF-1 feeder cells and one selection step using hESCs, as shown in Fig. 1A. The Tween concentration used in the wash buffer increased from 0.5% to 1% during the screening process. The phage pools exhibited an approximately 300-fold increase in phage yield rate from 1.13 × 10−7 to 3.87 × 10−5 (Table 1). This increase after two rounds of screening indicates that bio-panning facilitates the identification of phages that bind to hESCs. Although the panning procedure led to enrichment of specific phages, many nonspecific phages were still retained in the pool, therefore further re-selection was performed as described below.

Experimental scheme.
(A) Phages that specifically bind to human ESCs were enriched in the phage pool by two rounds of bio-panning. Each bio-panning round included two subtraction steps with dESCs, two subtraction steps with CF-1 cells and a selection step with ESCs. Monoclonal phages were enriched, purified and re-screened by enzyme linked immunosorbent assay (ELISA). Phages that showed high binding affinity and specificity were further confirmed by immunofluorescence microscopy and ELISA. The selected phages were then chemically linked to QDs and tested for hESC targeting. (B) Illustration of chemical conjugation between the phage and QDs. (C) This enlarged view of (B) shows how the –NH2 groups on the phage are conjugated to the free –COOH groups on the surface of the QDs via EDC.
Re-selection and confirmation of binding specificity
A total of 190 monoclonal phages were randomly selected from the phage pool. These phages were amplified and titered and their binding affinities to hESCs, CF-1 cells and d-ESCs were tested by whole-cell ELISA (data not shown). The ELISA assay was the second step in the double selection screening procedure. Among these phage clones, 22 monoclonal phages (H3, H48, H66, H68, H123, H134, H138, H143, H146, H149, H150, H151, H158, H161, H163, H166, H173, H175, H178, H179, H182 and H185) were chosen because of their high binding affinity for hESCs and relatively low binding affinity for the CF-1 feeder cells and d-ESCs, as shown in Fig. 2A (the experiment was performed in triplicate). The phage clones identified by this double selection procedure specifically bound hESCs. The phage H21, which showed a high binding affinity for both the CF-1 feeder cells and the hESCs, was used as a positive control for MEFs and BSA was used as a negative control.

Binding affinity confirmation.
(A) Binding affinity of selected phages. The 22 selected phage clones were added to human ESCs, dESCs and CF-1 feeder cells to perform the whole-cell ELISA assay. The H21 phage was used as the MEF positive control and BSA was used as the negative control. The results are presented as the percent of binding (absorption value of a selected phage at 492 nm/absorption value of BSA control at 492 nm), with the control (BSA) value set as 100%. The error bars represent 95% confidence intervals. ANOVA test: **P<0.01; *P<0.05. (B) Immunofluorescence assay. The cell nuclei are shown in blue (Hoechst 33258) and the phages are shown in green. Scale bar, 100 μM. (C) Fluorescence signal intensity analysis. Values were calculated as the average green fluorescence intensity. Three fields of view per well were measured for each experiment. Values are shown as the mean ± standard deviation.
Immunocytochemistry assays showed that the selected phage clones directly interacted with hESCs, therefore confirming the results from the binding assay. Staining with a FITC-conjugated anti-M13 antibody showed that all of the selected phages bind to hESCs (Fig. 2B). In contrast, the BSA control (in which BSA and the anti-M13 antibody were added to the hESCs) showed very little fluorescence (Fig. 2B, control). These results demonstrate that the selected phages can bind to hESCs. As the phage clones showed different intensities of staining, we performed a fluorescence signal intensity analysis (three fields of view were examined per well). The obtained images were visualized using two channels, one for green fluorescence (phage) and one for blue fluorescence (cell nuclei stained with Hoechst 33258), as shown in Fig.S2. The intensity of the green fluorescence signal in a defined area was calculated using OriginPro software and the values are shown in Fig. 2C. The binding affinity of the phage clones to the targeted cells, the phage-hESC binding ratios (Fig. 2A) and the fluorescent signal intensity analysis confirmed that the selected phage clones differ in their ability to interact with hESCs. We therefore performed sequences analyses to determine the peptide sequences displayed by the selected phage.
Comparison of binding ability between different species and DNA sequences analyses
To identify the phages that target only hESCs, the binding ability of 22 selected phages was assessed in the hESC cell line X-01, the monkey ESC cell line RS366.4 and the mouse ESC cell lines R1 and E14. Virtually identical phage binding rates were observed in the R1 and E14 cell lines (Fig. 3A). However, some of the peptides, namely, H149, H151, H158, H161, H163, H166, H173, H175, H178, H179, H182 and H185, showed significantly different binding rates (P<0.01) in monkey ESCs, hESCs and mouse ESCs. These results suggest that differences between species may affect ESC recognition by phages and that the receptor molecule may not be the same in different species.

Re-selection and homology analysis.
(A) The 22 selected phage clones were added to human ESCs, monkey ESCs (RS366.4) and mouse ESCs (R1 or E14). The H21 phage and BSA were used as negative controls. The results are presented as the percent of the absorption value (mean ± standard deviation) in the ELISA assay compared to the controls, which were set as 100%. The error bars represent 95% confidence intervals. ANOVA test: **P<0.01; *P<0.05. (B) Five types of homologous sequences were identified. The putative consensus motifs are shown beneath each type. (C) Structural models of the H166 peptide (a) and the h178 peptide (b). The peptides exhibited structural similarity at the N-terminus and the H178 peptide contained a coil domain. The peptides were attached to the phage by their C-termini. The PDB models were generated using PEP-FOLD34.
The selected phage clones were extracted and sequenced as shown in Table 2. All of the sequences were unique; that is, no sequence was isolated more than once. A homology analysis revealed five types of homologous sequences (HL+T, VPHSS-TP, YA+N-LM-A and ND-VA motifs), as shown in Fig. 3B. Two phages displaying the sequences HGAAWGTRTGHV (H166) and VGGEAWSSPTDL (H178) shared the G-AW—T motif, as shown in Fig. 3B (highlighted with a red rectangle). These two phages also exhibited a similarly high binding affinity for hESCs and low binding affinity for mESCs and rESCs. Structural modeling showed that the H166 and H178 peptides have a similar structure at their N-terminus (Fig. 3C,a) and that there is a coil structure in the middle of H178 (Fig. 3C,b). Therefore, the slight differences in binding ability between these two phages may reflect the different spatial structures of the peptides. These results also suggest that the G-AW motif in the N terminus of these two peptides may be an important core structure for hESC binding. The coil in the H178 peptide may contribute to its higher binding affinity for hESCs as compared to the H166 phage.
We next used chemically synthesized peptides to test whether the HGAAWGTRTGHV and VGGEAWSSPTDL sequences could compete with the H166 phage and H178 phage for binding to hESCs. Both synthetic peptides competed with the H166 and H178 phages, achieving 50% inhibition at approximately 600 nM (Fig. 4). Two synthesized peptides with randomly scrambled sequences (THAGHAGVGRWT and DTSAGVLSWGEP), which shows no significant homology to the two peptides tested here, did not compete with the H166 phage and H178 phage in binding to hESCs. This demonstrates that the binding we observed was sequence-specific.

Competitive binding assay with the H166 and H178 phage and free peptide.
The chemically synthesized H166 (HGAAWGTRTGHV) and H178 (HGAAWGTRTGHV) peptides were used in a competitive binding assay with the H166 phage and the H178 phage. The phages were added to hESCs, followed by the peptides at concentrations ranging from 10 nM to 10,000 nM. THAGHAGVGRWT and DTSAGVLSWGEP peptides were used as the control. The binding ratio was calculated by dividing the number of phages recovered in the presence of the peptide by the number of phages recovered in the absence of peptide. The values are shown as the mean ± standard deviation.
Homology analysis
Previous studies have shown that peptides identified by bio-panning often interact specifically with the active ligand binding sites of the membrane receptors, most likely due to similarities between the identified peptide and the native ligand23,24. A search of GenBank identified several Homo sapiens proteins that may be related to the peptide sequences investigated here. The peptide HGAAWGTRTGHV, displayed by phage H166 (Supplementary Table S2), shows homology to many proteins, including membrane receptors (T-cell receptor beta chain, G protein-coupled receptor PGR6, CD163 antigen, transmembrane protein 200B and scavenger receptor cysteine-rich type 1) and ECM proteins (fibrillin, M130 antigen extracellular variant and annexin A9), as well as some proteins with unknown functions. Among these proteins, several secreted polypeptides, the M130 antigen extracellular variant and the CD163 antigen and its isoforms share the HG-WGT motif and contain AWGT repeats. One of the proteins with an unknown function contains several GTRT repeats. Other non-repeating motifs are also present in these homologous proteins. For example, PGR6 contains an HGAAW motif, fibrillin contains a GAAWGT motif and the T-cell receptor beta chain and CDR3 proteins both contain an AW-TRTG motif. The list of proteins homologous to the H178 peptide (VGGEAWSSPTDL), shown in Supplementary Table S3, contains many membrane receptors and ECM receptors, including lectin, immunoglobulin receptors, the T-cell receptor, FRAS1-related extracellular matrix 1 and collagen alpha-1. Lectin and the galectin-3-binding protein precursor share the EAW-S-PTDL motif, the T-cell receptor beta chain contains the AWS-PT motif, the mucin-17 precursor has many repeating EA-SSPT and SSPT motifs and the complement receptor type 1 isoform S precursor contains WSSP repeats. Therefore, our homology analysis suggests that the H166 and H178 peptides may interact with proteins in the hESC membrane or extracellular matrix (ECM).
Targeting ESCs with quantum dot (QD)-conjugated phages
Free -COOH groups on the surface of the QDs can be conjugated with phage coat proteins containing -NH2 groups via EDC, as shown in Fig. 1B,1C. In this method, non-conjugated QDs and phages are removed by centrifugation and washing and non-specific fragments are removed using an enhanced wash step during the labeling process. To evaluate whether a QD-phage conjugate can effectively target hESCs, H178 phage were conjugated to water-soluble CdSe-ZnS QDs. The resulting QD-phage conjugate was then incubated with cells and visualized using confocal laser scanning microscopy. The conjugates bound efficiently to the hESCs, but did not significantly bind to the rESCs, mESCs and MEFs. As shown in Fig. 5a,5b, we detected red signals indicating the presence of the QD-phage conjugates on the hESCs, but not on the rESCs (monkey ESCs) (Fig. 5c), mESCs (mouse ESCs R1) (Fig. 5d), or CF-1 MEFs (Fig. 5e). The QD signals specifically appeared on the borders of the hESCs (arrow) where no blue fluorescence (indicating the cell nucleus) was detected. No binding signals were observed when peptide-free QDs were used (Fig. 5B,f).

Targeting hESCs with QD-labeled phages.
(a, b) QD-labeled H178 phage showed strong red fluorescence signals when added to hESCs, whereas the rESC (rhesus monkey ESCs) (c) and CF1-MEFs (e) showed no significant red fluorescence. (d) The mESCs (mouse cell line ESC-R1) showed non-specific staining. Arrows show red fluorescence staining around the borders of the hESC clone and the red fluorescence does not overlap with any blue signal (Hoechest 33258), which indicates the location of the cell nuclei. (f) QDs added to hESCs was used as the control. Scale bar, 100 μM (with the exception of (b) and (e), in which the scale bar represents 50 μm).
Discussion
Phage display is an effective method for identifying unique peptide ligands that are specific to ESCs. The method used in this study eliminated the possibility of selecting non-relevant phages and generated a pool of phages that bind specifically to hESCs. Two subtraction steps were performed using CF-1 feeder cells and d-ESCs before each selection step. Recent studies show that two problems may be encountered when performing phage display-based bio-panning. One is the decreased diversity of phage pools during the amplification of phages in bacteria25,26,27. Another is the additional rounds of screening that are required, as this increases the risk of losing promising clones, which propagate slower than non-binding phages28. Other difficulties include recovering hESCs from their frozen state and maintaining hESC pluripotency during long-term in vitro culture29. Therefore, a shorter culture time and fewer rounds of selection are preferable during the screening process. It has been reported that one round of screening is sufficient for phage pool enrichment28. Therefore, improved results are obtained by reducing the number of panning rounds and using an ELISA assay for the double selection of phage clones. All of the sequences described here are novel and have not been previously reported30.
Differences in ESC markers exist in different species31,32. Our ELISA results show that the selected phage clones had similar binding rates for the two mouse ESC lines but distinct binding affinities between human, mouse and macaque ESC cell lines. These results may be due to differences in membrane receptors, ECM content and protein glycosylation among the ESCs from these species. The H166 and H178 peptides described here could be used as tools for specifically targeting hESCs. The identification of these two phages may lead to the discovery of the corresponding hESC receptors in the future.
Membrane receptors are composed of extracellular, transmembrane and intracellular domains. Each domain is a spatially distinct part of the molecule with a unique function. The most important part of a membrane receptor in terms of drug or cargo delivery is the extracellular domain (ectodomain), which contains one or more ligand binding sites12. Our homology analysis suggests that the H166 and H178 peptides may interact with proteins in the hESC membrane or extracellular matrix (ECM). The QD-phage labeling results suggest that the H178 peptide interacts with the edge parts of the HESCs clones, which are the domains of ECM. The identification of these two novel peptide-ESC ligands provides useful tools for future hESC research. Further study is needed to understand the biological functions of these peptides and identify their membrane receptors.
Methods
Cell lines and cell culture
The hESC cell line X-0133, provided by the Stem Cell Bank, Chinese Academy of Sciences, was cultured on a monolayer of irradiated CF1 feeder cells (Sidansai Biotechnology, Shanghai) in hESC growth medium consisting of Dulbecco's modified Eagle's medium (DMEM)/F12 with GLUTAMAX (Invitrogen), 20% knock-out serum replacement (Invitrogen), 4 ng/mL basic fibroblast growth factor (bFGF; Invitrogen), 1% non-essential amino acids (Invitrogen) and 0.1 mmol/L β-mercaptoethanol (Sigma). The cells were grown at 37°C with 5% CO2. Irradiated CF1 feeder cells were seeded to matrix gel-coated culture flasks at a density of 30,000 cells/cm2 two days before hESC passage. The hESCs were passaged approximately once a week. Collagenase IV (5 mg/mL, Invitrogen) was used to dissociate the human ES cell clumps from the feeder cells. The rhesus monkey ESC line RS366.4, kindly provided by the Wisconsin Regional Primate Research Center (University of Wisconsin, USA), was cultured on mitomycin C (10 μg/mL, Roche)-treated ICR feeder cells, which were seeded to 0.1% gelatin-coated flasks. The rESC medium consisted of 85% Dulbecco's modified Eagle's medium (Hyclone, Logan, UT, USA), 15% fetal bovine serum (FBS) (Gibco BRL), 1% non-essential amino acids (Gibco BRL, USA), 1 mM L-glutamine (Gibco BRL) and 0.1 mM β-mercaptoethanol (Amresco, Solon, OH, USA). The cells were grown at 37°C with 5% CO2. The rESCs were passaged approximately every four days. Dispase (5 mg/mL, Invitrogen) was used to dissociate the macaque ES cell clumps from the feeder cells. The mouse ESC cell lines E14 and R1 were obtained from the Joint Laboratory of Stem Cell Research (Shenzhen, China) and the Stem Cell Bank, Chinese Academy of Sciences, respectively. The cells were cultured on mitomycin C (10 μg/ML, Roche, Shanghai)-treated ICR feeder cells, which were seeded to 0.1% gelatin-coated flasks. The mESC growth medium consisted of knockout DMEM (Gibco) containing 15% fetal bovine serum, 0.5% 200 mM L-glutamine solution, 1% non-essential amino acids, 0.2% 55 mM 2-β-mercaptoethanol solution, 1% penicillin–streptomycin (Gibco 100× solution) and 1000 units/ml LIF (Chemicon). The cells were grown at 37°C with 5% CO2. The mESCs were passaged approximately every three days. Trypsin (0.25%) was used to remove the mouse ES cells and the ICR cells and the colonies were dissociated into a single cell suspension by vigorous pipetting. The single cell suspension was then plated to a 100 mM tissue culture dish and incubated at 37°C for 45 to 60 min to remove the MEF feeder cells. The non-adherent cells were collected, centrifuged and plated on top of the ICR feeder cells.
Cell preparation
The hESC clones were recovered by scraping and collected in 15 ML tubes for splitting or phage display screening. The remaining cells were cultured with feeder cells in 90% DMEM (Gibco) and 10% fetal bovine serum at 37°C with 5% CO2. After three days of culture, the cells were split directly into cell culture flasks and were cultured continuously for 21 days.
Alkaline phosphatase (AKP) assay
Undifferentiated ESCs were added to a 24-well microtiter plate (Falcon) pre-coated with matrix gel (for human ESCs) or CF1 feeder cells and 0.1% gelatin (for monkey and mouse ESCs) and incubated for 24 h. The plates were then washed twice with PBS and the cells were fixed in 4% paraformaldehyde (in PBS) for 10 min. The plates were then washed three times with PBS. Next, the cells were incubated with a colorizing solution (containing 6.6 μL of nitrotetrazolium blue chloride solution, made by dissolving 5 mg of NBT in 100 μL of 70% DMSO; 3.3 μL of BCIP, made by dissolving 5 mg of BCIP in 100 μL of 100% DMSO; and 1 mL of Tris-HCl containing 100 mM Tris, 100 mM NaCl, and1 mM MgCl2, pH 9.5) at 37°C for 1 h. The plates were then washed three times with PBS and the ESCs were collected and washed twice with PBS. The cells were then lysed in cell lysis buffer (50 mM pH 8.0 Tris-HCl, 4 M urea, 1% of a 20% Triton X-100 solution and one Complete Protease Inhibitor Cocktail tablet (Roche) in 10 mL of solution) on ice for 20 min and assayed using the AKP kit (Millipore, Merck).
Immunostaining
Undifferentiated human ESCs were added to a 24-well microtiter plate (Falcon) pre-coated with matrix gel and cultured in conditioned ESC medium (hESC medium collected from irradiated CF1 feeder cells after 24 h of growth). Antibodies to Nanog (1:150; R&D Systems, USA), Oct4 (1:100; Santa Cruz, USA), SSEA-4 (Ascites; 1:400; Developmental Studies Hybridoma Bank) and TRA-1-60 (1:150; Chemicon, USA) were used to analyze the pluripotency. The ESC colonies grown on feeder cells were first washed with phosphate buffered saline (PBS) and then fixed for 30 min at room temperature in 4% paraformaldehyde. After permeabilization, the cells were washed once in PBS and twice in an antibody dilution buffer (PBS with 0.2% BSA and 0.1% Triton X-100; 5 min for each wash) and blocked in a blocking solution (PBS with 1% BSA, 4% normal serum and 0.4% Triton X-100). The cells were incubated with the primary antibodies in antibody dilution buffer overnight at 4°C. The cells were washed three to five times with PBS containing 0.1% Triton X-100 (PBS-T) and then incubated with secondary antibodies in antibody dilution buffer for 1 h at room temperature in the dark. Next, the cells were washed three times with PBS-T and incubated with DAPI at a final concentration of 1 mg/mL in PBS for 5 min. Finally, the cells were washed twice (5 min each) with PBS and visualized with a fluorescent microscope (Olympus, Japan).
RT-PCR for ESC marker genes
Total RNA was extracted from ESCs with Trizol (Gibco BRL) and reverse transcribed into cDNA using a One-Step RNA PCR Kit (Takara). The cDNA was then amplified in a 20 μL reaction using the primers described in Supplementary Table S1. After 35 amplification cycles, the PCR products were subjected to gel electrophoresis on a 1.3% agarose gel containing 0.1 μL/mL of an ethidium bromide alternative (Applygen Co.).
Biopanning in vitro
The hESCs clumps were quickly digested with 0.05% trypsin in 0.53 mM EDTA to dissociate them into a single cell suspension. The cells were then washed twice in hESC medium and resuspended in blocking buffer (3% BSA in PBS) for use in the phage display screening assay. Differentiated hESCs and CF1 cells were digested with 0.25% trypsin in 0.53 mM EDTA, washed twice in PBS and resuspended in blocking buffer. A Ph.D-12 peptide phage display library was used for bio-panning (New England Biolabs, Beijing, China). The kit includes a combinatorial library of random peptide 12-mers fused to the N-terminus of a minor coat protein (PIII) in the M13 phage. A negative/positive screening process was performed to enrich for hESC-targeting phage. For the first round of panning, a 10 μL phage library containing 1.5 × 1011 phages was added to 2 mL of dESCs (differentiated hESCs) in suspension (2 × 106), prepared as described above. The mixture was incubated at room temperature (RT) for 1 h with mild agitation. The supernatant, which contained the phages that did not bind to the dESCs, was recovered by centrifugation (3000 rpm for 3 min), added to the prepared CF-1 feeder cells (2 × 106) and incubated at RT for 1 h with mild agitation. The supernatant, which contained the phages that did not bind to the CF-1 cells, was then added to 2 × 107 hESCs and incubated at RT for 90 min with mild agitation. The cells were then washed five times with wash buffer (PBS, 0.5% Tween-20) and the phages bound to the hESCs were eluted using 1 mL of elution buffer (0.1 M glycine–HCl, 0.1% BSA, pH 2.2). The sample was then neutralized with 188 μL of a neutralizing buffer solution (1 M Tris-HCl, pH 9.1). The eluted phages were titered and amplified according to the manufacturer's instructions, using the E. coli strain ER2738. This procedure was performed twice. The phage pool binding efficiency was calculated as the ratio of the phage recovered to the input titer for each round of screening.
Whole-cell enzyme-linked immunosorbent assay
A whole-cell ELISA assay was used for further selection of the randomly picked phage clones. ESCs were pre-seeded in 96-well plates (hESCs were added to a matrix-gel coated plate with conditioned hESC medium, rESCs to a 0.1% gelatin-coated plate with rESC medium, mESCs to a 0.1% gelatin coated plate with mESC medium and MEFs were seeded directly to a plate with MEF medium). The cells were grown to confluency over 24 hours and fixed with 4% paraformaldehyde for 15 min at RT. The cells were then washed three times with PBS for 5 min each. A 250 μL volume of blocking buffer (3% BSA-PBS) was then added to each well and the plate was incubated for 2 h at 37°C. The cells were then washed three times for 5 min per wash using PBS. The selected phage clones (1 × 109), suspended in 200 μL blocking buffer, were added to each well and the plates were incubated at RT for 90 min with mild agitation. The cells were then washed three times (5 min per wash) using 0.1% Tween-20-PBS, washed three times with PBS and incubated with an HRP–conjugated mouse anti-M13 antibody (diluted 1:1000 in a blocking buffer) at RT for 90 min. After repeated 5 min washes in PBS, the supernatants were aspirated and the color reaction was developed by incubating the cells in 100 μL/well o-Phenylenediamine for 20 min in the dark at RT. The color reaction was stopped by adding 50 μL/well 2 M H2SO4. The supernatants were transferred to high-binding ELISA plates (BBI, China) and the absorbance at 492 nm was read on a GENios microplate reader (Tecan, Männedorf, Switzerland). The absorbance at 405 nm was taken as a reference. Each experiment was performed in triplicate. Data analysis was performed using OriginPro software. The results are presented as the percent of binding (absorption value of the selected phages at 492 nm/absorption value of BSA control at 492 nm), with the control (BSA) value set as 100%. The results presented are from three independent experiments (n = 3). A statistical analysis was performed using ANOVA test with statistical significance of P < 0.05 and P< 0.01.
Phage DNA sequencing and homology analysis
Phage plaques from the final round of bio-panning were amplified and purified according to the instructions provided by the manufacturer of the PH.D-12 library. After re-selection by whole-cell ELISA, the selected monoclonal DNA phages were sequenced at Invitrogen, Guangzhou, China. Multiple sequence alignments and the identification of small peptide motifs were performed using CLUSTALX 2.0.10 (EMBL-EBI, http://www.ebi.ac.uk/Tools/clustalw2). The 3D peptide model was constructed using PEP-FOLD34. NCBI protein-protein BLASTP 2.2.19 was used for the GenBank search35.
Immunocytochemistry of the selected phage
Human ESCs were added to a 24-well microtiter plate (Falcon) pre-coated with matrix gel and cultured in conditioned medium for 24 h. The cells were then fixed in 4% paraformaldehyde (in PBS) for 15 min and the plate was washed three times with PBS. Next, the cells were blocked with 3% BSA in PBS for 1 h at 37°C and incubated with positive phage particles (approximately 4 × 109 cfu) and an HRP-conjugated anti-M13 phage antibody at 37°C for 1.5 h. The cells were washed five times with PBS and three times with 0.1% PBS-T and incubated with a FITC-IgG antibody (Sigma) for 1 h in the dark. The cells were also incubated with Hoechst 33258 (5 μg/ml, Sigma) for 15 min. The cells were then visualized using an Eclipse TE2000 fluorescence microscope (Nikon). The images were separated into a green channel (phage fluorescence) and a blue channel (nuclear fluorescence) using Photoshop (Fig.S2). The signal intensity of the green fluorescence was calculated by averaging the intensity of three fields of view per well using Image-Pro Plus 6.3 (Media Cybernetics).
Determination of binding efficiency and specificity by competition with free peptides
The H166 peptide (HGAAWGTRTGHV), the H178 peptide (VGGEAWSSPTDL), two synthesized peptides with randomly scrambled sequences (THAGHAGVGRWT and DTSAGVLSWGEP) were synthesized at Sinoasis Pharmaceuticals, Guangzhou, China. The synthetic peptides were added to the cells at various concentrations ranging from 1 nM to 10 μM before phage was added during the biopanning process, as described above. The ratio of phage binding to target cells was calculated as the number of phages recovered after peptide competition divided by the number of phages recovered with no peptide competition. The random (control) peptide was used as a negative control. The results presented represent the mean ± standard error.
Phage labeling with QDs
The prepared QDs36,37 were conjugated to the phage using an EDC/NHS-mediated method. Approximately 5.8 mg of water-soluble QDs was mixed with 2 mM NHS and 5 mM EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) in MES-buffered saline at pH 4.7 at 37°C for 30 min to form an amine-reactive sulfo-NHS ester. All chemicals were purchased from Sigma-Aldrich. The phages were then washed with 50 mM borate buffer (pH 8.5) and centrifuged at 10,000 × g for 30 min at 4°C (Beckman Avanti 30, F1202 rotor). The activated water-soluble QDs were suspended in the same buffer. Then, 1 mL of phage (109 PFU/μl) was added. The solution was incubated at RT for 2 h to allow for conjugation. Next, the conjugates were blocked in a 5% BSA solution at 37°C for 30 min. The resulting QD-phage conjugate was washed several times and stored at 4°C, pH 7.4 before use. Additional detail regarding the water-soluble QDs and the chemical conjugation process will be published elsewhere.
Cell targeting assay
Human ESC clumps were seeded on matrix-gel coated chamber well slides (Shanghai Sango Biological Engineering Technology, Shanghai, China) in conditioned media and cultured overnight at 37°C with 5% CO2. The chambers were then washed twice with PBS and the cells were fixed with 4% paraformaldehyde for 15 min at RT, washed three times with PBS and incubated in blocking buffer (1% BSA/PBS) for 2 h at 37°C. Next, the cells were incubated with QD-labeled phage (10 μL diluted with 200 μL of 1% BSA/PBS) and QD-labeled peptides for 1 h at 37°C, followed by three washes with PBS, three washes with PBS containing 0.1% Tween-20 and three washes with PBS containing 0.5% Tween-20. The cells were then counterstained with Hoechst 33258 (Invitrogen), mounted in PBS and visualized by confocal laser scanning microscopy (FV1000, OLYMPUS).
References
Biswas, A. & Hutchins, R. Embryonic Stem Cells. Stem Cells Dev. 16, 213–222 (2007).
Pouton, C. W. & Haynes, J. M. Embryonic stem cells as a source of models for drug discovery. Nat Rev Drug Discov. 6, 605–616 (2007).
Wobus, A. M. & Boheler, K. R. Embryonic stem cells: prospects for developmental biology and cell therapy. Physiol Rev. 85, 635–678 (2005).
Smith, G. P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 228, 1315–1317 (1985).
Derda, R. et al. High-Throughput Discovery of Synthetic Surfaces That Support Proliferation of Pluripotent Cells. J Am Chem Soc. 132, 1289–1295 (2010).
Lu, S. et al. Targeting of Embryonic Stem Cells by Peptide-Conjugated Quantum Dots. Plos One. 5, e12075 (2010).
Nowakowski, G. S. et al. A specific heptapeptide from a phage display peptide library homes to bone marrow and binds to primitive hematopoietic stem cells. Stem Cells. 22, 1030–1038 (2004).
Zhao, S. J., Zhao, W. X. & Ma, L. Novel peptide ligands that bind specifically to mouse embryonic stem cells. Peptides. 31, 2027–2034 (2010).
Zhao, W., Yuan, H., Xu, X. & Ma, L. Isolation and initial application of a novel peptide that specifically recognizes the neural stem cells derived from rhesus monkey embryonic stem cells. J Biomol Screen. 15, 687–694 (2010).
Daquinag, et al. An Isoform of Decorin Is a Resistin Receptor on the Surface of Adipose Progenitor Cells. Cell Stem Cell. 9, 74–86 (2011).
Soendergaard, M. & Northup, J. R. N. Peptide Phage Display for Discovery of Novel Biomarkers for Imaging and Therapy of Cell Subpopulations in Ovarian Cancer. J Molecular Bio. 01, s2–004 (2012).
Molek, P., Strukelj, B. & Bratkovic, T. Peptide Phage Display as a Tool for Drug Discovery: Targeting Membrane Receptors. Molecules. 16, 857–887 (2011).
Ye, L. et al. Generation of induced pluripotent stem cells using site-specific integration with phage integrase. Proc Natl Acad Sci U S A. 107, 19467–19472 (2010).
Ng, S., Jafari, M. R. & Derda, R. Bacteriophages and viruses as a support for organic synthesis and combinatorial chemistry. ACS Chem Biol. 7, 123–138 (2012).
Edgar, R. et al. High-sensitivity bacterial detection using biotin-tagged phage and quantum-dot nanocomplexes. Proc Natl Acad Sci U S A. 103, 4841–4845 (2006).
Mosier-Boss, P. A. et al. Use of fluorescently labeled phage in the detection and identification of bacterial species. Appl Spectrosc. 57, 1138–1144 (2003).
Minikh, O., Tolba, M., Brovko, L. Y. & Griffiths, M. W. Bacteriophage-based biosorbents coupled with bioluminescent ATP assay for rapid concentration and detection of Escherichia coli. J Microbiol Methods. 82, 177–183 (2010).
Yim, P. B. et al. Quantitative characterization of quantum dot-labeled lambda phage for Escherichia coli detection. Biotechnol Bioeng. 104, 1059–1067 (2009).
Souza, G. R. et al. Networks of gold nanoparticles and bacteriophage as biological sensors and cell-targeting agents. Proc Natl Acad Sci U S A. 103, 1215–1220 (2006).
Carrico, Z. M. et al. N-terminal labeling of filamentous phage to create cancer marker imaging agents. ACS Nano. 6, 6675–6680 (2012).
Lin, S. et al. Quantum dot imaging for embryonic stem cells. BMC Biotechnol. 7, 67 (2007).
Xing, Y. & Rao, J. Quantum dot bioconjugates for in vitro diagnostics & in vivo imaging. Cancer Biomark. 4, 307–319 (2008).
Arap, W., Pasqualini, R. & Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 279, 377–380 (1998).
Pasqualini, R. & Ruoslahti, E. Organ targeting in vivo using phage display peptide libraries. Nature. 380, 364–366 (1996).
Derda, R. et al. Diversity of Phage-Displayed Libraries of Peptides during Panning and Amplification. Molecules. 16, 1776–1803 (2011).
Thomas, W. D., Golomb, M. & Smith, G. P. Corruption of phage display libraries by target-unrelated clones: Diagnosis and countermeasures. Anal Biochem. 407, 237–240 (2010).
Vodnik, M., Zager, U., Strukelj, B. & Lunder, M. Phage Display: Selecting Straws Instead of a Needle from a Haystack. Molecules. 16, 790–817 (2011).
PAC. t. Hoen et al. Phage display screening without repetitious selection rounds. Anal Biochem. 421, 622–631 (2012).
Thomson, J. A. et al. Embryonic stem cell lines derived from human blastocysts. Science. 282, 1145–1147 (1998).
Ru, B. et al. MimoDB: a new repository for mimotope data derived from phage display technology. Molecules. 15, 8279–8288 (2010).
Zhao, W., Ji, X., Zhang, F., Li, L. & Ma, L. Embryonic stem cell markers. Molecules. 17, 6196–6236 (2012).
Adewumi, O. et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat Biotechnol. 25, 803–816 (2007).
Wu, Z. et al. Derivation and characterization of human embryonic stem cell lines from the Chinese population. J Genet Genomics. 38, 13–20 (2011).
Maupetit, J., Derreumaux, P. & Tuffery, P. PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res. 37, W498–503 (2009).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Changhua Zhou, et al. Synthesis of size-tunable photoluminescent aqueous CdSe/ZnS microspheres via a phase transfer method with amphiphilic oligomer and their application for detection of HCG antigen. J. Mater. Chem. 21, 7393–7400 (2011).
Zhou, C. et al. A versatile method for the preparation of water-soluble amphiphilic oligomer-coated semiconductor quantum dots with high fluorescence and stability. J Colloid Interf Sci. 344, 279–285 (2010).
Acknowledgements
Financial support was provided by the Hi-tech Research and Development Program of China (No. 2006AA03Z359). We thank Sidansai Biotechnology (Shanghai) for technical assistance. We are also grateful to Dr. Xu Zhang (Mount Sinai School of Medicine) for suggesting the three-dimensional modeling, Dr. Ke Yang for data management and Dr. Xun Zhang (Harvard medical school) for critical reading and revision of the manuscript.
Author information
Authors and Affiliations
Contributions
L.M., L.L. and W.Z. designed the research. W.Z., L.J. and Z.T. performed the experiments, H.Y. and C.Z. prepared the quantum dots, L.M., W.Z. and L.J. analyzed the data and W.Z. wrote the main manuscript text and prepared all of the figures. All of the authors reviewed and approved the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Dataset 1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zhao, W., Jin, L., Yuan, H. et al. Targeting human embryonic stem cells with quantum dot-conjugated phages. Sci Rep 3, 3134 (2013). https://doi.org/10.1038/srep03134
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03134
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
