
Abstract
Matrix metalloproteinase (MMP) family is considered to be associated with chronic obstructive pulmonary disease (COPD) pathogenesis, however, no consistent results have been provided by previous studies. In this report, we performed Meta analysis to investigate the association between four kinds of MMP single nucleotide polymorphisms (SNP, MMP1 -1607 1G/2G, MMP3 -1171 5A/6A, MMP9 -1562 C/T, MMP12 -82 A/G) and COPD risk from 21 studies including 4184 cases and 5716 controls. Both overall and subgroup association between SNP and COPD susceptibility were tested. There was no evident association between MMP polymorphisms and COPD susceptibility in general population. On the other hand, subgroup analysis suggested that MMP9 -1562 C/T polymorphism was related to COPD, as we found that C allele carriers were at lower risk in some subgroups stratified by lung function, age and genotype identification method, compared with TT homozygotes. Our results indicated the genotype TT might be one genetic risk factor of severe COPD.
Similar content being viewed by others

Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic airway inflammatory disease, which is characterized by not fully reversible airflow limitation, inflammatory cells infiltration, mucus overproduction and airway remodeling1. As a complex disease, the precise molecular mechanism of COPD is still unknown. However, it was widely accepted that the occurrence of COPD relied on the interaction of gene and environment.
Protease and anti-protease imbalance was considered as an important mechanism involved in the pathogenesis of COPD. Since the discovery of relationship between a α 1-antitrypsin and COPD, no other proteases or anti-proteases have been confirmed to be associated with this disease2. Matrix metalloproteinase (MMP) is a group of protease, which mediate various physiological and pathological processes. So far, at least 24 kinds of MMPs have been identified in human3. Increasing evidence from animal experiments suggested that MMPs played a pivotal role in COPD4,5,6,7,8,9,10,11.
The activity of MMPs is dependent on the gene encoding them. The existence of gene polymorphism determines the different expression level of these genes among individuals, which ultimately result in different phenotype of disease in a population. In the past decade, considerable efforts have been made to find out the relationship between MMP single nucleotide polymorphism (SNP) and COPD risk in several populations12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32. However, the results of different researches were not consistent. Some reports showed that specific MMP genotype was related to occurrence of COPD13,14,19,23,27,31,32, while other reports did not support the association between MMP polymorphism and COPD susceptibility12,15,16,17,18,20,21,22,24,25,26,28,29,30. These contradictory findings may be partly due to limited sample size, false-positive results and publication bias. In order to identify which MMP polymorphism play the key role in COPD occurrence, we conducted a comprehensive meta-analysis to quantify the overall risk of MMP polymorphisms on COPD.
Results
Characteristics of included studies
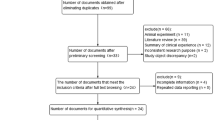
We identified 123 related articles, of which 30 studies were potentially appropriate. 7 studies did not examine any MMP single nucleotide polymorphism (SNP) mentioned above. 3 studies were excluded because of lack of proper control. Further more, 2 repeated studies were also ruled out. Finally, 3 additional studies by manual search were added to analysis. Thus, 21 studies including 4184 cases and 5716 controls met the including criteria (Figure 1). MMP1 SNP and MMP9 SNP were both mentioned in 12 studies, while 5 and 6 studies provided the association of MMP3 and MMP12 SNPs with COPD, respectively. The study characteristics were listed in Table 1. COPD patients were diagnosed through lung function index in all studies expect two by Enewold et al.15 and Minematsu et al.24, respectively. Population and hospital based controls were involved in different studies. In addition, frequency-matched controls to the cases by ethnicity, sex, age and smoking status were applied in some studies. A classical polymerase chain reaction – restriction fragment length polymorphism (PCR-RFLP) assay was performed in 15 of the 21studies.

Study identification, inclusion and exclusion for meta-analysis.
Overall effects for alleles and genotypes
The distribution of alleles and genotypes of each study was listed in Table 2. Due to existence of moderate to high heterogeneity among studies, the random effect model was used to evaluate the odds ratio. For all the four MMP polymorphisms, we did not find the distribution of alleles was related to COPD risk (i.e. MMP1 1G versus 2G: OR 0.99, 95% CI 0.89–1.10, p = 0.81) (Table 3). On the other hand, we did not observe a significant association between genotypes and COPD risk through a series of comparisons, including dominant model (i.e. MMP9 CC versus CT + TT: OR 0.80, 95% CI 0.57–1.14, p = 0.22), recessive model (i.e. MMP9 CC + CT versus TT: OR 0.75, 95% CI 0.45–1,24, p = 0.26) and homozygote model (i.e. MMP9 CC versus TT: OR 0.71, 95% CI 0.40–1.24, p = 0.23) (Table 3).
Subgroup analysis
To exclude the effect of confound factors, such as genetic background, smoking index, lung function, detection methods etc., subgroup analysis was introduced to further elucidate the relationship between MMP SNPs and the risk of COPD. We divided included studies into 2 to 3 subgroups according to ethnicity (Asian or Caucasian), smoking index (matched or not), age (matched or not), lung function (FEV1/prediction%) and genotype detection method (RFLP or not).
For the MMP9 -1562C/T polymorphism, ethnicity and smoking index did not affect the distribution of alleles and genotypes between COPD patients and controls. Although age did not affect the distribution of alleles in these two subpopulations, it did affect genotypes distribution, especially for TT genotype. In age matched subgroup, carriers of C allele had much lower susceptibility of COPD, compared with TT homozygotes (CC + CT vs. TT: OR 0.56, 95% CI 0.33–0.96, p = 0.03; CC vs. TT: OR 0.44, 95% CI 0.25–0.77, p = 0.004) (Figure 2a–b). Just like age, lung function also affected TT genotype distribution in two subpopulations. In one subgroup where COPD patients with relatively worse lung function (FEV1 < 50% prediction), carriers of C allele were at much lower risk of COPD, compared with TT homozygotes (CC + CT vs. TT: OR 0.60, 95% CI 0.37–0.99, p = 0.05; CC vs. TT: OR 0.53, 95% CI 0.31–0.88, p = 0.01) (Figure 2 c–d). The method of genotype identification seemed to be another factor that related to the genotype distribution in the two subpopulations. The risk of COPD was lower among carriers of C allele than that among TT homozygotes in the subgroup where RFLP was used to detect genotype (CC + CT vs. TT: OR, 0.61, 95% CI 0.41–0.92, p = 0.02; CC vs. TT: OR0.54, 95% CI 0.35–0.82, p = 0.004) (Figure 2 e–f).

Subgroup analysis of correlation between MMP9 polymorphism and COPD risk under two pairs of comparisons (CC + CT vs. TT; CC vs. TT).
(a–b) The studies were divided into two groups according to age (unmatched or matched) under the comparison of CC + CT vs. TT (a) and CC vs. TT (b); (c–d) The studies were divided into three groups according to lung function (FEV1 > 50% prediction, FEV1 < 50% prediction or unknown) under the comparison of CC + CT vs. TT (c) and CC vs. TT (d); (e–f) The studies were divided into two groups according to genotype identification method (RFLP or Non-RFLP) under the comparison of CC + CT vs. TT (e) and CC vs. TT (f).
For the MMP1 -1607 1G/2G polymorphism, all the factors mentioned above seemed not to be associated with the distribution of both alleles and genotypes between COPD patients and controls (Supplementary Table S1).
For the MMP3 and MMP12 polymorphisms, we did not carry out subgroup analysis because of limited number of studies.
Heterogeneity analysis
For MMP9 -1562C/T polymorphism, I2 showed a moderate variation under most comparisons when we performed overall and subgroup analysis. However, in the subgroup of Caucasian, there was almost no variation. Moreover, in the age matched, worse lung function and RFLP subgroup, no heterogeneity was detected except for the comparison between CC and CT + TT (I2 > 80% in these subgroups) (Table 3, Supplementary Table S2).
For the overall and subgroup analysis of MMP1 -1607 1G/2G polymorphism, I2 showed a moderate variation under most comparisons. In the subgroup of Caucasian and non-RFLP, no variation was found except for the comparison between 1G1G and 1G2G + 2G2G (I2 = 29% in Caucasian subgroup and I2 = 31% in non-RFLP subgroup, respectively). For the subgroup analysis of age and lung function, I2 showed low or no heterogeneity under different comparisons in different subgroups (Table 3, Supplementary Table S1).
For overall analysis of MMP3 -1171 5A/6A and MMP12 -82A/G polymorphism, I2 showed a low or moderate variation under all comparisons (Table 3).
Sensitivity analysis
Sensitivity analysis was performed to test the effect of a specific study on the overall results. There were six studies not exhibiting Hardy–Weinberg equilibrium (HWE) (5 for MMP1 -1607 1G/2G and 1 for MMP9 -1562 C/T). However, omitting these studies did not significantly alter the pooled OR value in both polymorphisms. Although the genotypes distribution of MMP9 -1562 C/T in the study reported by Lee et al. was not deviate from HWE, we found this study affected the general results obviously. If this study was excluded, the pooled OR decreased from 0.75 to 0.61 under the comparison of CC + CT vs. TT (95% CI: 0.41–0.92, p = 0.02) and from 0.71 to 0.54 under the comparison of CC vs. TT (95% CI: 0.35–0.82, p = 0.004). (Supplementary Figure S1)
Publication bias
Publication bias was detected by Begg's and Egger's test. These tests did not show significant results in almost all comparisons (Table 3). The shape of funnel plots did not reveal evidence of obvious asymmetry (Figure 3). These results indicated little publication bias.

Publication bias on MMP polymorphism.
(a) Begg's funnel plot of the 12 eligible studies assessing MMP1 -1607 1G/2G polymorphism; (b) Begg's funnel plot of the 11 eligible studies assessing MMP9 -1567 C/T polymorphism.
Discussion
In order to seek out the genetic variants related to COPD, much effort has been made to explore the association between gene polymorphism via case-control study or cohort study. Recently, several genome-wide association studies (GWAS) have identified novel SNPs located in chromosomal loci 15q25 with genome-wide significance for association with COPD33,34,35. However, we have not found any data about the association between MMP and COPD based on GWAS.
Matrix metalloproteinase is a family of zinc-dependent endopeptidases degrading all the main protein components of the extracellular matrix and playing an essential role in tissue remodeling and repair associated with development and inflammation36. MMP-1, MMP3 and MMP-12 are located in close proximity on chromosome 11 while MMP-9 is located on chromosome 20 3. Many clinical studies suggested that MMPs were involved in COPD as concentration of MMPs in serum or induced sputum in patients was higher than that in non-COPD patients or healthy volunteers37,38,39,40. The degree of COPD severity is dependent on lung function. Joos et al. reported that MMP1 -1607 1G/2G polymorphism was associated with the rate of decline in lung function and the haplotypes consisting of MMP1 -1607 1G/2G and MMP12 + 357Asn/Ser polymorphism were also related to lung function decline rate41. On the other hand, Hunninghake et al. found that the minor allele (G) of a functional variant in the promoter region of MMP12 was associated with a reduced risk of COPD in the NAS cohort consisting of initially healthy adult men and a cohort of smokers42. Variation in gene promoter region might cause the change of protein activity, which was considered to be associated with COPD susceptibility.
However, from the results of present analysis, we failed to find correlation between genotypes of various MMP family members and the risk of COPD. It was a negative result, but was in accordance with the results of majority studies included in this analysis. Although the remaining studies showed significant association between certain genotype and COPD susceptibility, it could not be ruled out the existence of false positive results due to the reasons as follows. First, some studies contained a small sample size, so the results might not stable enough. Second, different detection methods in different studies were the source of deviation, which affected overall results. Third, the positive results reported by some authors were contradictory. For example, Minematsu24 and Cheng13 reported that CC was a protective genotype, while Lee23 and Zhou32 suggested that CC was a susceptible genotype. Due to these paradoxical results, no significant overall effect could be obtained.
The great discrepancy among different studies indicated the existence of heterogeneity. In fact, moderate to high heterogeneity was detected in almost all comparisons. Subgroup analysis was introduced to further seek out the source of heterogeneity. For the analysis of MMP9 and MMP1 polymorphism, all the studies were divided into subgroups according to ethnicity, age, smoking index, lung function and methods for genotype identification. After stratification, the heterogeneity of at least one subgroup reduced or disappeared. It demonstrated that these factors were at least part source of heterogeneity. In addition, we also found in some subgroups, including age matched subgroup, worse lung function subgroup and RFLP subgroup, the carriers of allele C had much less risk of COPD. It was worth to note that the distribution of MMP9 genotypes in COPD patients with worse lung function is different from that in normal people and patients with better lung function. In fact, one report by Korytina (2008) et al.21 showed that in very sever COPD patients (FEV1 < 30% prediction) the allele T was more frequency. So it may provide us the direction to selecting proper subpopulation of COPD.
Hardy–Weinberg equilibrium (HWE) results showed the p values of several studies were less than 0.05, which suggested the potential to influence the overall effect. Sensitivity analysis showed that these studies had minor effect on OR values, which indicated the stability of present work. Unexpectedly, another report greatly affected the main results. For MMP9 polymorphism, removing Lee's report would contribute to evident change of results. Apart from the inconsistency of some demography indexes between COPD cases and control subjects, this study used a unique method for genotype identification. It was the only study that did not use PCR-RFLP to analyze MMP9 genotype. This deficiency might partly explain the results mentioned above, but further research was needed to elucidate the discrepancy among these studies.
There were some limitations in the present analysis. First, the sample size was too small. The quantity of patients in these studies was just more than 4000. However, it was estimated that there were about 200 million COPD patients in the world. In other words, only a small part of patients were recruited to study. Second, the existence of confound factors severely affected the ultimate results. As discussed above, age, lung function and even detection method were demonstrated to be confounders in present analysis. These confounders were present in almost every study we selected. Third, the data we obtained now were not comprehensive. Almost all the studies were either on Asian or Caucasian. African was one of the three largest ethnics on the earth, but so far we have found only one article on African. Moreover, the occurrence of COPD is not depend on one gene but a cluster of genes, so the gene linkage is important for exploring disease pathogenesis. However, we are lack of related data, which may prevent us from further research.
In conclusion, our present analyses did not show significant association between MMP SNPs and COPD risk in the whole population. However, we found TT genotype of MMP9 was a risk factor in the subpopulation with relatively worse lung function. So, it may be helpful to screen out potential severe COPD patients by detecting MMP9 genotype. In light of various deficiencies in present studies, there is a great need of further studies including large population, selecting appropriate control subjects, unifying detection method and paying much attention to gene linkage in order to confirm the role of MMP SNPs in the COPD susceptibility.
Methods
Search strategy
We carried out a comprehensive search strategy in various databases including Pubmed, Embase, Cochrane Library and China National Knowledge Infrastructure (CNKI) to seek out the articles which were about the association between MMP polymorphisms and the risk of COPD. The terms we used as follows: “chronic obstructive pulmonary disease”, “COPD”, “emphysema”, “chronic bronchitis”; “matrix metalloproteinase”, “interstitial collagenase”, “stromelysin”, “gelatinase”, “macrophage elastase”; and “genetic polymorphism”, “variant”, “variation”, “association”. Additional studies were identified by a manual search from references of original studies or review articles on this topic. Only studies with full text articles published until October 2012 were included.
Study selection
The criteria for the papers selection were as follows: (1) studies with case-control or prospective longitudinal cohort design; (2) COPD as the outcome, with at least two comparison groups (COPD vs. healthy control groups); (3) the study including at least one of the four kinds of MMP polymorphisms (MMP1 -1607 1G/2G, MMP3 -1171 5A/6A, MMP9 -1562 C/T, MMP12 -82 A/G) in COPD cases and controls; (4) provide the available genotype frequency in COPD cases and healthy controls.
Data extraction
Information was carefully extracted from all eligible publications independently by two authors according to the inclusion criteria listed above. Once encountering disagreements, we resolved them by discussions with the third person. The data we extracted from papers contain basic information of study (author, publication year), population (sample size, ethnicity, age, lung function and smoking index), COPD definition, genotype distribution in cases and controls, genotype identification method, Hardy-Weinberg equilibrium (HWE) test etc.
Data analysis
Odds ratio (OR) and 95% CIs were used to assess the strength of association between all kinds of MMP polymorphisms (MMP1 -1607 1G/2G, MMP3 -1171 5A/6A, MMP9 -1562 C/T, MMP12 -82 A/G) and COPD risk. Dominant, recessive and homozygote model were applied for genotype analysis.
Heterogeneity assumption was checked by the Cochran Q test. If p value for the Q test is over 0.10, we consider that there is lack of heterogeneity. We also used the statistic of I2 to detect the degree of heterogeneity, with I2 < 25%, 25%–75% and >75% to represent low, moderate and high degree of inconsistency, respectively43,44. In the analysis of pooled data, we used two different models according to the trait of the included studies: If no heterogeneity was found, a fixed effect model was adopted to determine the gene effect or the random effect model was used45,46. What's more, if heterogeneity across studies existed, subgroup analysis was performed to seek out the source of heterogeneity. Studies were subdivided by ethnicity (Caucasian versus Asian), genotyping methods (RFLP versus others), age (unmatched versus matched), smoking index (unmatched versus matched), lung function of COPD cases (FEV1 > 50% prediction versus FEV1 < 50% prediction) to find the source of any heterogeneity.
Hardy–Weinberg equilibrium (HWE) was tested in healthy control within each study. Deviation from HWE was tested using the χ2 test. Studies with controls that depart from HWE (p < 0.05) were subjected to a sensitivity analysis in order to check the consistency of the overall effect.
We made use of Begg's funnel plot to examine the underlying publication bias and also used Egger's weighted regression method to calculate P for bias47,48. If no publication bias existed, the funnel plot looked symmetrical.
All analyses were conducted with the use of Review Manager, V.5.0 (Revman, The Cochrane Collaboration) or STATA software, V.10.0 (STATA Corp).
References
Decramer, M., Janssens, W. & Miravitlles, M. Chronic obstructive pulmonary disease. Lancet 379, 1341–1351 (2012).
Foreman, M. G., Campos, M. & Celedón, J. C. Genes and chronic obstructive pulmonary disease. Med Clin North Am 96, 699–711 (2012).
Zitka, O. et al. Matrix metalloproteinases. Curr Med Chem 17, 3751–3768 (2010).
Churg, A. et al. An MMP-9/-12 inhibitor prevents smoke-induced emphysema and small airway remodeling in guinea pigs. Thorax 62, 706–713 (2007).
Churg, A. et al. Alpha1-antitrypsin suppresses TNF-a and MMP-12 production by cigarette smoke-stimulated macrophages. Am J Respir Cell Mol Biol 37, 144–151 (2007).
Le Que'ment, C. et al. The selective MMP-12 inhibitor, AS111793 reduces airway inflammation in mice exposed to cigarette smoke. Br J Pharmacol 154, 1206–1215 (2008).
Mercer, B. A., Wallace, A. C., Brinckerhoff, C. E. & D'Armiento, J. M. Identification of a cigarette smoke-responsive region in the distal MMP-1 promoter. Am J Respir Cell Mol Biol 40, 4–12 (2009).
Atkinson, J. J. et al. The role of matrix metalloproteinase-9 in cigarette smoke-induced emphysema. Am J Respir Crit Care Med 183, 876–884 (2011).
D'Armiento, J., Dalal, S. S., Okada, Y., Berg, R. A. & Chada, K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell 71, 955–961 (1992).
Hautamaki, R. D., Kobayashi, D. K., Senior, R. M. & Shapiro, S. D. Macrophage elastase is required for cigarette smoke-induced emphysema in mice. Science 277, 2002–2004 (1997).
Nerusu, K. C. et al. Matrix metalloproteinase-3 (stromelysin-1) in acute inflammatory tissue injury. Exp. Mol. Pathol 83, 169–176 (2007).
Cai, J., Wang, L. & Wang, A. MMP-1 gene polymorphsim and COPD susceptibility in Han nationality in Jiangxi province. Shandong Medical Journal 50, 76–77 (2010).
Cheng, S. L., Yu, C. J. & Yang, P. C. Genetic polymorphisms of cytochrome p450 and matrix metalloproteinase in chronic obstructive pulmonary disease. Biochem Genet 47, 591–601 (2009).
van Diemen, C. C. et al. Genetic variation in TIMP1 but not MMPs predict excess FEV1 decline in two general population-based cohorts. Respir Res 12, 57 (2011).
Enewold, L., Mechanic, L. E., Bowman, E. D., Platz, E. A. & Alberg, A. J. Association of Matrix Metalloproteinase-1 Polymorphisms with Risk of COPD and Lung Cancer and Survival in Lung Cancer. Anticancer Res 32, 3917–3922 (2012).
Han, W., Kuang, J. & Rao, W. MMP-9 polymorphisms and susceptibility to chronic obstructive pulmonary disease. Chin J Tuberc Respir Dis 29, 277–278 (2006).
Haq, I. et al. Association of MMP-2 polymorphisms with severe and very severe COPD: a case control study of MMPs-1, 9 and 12 in a European population. BMC Med Genet 11, 7–12 (2010).
Hersh, C. P. et al. Attempted replication of reported chronic obstructive pulmonary disease candidate gene associations. Am J Respir Cell Mol Biol 33, 71–78 (2005).
Hua, D. et al. Association of MMP9 polymorphisms with susceptibility to chronic obstructive pulmonary disease in Ethnic Tibetan population. Int J Respir 30, 1157–1160 (2010).
Ito, I. et al. Matrix metalloproteinase-9 promoter polymorphism associated with upper lung dominant emphysema. Am J Respir Crit Care Med 172, 1378–1382 (2005).
Korytina, G. F., Akhmadishina, L. Z., Ianbaeva, D. G. & Viktorova, T. V. Polymorphism in Promoter Regions of Matrix Metalloproteinases (MMP1, MMP9 and MMP12) in Chronic Obstructive Pulmonary Disease Patients. Russian Journal of Genetics 44, 202–208 (2008).
Korytina, G. F. et al. Association of MMP3, MMP9, ADAM33 and TIMP3 Polymorphisms with Chronic Obstructive Pulmonary Disease and Its Progression. Molecular Biology 46, 438–449 (2012).
Lee, S. Y. et al. Polymorphisms in matrix metalloproteinase-1,-9 and -12 genes and the risk of chronic obstructive pulmonary disease in a Korean population. Respiration 80, 133–138 (2010).
Minematsu, N., Nakamura, H., Tateno, H., Nakajima, T. & Yamaguchi, K. Genetic polymorphism in matrix metalloproteinase-9 and pulmonary emphysema. Biochem Biophys Res Commun 289, 116–119 (2001).
Santus, P. et al. Stromelysin_1 polymor_ phism as a new potential risk factor in progression of chronic obstructive pulmonary disease. Monaldi Arch Chest Dis 71, 15–20 (2009).
Schirmer, H., Basso da Silva, L., Moreira, J. S., Moreira, A. L. & Simon, D. Matrix metalloproteinase gene polymorphisms: lack of association with chronic obstructive pulmonary disease in a Brazilian population. Genet Mol Res 8, 1028–1034 (2009).
Sun, P., Chen, J., Chen, R., Jin, J. & Li, Y. The relevance of matrixmetalloproteinase-1 gene polymorphism with chronic obstructive pulmonary disease susceptibility in Han nationality of northern China. Chin J Respir Crit Care Med 4, 342–344 (2005).
Sun, C., Wang, W., Yang, J., Siqingaowa & Wang, L. The association of matrix metalloproteinase 3,12 polymorphism with susceptibility of chronic obstructive pulmonary disease. Chin J CIinicians (EIectronic Edition) 6, 5826–5830 (2012).
Tesfaigzi, Y. et al. Genotypes in matrix metalloproteinase 9 are risk factors for COPD. Int J Chron Obstruct Pulmon Dis 1, 267–278 (2006).
Zhang, R. et al. Matrix metalloproteinase polymorphisms and susceptibility to chronic obstructive pulmonary disease in Han nationality of north China. Journal of Cardiovascular and Pulmonary Diseases 23, 240–243 (2004).
Zhang, R., He, Q., Yang, R., Lu, B. & Liu, Y. Study on matrix metalloproteinase 1, 9, 12 polymorphisms and susceptibility to chronic obstructive pulmonary disease among Han nationality in Northern China. Chin J Epidemiol 26, 907–910 (2005).
Zhou, M. et al. Genetic polymorphism in matrix metalloproteinase-9 and the susceptibility to chronic obstructive pulmonary disease in Han population of south China. Chin Med J (Engl) 117, 1481–1484 (2004).
Pillai, S. G. et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet 5, e1000421 (2009).
Cho, M. H. et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet 42, 200–202 (2010).
Kong, X. et al. Genome-wide association study identifies BICD1 as a susceptibility gene for emphysema. Am J Respir Crit Care Med 183, 43–49 (2011).
Churg, A., Zhou, S. & Wright, J. L. Matrix metalloproteinases in COPD. Eur Respir J 39, 197–209 (2012).
Demedts, I. K. et al. Elevated MMP-12 protein levels in induced sputum from patients with COPD. Thorax 61, 196–201 (2006).
Vornooy, J. H. J., Lindeman, J. H. N., Jacobs, J. A., Hanemaaijer, R. & Wouters, E. F. Increased activity of matrix metalloproteinase -8 and matrix metalloproteinase -9 in induced sputum from patients with COPD. Chest 126, 1802–1810 (2004).
Culpitt, S. V., Rogers, D. F., Traves, S. L., Barnes, P. J. & Donnelly, L. E. Sputum matrix metalloproteases: comparison between chronic obstructive pulmonary disease and asthma. Respir Med 99, 703–710 (2005).
Louhelainen, N. et al. Elevation of sputum matrix metalloproteinase-9 persists up to 6 months after smoking cessation: a research study. BMC Pulm Med 10, 13 (2010).
Joos, L. et al. The role of matrix metalloproteinase polymorphisms in the rate of decline in lung function. Hum Mol Genet 11, 569–576 (2002).
Hunninghake, G. M. et al. MMP12, lung function and COPD in high-risk populations. N Engl J Med 361, 2599–2608 (2009).
Higgins, J. P. & Thompson, S. G. Quantifying heterogeneity in a meta analysis. Stat Med 21, 1539–1558 (2002).
Higgins, J. P., Thompson, S. G., Deeks, J. J. & Altman, D. G. Measuring inconsistency in meta-analyses. BMJ 327, 557–560 (2003).
Wang, J. H. et al. Polymorphisms of matrix metalloproteinases in myocardial infarction: a meta-analysis. Heart 97, 1542–1546 (2011).
Yan, F. G. et al. Association between polymorphism of glutathione S-transferase P1 and chronic obstructive pulmonary disease: A meta-analysis. Respiratory Medicine 104, 473–480 (2010).
Begg, C. B. & Mazumdar, M. Operating characteristics of a rank correlation test for publication bias. Biometrics 50, 1088–1101 (1994).
Egger, M., Davey, S. G. & Schneider, M. Bias in meta-analysis detected by a simple, graphical test. BMJ 315, 629–634 (1997).
Acknowledgements
This work was supported in part by grants from the State Key Program of National Natural Science Foundation of China (No.81130001), National Basic Research Program of China (No.2009CB522103), National Key Technologies R&D Program for the 12th Five-year Plan (No.2012BAI05B01).
Author information
Authors and Affiliations
Contributions
H.Z. have contributed to the design of the study, analysis and interpretation of data and drafting a part of manuscript. Y.W. also took part in collecting and analyzing data and drafting a part of manuscript. Y.J., J.Z., C.Z. and L.C. searched the related papers and extracted data. J.J. carried out statistical analysis and revised manuscript. Z.C. prepared all figures. W.L. and H.S. designed this study and revised manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Support information of sensitivity analysis and subgroup analysis
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zhou, H., Wu, Y., Jin, Y. et al. Genetic Polymorphism of Matrix Metalloproteinase Family and Chronic Obstructive Pulmonary Disease Susceptibility: a Meta-analysis. Sci Rep 3, 2818 (2013). https://doi.org/10.1038/srep02818
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02818
This article is cited by
-
Transgenic Mice Overexpressing Vitamin D Receptor (VDR) Show Anti-Inflammatory Effects in Lung Tissues
Inflammation (2017)
-
Matrix Metalloproteinase-3 -1171 5A/6A Polymorphism (rs35068180) is Associated with Risk of Periodontitis
Scientific Reports (2015)
-
The association between the rs6495309 polymorphism in CHRNA3 gene and lung cancer risk in Chinese: a meta-analysis
Scientific Reports (2014)
-
Association between the IL1B, IL1RN polymorphisms and COPD risk: A meta-analysis
Scientific Reports (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
