
Abstract
Recently, smart polymer vesicles have attracted increasing interest due to their endless potential applications such as tunable delivery vehicles for the treatment of degenerative diseases. However, the evolution of stimuli-responsive vesicles from bench to bedside still seems far away for the limitations of current stimuli forms such as temperature, light, redox, etc. Since ultrasound combined with chemotherapy has been widely used in tumor treatment and the pH in tumor tissues is relatively low, we designed herein a novel polymer vesicle that respond to both physical (ultrasound) and chemical (pH) stimuli based on a PEO-b-P(DEA-stat-TMA) block copolymer, where PEO is short for poly(ethylene oxide), DEA for 2-(diethylamino)ethyl methacrylate and TMA for (2-tetrahydrofuranyloxy)ethyl methacrylate. These dually responsive vesicles show noncytotoxicity below 250 μg/mL and can encapsulate anticancer drugs, exhibiting retarded release profile and controllable release rate when subjected to ultrasound radiation or varying pH in tris buffer at 37°C.
Similar content being viewed by others

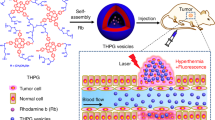

Introduction
Smart polymer vesicles that respond to stimuli have been suggested to be promising delivery vehicles for controlled encapsulation and release1,2. To effectively achieve this, it is important that the polymer vesicles respond to external stimuli which could be classified as either chemical stimuli or physical stimuli1. Chemical stimuli such as changes in pH3, oxidation/reduction4 may change polymer structure, accompanying unwanted deformation or leakage of polymer vesicles. In contrast, physical stimuli are much convenient and clinically safe since no by-products with uncertain biohazard generated throughout the response procedure5. Typical physical stimuli include variation of temperature6, light7, electrical field8, etc. However, there are still some disadvantages for the physical stimuli mentioned above when polymer vesicles are used for drug delivery1. For example, there are no temperature or electrical field responsive polymer vesicles used for the drug delivery in clinic due to the lack of appropriate polymers; UV-responsive polymers are not ideal for the biomedical applications due to the risk of UV light to the skin.
Compared to the above stimuli, noninvasive ultrasound offers the following advantages: (1) penetrating deeply into the interior of the body; (2) capable of focusing and controllable; (3) providing high resolution images of the soft tissues; (4) easily accessible and low cost; and (5) providing relatively easy dynamic examination compared with other radiologic modalities9. Ultrasound has been widely used in molecular imaging10, diagnosis and treatment11. Recently, ultrasound triggered release from microemulsions12, polymer micelles13, liposomes14 and multilayered capsules15 have been reported. Wang et al. made use of focused ultrasound with high intensity as a rational means to control polymer micellar disruption and showed that the focused high-frequency ultrasound beam could induce the hydrolysis reaction of the copolymer at room temperature16. For example, block copolymer micelles in aqueous solution could be disrupted by 1.1 MHz of ultrasound17. However, as far as we are aware, block copolymer vesicles that respond to ultrasound for drug delivery have not been reported yet. Polymer vesicle is one of the most promising systems for drug delivery applications and may offer many advantages compared to liposomes and polymer micelles18,19. Typical polymer vesicles are water dispersible with great temporal-spatial stability and the hollow cavity enables the vesicles encapsulate hydrophilic drug whereas the hydrophobic vesicle membrane could carry hydrophobic drugs simultaneously20. Moreover, the application of ultrasound combined with chemotherapy in tumour treatment has been well-established and the relatively low pH of tumour tissues makes pH-sensitive polymer vesicles the potential drug carriers for drug delivery1,21. Therefore, the design and synthesis of novel polymer vesicles that respond to both ultrasound and pH stimuli will provide dual controls for tumour therapy, especially with the help of ultrasound to achieve synergistic effect.
Herein we report a novel polymer vesicle that is responsive to both physical (ultrasound) and chemical (pH) stimuli and explore their drug entrapment and release abilities under different conditions such as ultrasound radiation and solution pH. Figure 1 shows the schematic representation of controlled drug release for PEO43-b-P(DEA33-stat-TMA47) block copolymer vesicles. Potential advantages of this dually responsive polymer vesicle are: (i) the potential for encapsulating both hydrophilic and hydrophobic actives simultaneously and (ii) the dual triggers (ultrasound and pH) controlled release of encapsulated drugs, which may be realized by simply adjusting ultrasound radiation time and solution pH.

Formation of ultrasound and pH dually responsive PEO43-b-P(DEA33-stat-TMA47) vesicle and controlled drug release triggered by ultrasound radiation or decreasing pH value.
Upon ultrasound radiation, the disruption and re-self-assembly lead to smaller vesicle. Upon decreasing the solution pH, the shrinkage of vesicles by the recrystallization of PTMA chains surpasses the swelling of vesicles by the partial protonation of DEA, leading to smaller vesicles. Further decreasing the solution pH will lead to the complete protonation of DEA and finally the disassembly of vesicles. Both ultrasound radiation and decreasing pH can lead to faster drug release.
Results
Syntheses of block copolymers by ATRP
To synthesize the ultrasound and pH dually responsive copolymers, TMA monomer was firstly synthesized by the addition reaction between 2-hydroxyethyl methacrylate and 2,3-dihydrofuran in methanol with poly(4-vinylpyridine) hydrochloride (P4VP·HCl) as the out-phase catalyst. The synthetic route is shown in Fig. S1, supplementary information (SI).1H NMR spectrum of TMA confirmed that TMA monomer was successfully prepared with a purity of ~99.5%, as shown in Fig. S2, SI. Table 1 shows four block copolymers synthesized by ATRP. Polymers 1 and 2 are control copolymers, which are only responsive to pH and ultrasound, respectively. Polymers 3 and 4 are pH and ultrasound dually responsive copolymers with different monomer compositions. In this paper we mainly investigate polymer 3 unless specially noted. To prepare these copolymers, the macroinitiator, PEO43-Br, is firstly synthesized by the reaction of 2-bromoisobutyryl bromide with polyethylene glycol monomethylether (MeO-PEO-OH, Mn = 1900) in the presence of triethylamine in anhydrous toluene22. PEO43-Br is then chain-extended using TMA and DEA to produce P(DEA-stat-TMA), PTMA and PDEA blocks under different conditions (see Fig. S1, Fig. S3–Fig. S5 and Fig. S6 in SI for the synthetic route, 1H NMR spectra and GPC curves of block copolymers, respectively).
Self-assembly of block copolymers into vesicles
The water dispersible polymer vesicles were obtained by simply adding water into copolymer solution in THF (67/33, w/w) at pH 7.4 followed by dialysis against water at pH 7.4 to thoroughly remove THF. In the process of self-assembly, the colorless solution turned into bluish when the water content was about 50 wt%, indicating the formation of vesicles. The hydrophilic PEO chains form vesicle coronas, while the hydrophobic PDEA, PTMA or P(DEA-stat-TMA) chains form vesicle membrane. Figure S7 in SI shows the typical hydrodynamic diameter (DH) distributions of vesicles self-assembled under the same conditions by polymers 1–4. The vesicles obtained from polymers 3 and 4 have the intensity-averaged mean diameters of 415 nm (curve a) and 210 nm (curve d), respectively. The smaller DH of polymer 4 vesicles resulted from the lower molecular weight and the smaller hydrophobic to hydrophilic ratio of the copolymer. This suggested that the sizes of ultrasound and pH dually responsive polymer vesicles can be easily tuned by varying the block copolymer composition. On the other hand, the mean diameters of PEO43-b-PDEA41 (polymer 1) and PEO43-b-PTMA60 (polymer 2) vesicles are 212 and 363 nm, respectively. The transmission electron microscopy (TEM) images in Fig. 2 A and B clearly show the formation of PEO43-b-P(DEA33-stat-TMA47) (polymer 3) vesicles with collapsed morphology at dry state. The mean vesicle diameter estimated from Fig. 2 A is 391 ± 111 nm. This value is slightly greater than the hydrodynamic diameter (DH) of 377 nm from dynamic light scattering (DLS) for the same vesicles in aqueous solution at pH 7.4 and 25°C, as shown in curve (a) of Fig. 2 D. This discrepancy indicates the flattening of vesicles adsorbed onto the TEM grid19,23. This buckling effect makes it very difficult to determine the mean membrane thickness of larger vesicles (see Fig. 2 B). However, a mean membrane thickness of ca. 25 nm can be estimated by TEM (see the close-up in Fig. 2 A) for smaller vesicles (<200 nm) since they are less prone to buckling during drying process24. According to earlier studies19,22, the vesicle membrane thickness should be essentially independent of the vesicle diameter. This membrane thickness of ca. 25 nm is comparable to the contour length of 19.8 nm calculated for the hydrophobic P(DEA33-stat-TMA47) block, suggesting that these chains are interdigitated within the vesicle membranes.

TEM (A–C) and DLS (D) studies of PEO43-b-P(DEA33-stat-TMA47) (polymer 3) vesicles.
The TEM images are correlated to the DLS samples: A and B → (a) (at pH 7.4 without sonication); C → (b) (at pH 7.4, sonication for 90 min).
The effect of ultrasound radiation time on the size of polymer vesicles
Ultrasound with the power of 180 W and the frequency of 40 kHz was applied to investigate its effect on the size of polymer vesicle. Figure 3 shows the effect of ultrasound irradiation time on the hydrodynamic diameters (DH's) of PEO43-b-PDEA41 (polymer 1, only pH-responsive), PEO43-b-PTMA60 (polymer 2, only ultrasonic-responsive) and PEO43-b-P(DEA33-stat-TMA47) (polymer 3, both pH- and ultrasonic-responsive) vesicles.

The effect of the ultrasound radiation time on the size of polymer vesicles in aqueous solution at pH 7.4 and 25°C.
(A) PEO43-b-PDEA41, (B) PEO43-b-PTMA60 and (C) PEO43-b-P(DEA33-stat-TMA47) copolymer vesicles.The black curves and blue symbols stand for the hydrodynamic diameters and polydispersity index (PDI) of copolymer vesicles, respectively. The PDI remains low during the ultrasound radiation process, which makes the data reliable. Experimental conditions: the concentration of vesicle solution: 0.15 mg/mL; volume: 15 mL. Ultrasound power: 180 W; frequency: 40 kHz.
The average DH's of both polymer 2 and polymer 3 vesicles (with PTMA composition) decrease upon ultrasound irradiation time, while polymer 1 vesicles (without PTMA composition) show no obvious change even being subjected to sonication for 150 min. This confirms that the PTMA chain is sensitive to the ultrasound irradiation. The diminution of the hydrodynamic diameters of polymers 2 and 3 vesicles might be caused by ultrasonic cavitation. When an ultrasonic wave passed through the solution, a large number of micro-bubbles formed, grew and collapsed in a very short time (about a few microseconds)24, producing a relatively strong instantaneous energy which might cause the disruption and rearrangement of polymer vesicles containing ultrasound-sensitive PTMA chains.
The composition of polymer 3 hardly changed after sonication for 150 min as confirmed by 1H NMR analysis of freeze-dried vesicles upon sonication (see curves C and D in Fig. S4, SI), indicating a physical rather than chemical process occurred during this rearrangement process of polymer vesicles.
Moreover, TEM image in Fig. 2 C shows that smaller vesicles (148 ± 74 nm) of polymer 3 were obtained after ultrasound radiation for 90 min. This value is slightly greater than the hydrodynamic diameter (DH) of 124 nm from DLS for the same vesicles in aqueous solution at pH 7.4 and 25°C, as shown in curve (b) in Fig. 2 D, as a result of the flattening effect mentioned above.
There were two processes happened when subjected to ultrasound radiation for polymer 3 vesicles: ultrasound disruption and re-self-assembly of vesicles. Upon sonication, the volumes of polymer 2 and polymer 3 vesicles are shrunk by 85% and 96% (calculated from the DLS results), respectively. Therefore, this significant volume diminution is not resulted from the minor local adjustment of the ultrasound-responsive PTMA chains, but from the fast vesicle disruption and re-self-assembly.
Why polymer 3 vesicles are more sensitive to ultrasound radiation than polymer 2?
As shown in Fig. 3 B and C, the average DH of polymer 3 vesicles decreases faster than polymer 2 vesicles upon ultrasound radiation. Generally, one may ascribe it to the better chain motion ability of polymer 3 than polymer 2. That is to say, better motion abilities of the P(DEA33-stat-TMA47) elastic chains in polymer 3 than the individual PTMA chains in polymer 2 may result in a more sensitivity to ultrasound radiation of polymer 3 vesicles compared to polymer 2 vesicles.
In order to verify if this assumption is right, DSC measurements were adopted to determine the glass transition temperatures (Tg′s) of polymer 2 and polymer 3 copolymers (see Fig. S10, SI). Polymer 2 had two Tg's at −6.6 and 39.1°C that belong to PTMA60 and PEO43 chains, respectively, while polymer 3 also had two Tg's at 7.7 and 42.2°C that belong to P(DEA33-stat-TMA47) and PEO43 chains, respectively. This seems to go against with the expectation that the introduction of PDEA chains into polymer 3 may decrease its Tg so that the hydrophobic P(DEA33-stat-TMA47) chains should have better motion ability than PTMA60 chains. Actually, the introduction of PDEA chains increases the Tg of polymer 3. However, we note that the Tg's belonging to the hydrophobic chains of both polymers 2 and 3 are much lower than room temperature at which the experiments are carried out. Above Tg, both polymers 2 and 3 should have similar chain motion ability. Therefore, different Tg's between polymers 2 and 3 can not explain why polymer 3 vesicles are more sensitive to ultrasound radiation than polymer 2 vesicles.
Unexpectedly, the DSC results show that polymer 2 has a wide crystallization temperature (Tc) range from 20 to 30°C (see curve b in Fig. S10, SI) while polymer 3 has not (see curve a in Fig. S10). This is because the introduction of DEA to form statistical P(DEA-stat-TMA) chains breaks up the steric regularity of PTMA chains that leads to the disappearance of Tc of polymer 3. As a result, the crystallization of polymer 2 restricts the motion ability of polymer chains at room temperature that makes it less sensitive to ultrasound radiation.
The effect of solution pH on the size and morphologies of self-assemblies
It is known that DEA homopolymer (PDEA) is pH-responsive22. It dissolves in water at low pH as a weak cationic polyelectrolyte, but it becomes insoluble above pH 7.3 due to deprotonation of its tertiary amine groups3. Therefore, the introduction of PDEA chains into polymer 3 makes it sensitive to solution pH, as judged by DLS study. Figure 4 shows the effect of solution pH on size distribution and hydrodynamic diameters (DH's) of PEO43-b-PDEA41 (polymer 1), PEO43-b-PTMA60 (polymer 2) and PEO43-b-P(DEA33-stat-TMA47) (polymer 3) vesicles. Polymer 2 vesicle is pH-independent (see Fig. 4 B) while both polymers 1 and 3 vesicles are pH sensitive (see Fig. 4 A and C). The DH of polymer 3 vesicles decreases slightly when the solution pH drops from 7.4 to 5.0, as shown in Fig. 4 C. This pH-responsive behavior is opposite to our previously reported PEO-b-P(DEA-stat-TMSPMA) polymer vesicles by self-crosslinking, where the DH increases when decreasing the solution pH from 7.0 to 2.022. We will discuss this issue in the Discussion section. Furthermore, both polymer 1 and polymer 3 vesicles completely dissociate at low pH due to protonation of the DEA-based blocks. The pH trigger points of polymer 1 and polymer 3 vesicles are pH 5.7 and 4.8, respectively. Macroscopically, the blue solution of polymer 1 vesicles turns to colorless one when the solution pH decreases to below 5.7 (see Fig. S13, SI). This phenomenon also appears at pH 4.8 for polymer 3, indicating the disassembly of polymer vesicles. TEM images in Fig. S12 A and C confirm that polymer 3 vesicles fall into pieces when the solution pH drops from 7.4 to 3.0, indicating the disassembly of polymer 3 vesicles.

The effect of solution pH on the size of vesicles in aqueous solution at 25°C.
(A) PEO43-b-PDEA41, (B) PEO43-b-PTMA60 and (C) PEO43-b-P(DEA33-stat-TMA47) copolymer vesicles.The black curves and blue symbols stand for the hydrodynamic diameters and PDI's of copolymer vesicles, respectively. The star-shaped symbols in (A) and (C) mean that the results have high polydispersity index (PDI) (>0.5) that make the data unreliable, which is consistent with the dissociation of polymer vesicles at low pH. The vesicle solution concentration is 0.15 mg/mL.
Blood compatibility and cytotoxicity tests
In order to investigate whether the dual responsive copolymer vesicles are suitable for drug delivery, we performed the hemolytic (with mice blood red cell) and cell viability (CCK-8 assay) experiments of polymer 3 vesicles. Figure S14 in SI shows the result of the haemolysis assay. Nearly 50% of erythrocytes lyses at the initial copolymer concentration of 1200 μg/mL. The haemolysis rate decreases with decreasing the concentration of polymer 3 vesicles and reaches to almost 0% when the concentration of polymer 3 vesicles is below 100 μg/mL (The critical vesicle formation concentration of polymer 3 is 13.5 μg/mL, Figure S15 in SI). This value is much larger than PEI-g-PEG polymers (rather than self-assemblies) reported by Thomas Kissel, in which a highest accredited polymer concentration of 14.0 μg/mL was investigated25.
The cytotoxicity tests were carried out by culturing polymer 3 vesicles with human HCCLM3 liver cancer cells and L02 liver cells in vitro for 48 h. The CCK-8 assay in Fig. 5 A confirmed polymer 3 vesicles (without DOX) do not significantly affect proliferation of HCCLM3 liver cell line up to a concentration of 1000 μg/mL. However, when the concentration of polymer 3 vesicles is higher than 500 μg/mL, L02 liver cell viability reduces by more than 25% compared to HCCLM3 cells. Besides, polymer 3 vesicle shows noncytotoxicity against both HCCLM3 liver cancer cells and L02 liver cells when its concentration is lower than 250 μg/mL. Moreover, the therapeutic efficacies of DOX-loaded polymer 3 vesicles and free DOX (as control) have been estimated in vitro by quantifying cell viability of human HCCLM3 liver cancer cells using the CCK-8 assay. It is confirmed in Fig. 5 B that encapsulation of DOX into polymer 3 vesicles effectively reduced cell viability of HCCLM3 cells as well as free DOX in a dose-dependent fashion.

Cytotoxicity tests.
(A) Polymer 3 vesicles with HCCLM3 liver cancer cell (black) and L02 liver cell (red); (B) DOX·HCl loaded polymer 3 vesicles and free DOX·HCl with HCCLM3 liver cancer cell. The cells with different materials were incubated for 48 h and the cell viability was detected by CCK-8 assay. Data are presented as the mean ± standard deviation (SD; n = 5). *p < 0.05 by t-test.
Drug loading/release of block copolymer vesicles
Because of their noncytotoxicity, the dually responsive polymer vesicles may be used in biomedical applications such as anticancer drug carrier. Figure S16 A in SI shows the stability of drug (DOX, an anti-cancer drug) loaded polymer 3 vesicles in 0.01 M tris buffer at pH 7.4 and 25°C. The DH of DOX-loaded vesicle is bigger than the DOX-free polymer 3 vesicles because the DOX·HCl salt affects the self-assembly behavior of the pH-responsive block copolymer26. The drug loaded polymer 3 vesicles are also sensitive to ultrasound radiation as shown in Fig. S16 B. Based on these studies, the in vitro drug release were carried out with DOX as a model anticancer drug and polymer vesicles as the drug carriers.
First, we evaluated the drug encapsulation/release ability of ultrasound and pH dually responsive polymer vesicles based on polymer 3. Figure 6 A and D show DOX encapsulation/release profiles obtained for polymer 3 vesicles at different conditions. The drug loading efficiency (DLE) was approximately 29.8 wt% and the drug loading content (DLC) was approximately 5.98 wt% (See Table S1 in SI) relative to the polymer vesicles. Curve a in Fig. 6 A and D obtained for control experiment utilizing an aqueous solution of 36 μg/mL of DOX in the absence of any vesicles indicates a rapid drug elution, as expected. The DOX release percentages is about 90% after 8 h. Curve b in Fig. 6 A and D shows that, after 48 h, the DOX release contents of DOX-loaded vesicles is 59% at pH 7.4 without sonication. In both cases, curve b shows retarded release of the drug due to its entrapment within the vesicles compared to curve a. Curves c and d in Fig. 6 A show the DOX-loaded polymer 3 vesicles, which subjected to ultrasound radiation for 20 and 60 min at the beginning of drug release experiments (after 20 or 60 minutes, the ultrasound radiation was terminated but the release experiments continued). The corresponding DOX release contents are 75% and 84%, respectively, after 48 h release. This means that the ultrasound radiation at the initial stage (20 min) has a significant impact on the faster drug release rate and a longer ultrasound radiation time (40 min) only leads to slightly higher (9%) release of DOX.

Cumulative drug release profiles of DOX-loaded copolymer vesicles at 37°C in tris buffer.
(A) and (D) for ultrasound and pH dually responsive PEO43-b-P(DEA33-stat-TMA47) (3) polymer vesicles; (B) and (E) for solely ultrasound-responsive PEO43-b-PTMA60 (2) vesicles; (C) and (F) for solely pH-responsive PEO43-b-PDEA41 (1) vesicles.The drug release experiments were carried out at different ultrasound radiation times (t = 0, 20 and 60 min) and different pH values (7.4, 5.5 and 4.0).
To evaluate the effect of the solution pH on the release profile of this dually responsive polymer vesicle, the DOX-loaded polymer 3 vesicles were also placed in acidic conditions. The DOX release contents of DOX-loaded polymer 3 vesicles at pH 4.0 and 5.5 are 79% and 64% without ultrasound radiation, respectively, as shown in curves c and d in Fig. 6 D. These results suggest that ultrasound-triggered and pH-triggered re-adjustments of vesicles are dominant drug release mechanism. Thus, controlled release can be achieved either by simply varying the solution pH or adjusting ultrasound irradiation time for the DOX/PEO43-b-P(DEA33-stat-TMA47) vesicles release system.
Second, as controls, the drug release experiments of solely pH-responsive polymer 1 vesicles and solely ultrasound-responsive polymer 2 vesicles were carried out under the same conditions as the dually responsive polymer 3 vesicles. The drug loading efficiencies (DLEs) and the drug loading contents (DLCs) were approximately 24.2% (DLE), 4.84% (DLC) for polymer 1 vesicles and 25.4% (DLE), 5.08% (DLC) for polymer 2 vesicles, as shown in Table S1 in SI.
To evaluate the ultrasound responsiveness, the release profiles of polymers 2 and 1 vesicles at different ultrasound radiation times are presented in Fig. 6 B and C. The DOX release content of DOX-loaded polymer 2 vesicles increases from 58% to 92% with increasing ultrasound radiation time (from 0 to 60 min) while that of polymer 1 vesicles shows no obvious increase (from 61% to 68%). This also confirms that polymer 2 vesicles are responsive to ultrasound radiation while polymer 1 vesicles are not. It is noteworthy that for ultrasound-responsive polymer vesicles, the drug release profiles for polymers 2 and 3 vesicles subjected to ultrasound radiation for 60 min (t = 60 min) showed that DOX release content of dually responsive polymer 3 vesicle (84%) is less than that of singular responsive polymer 2 vesicle (92%) after 48 h. This is possibly because the re-encapsulation of released DOX into the re-self-assembled smaller vesicles after ultrasound disruption of the original vesicles that slows down the release rate of polymer 3 vesicles.
To evaluate the pH responsiveness, the release profiles of polymers 2 and 1 vesicles at various pH values are presented in Fig. 6 E and F. The DOX release content of DOX-loaded polymer 1 vesicles increases from 61% to 89% with decreasing pH (from pH 7.4 to 4.0) while that of polymer 2 vesicles shows no obvious increase (from 58% to 64%) due to the absence of the pH-controlled valves in the membrane of the polymer 2 vesicles. This further confirms that polymer 1 vesicles are responsive to pH while polymer 2 vesicles are not.
Discussion
The ultrasound and pH dually responsive block copolymer vesicles have been successfully prepared based on novel PEO-b-P(DEA-stat-TMA) block copolymers. The hydrophilic PEO chains form vesicle coronas, while the hydrophobic P(DEA-stat-TMA) chains form vesicle membrane upon self-assembly in tetrahydrofuran (THF)/neutral water system. PTMA chains have been proven to be sensitive to ultrasound by comparing PEO43-b-P(DEA33-stat-TMA47) (polymer 3) vesicles with PEO43-b-PDEA41 (polymer 1) vesicles without PTMA chains because the latter did not show obvious size changes even subjected to ultrasound irradiation for 150 min. Both polymer 2 and polymer 3 vesicles are sensitive to ultrasound due to the ultrasonic cavitation effect. However, the mechanism of sonication effect is the disruption of polymer vesicles rather than the decomposition of polymer itself as evidenced by the unchanged 1H NMR spectra before and after sonication for 150 min of polymer 3 vesicles (Fig. S4 C and D, SI). Meanwhile, the self-healing properties of polymer vesicles contribute to the reassembly of disrupted vesicles during ultrasound radiation. That is, both the disruption and re-assembly of polymer vesicles exist in a competing way when subjected to ultrasound radiation. Moreover, the decrease of polymer vesicle diameters upon ultrasound radiation may result from faster disruption rate than reassembly rate when the polymer vesicles are big enough. However, as the vesicles become smaller, the disruption rate and reassembly rate reach equilibrium, leading to the smaller vesicles which are stable enough against further ultrasound radiation. We find that polymer 2 vesicle is less sensitive to ultrasound than polymer 3 vesicle since the crystallization restricts the motion ability of polymer 2 chains at room temperature.
The introduction of PDEA chains into polymer 3 makes the vesicles sensitive to solution pH as well. When the solution pH is higher than 7.3, polymer 3 vesicles shrunk a little bit due to the deprotonation of the tertiary amine groups of PDEA chain. It is noteworthy that when the solution pH drops from 7.3 to 5.0, the vesicles become smaller rather than swelling as normal22,23. This ‘abnormal’ phenomenon may be due to the competition between the protonation of PDEA and hydrophobic effect and recrystalline of PTMA. When decreasing the pH, the gradual protonation of PDEA chains in the polymer 3 vesicle membrane leads to a reverse of their role. In other words, when the solution pH changes from 7.3 to 5.0, partial protonated PDEA chains intend to run out from the hydrophobic membrane to form hydrophilic vesicle coronas. In principle, this process will lead to a swelling of polymer vesicles22. However, in the meanwhile, the crystallizable PTMA chains tend to gather together due to the hydrophobic effect and recrystalline. In principle, this rearrangement will lead to shrinkage of vesicle. Furthermore, the random arrangement of PDEA chains in the P(DEA-stat-TMA) chains of polymer 3 makes the contradictory competition more fiercely. Therefore, at the range of pH 7.3–5.0, it is the hydrophobic effect and the recrystalline of PTMA chains that dominate the readjustment of polymer 3 chains to form smaller vesicles. In contrast, in our previously reported normal pH-responsive behavior, there is only one protonation process, leading to an increase in the vesicle size when decreasing the solution pH22. However, when the solution pH drops to below 5.0 (e.g., pH 4.8), thorough protonation of PDEA chains enforces the whole polymer 3 chains to repulse reciprocally, resulting in the full disassembly of polymer 3 vesicles as confirmed by DLS studies (Fig. 4C in main text and Fig. S11C in SI) and TEM images in Fig. S12 A and C in SI.
The haemolysis result confirms that polymer 3 vesicles are compatible with blood when the concentration is less than 100 μg/mL. This is consistent with the well-proven biocompatibility of PEO chains, which just form the vesicle coronas. According to the cytotoxicity test, polymer 3 vesicles have no cytotoxicity against human L02 liver cells and HCCLM3 liver cancer cells when the concentration is less than 250 μg/mL (Fig. 5A). The proper concentration of DOX to realize the ideal therapeutic efficacy (cell viability of HCCLM3 cells is less than 20%) should be more than 2.5 μg/mL (Fig. 5B). The DLC of polymer 3 vesicles obtained from drug release experiment is 5.98%. Therefore, the concentration of polymer 3 vesicles needed to load that amount of DOX should be more than 41.8 μg/mL, which is much less than 250 μg/mL. On the other hand, DOX loaded polymer 3 vesicles are quite stable and sensitive to ultrasound radiation in tris buffer at pH 7.4. The DOX release experiments for all the three polymer vesicles also prove the concept that polymer 3 vesicles are pH and ultrasound dually responsive while polymers 1 and 2 vesicles are only responsive to pH and ultrasound, respectively. Consequently, polymer 3 vesicles are applicable to controlled (ultrasound and pH-triggered) drug release at the concentration ranges from 41.8 μg/mL to 250 μg/mL.
To conclude, the ultrasound-responsive polymer vesicles and ultrasound and pH dually responsive polymer vesicles have been successfully prepared based on novel PEO-b-PTMA and PEO-b-P(DEA-stat-TMA) block copolymers, respectively. The dually responsive polymer vesicles become smaller upon either ultrasound irradiation or by decreasing the solution pH. The ultrasound-induced significant size diminution of vesicles is resulted from the fast disruption and re-self-assembly of vesicles. The vesicles can also become smaller at low pH, as a result of the shrinkage of vesicles by the recrystallization of PTMA chains surpassing the swelling of vesicles by the partial protonation of DEA. During both processes, the controlled release of loaded anticancer drug can be reached. Moreover, the vesicles show noncytotoxicity at low concentration (<250 μg/mL), which makes the biomedical applications possible. The ultrasound-responsive block copolymer vesicle shows promising perspective on designing and developing new stimuli-responsive delivery vehicles in nanomedicine, etc.
Methods
Synthesis of PEO43-b-PDEA41 block copolymer by ATRP
2-(Diethylamino)ethyl methacrylate (DEA) (2.93 g, 15.8 mmol) and 2,2′-bipyridine (bpy) (92.6 mg, 0.590 mmol) were added to a solution of PEO43-Br (0.800 g, 0.400 mmol) in methanol (4.00 mL). The mixture was degassed by flushing with argon for 30 min at room temperature. Cu(I)Br (57.5 mg, 0.400 mmol) was quickly added into the flask before it was immersed in a preheated oil bath at 30°C for 24 h. Then the mixture was diluted by dichloromethane and passed through a neutral Al2O3 column with dichloromethane as the eluent to remove the catalyst. After concentration of the solution by solvent evaporation under reduced pressure and then dried in a vacuum oven at 30°C for 2 days, yield: 96%. The composition of PEO43-b-PDEA41 was determined from the 1H NMR spectrum (see Fig. S3, SI) by comparing the integrals of the resonance peaks of PEO (δ = 3.66) and PDEA (δ = 2.70 − 2.55).
1H NMR (400 MHz, CDCl3): δ 4.05 (broad, 83H, NCH2CH2O), 3.66 (broad, 170H, OCH2CH2O), 2.70 − 2.55 (broad, 248H, CH2N(CH2CH3)2), 1.80 − 1.63 (broad, 81H, CH2CCH3), 1.06 − 0.83 (broad, 361H, (CH3)2C, CH2CCH3, CH2CH3).
Synthesis of PEO43-b-PTMA60 block copolymer by ATRP
(2-Tetrahydrofuranyloxy)ethyl methacrylate (TMA) (3.20 g, 16.0 mmol) and N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA) (52.0 mg, 0.300 mmol) were added to a solution of PEO43-Br (0.400 g, 0.200 mmol) in anisole (4.00 mL). The mixture was degassed by passing argon through for 30 min at room temperature. Cu(I)Br (30.0 mg, 0.200 mmol) was quickly added into the flask. Then it was immersed in a preheated oil bath at 50°C for 8 h. The mixture was passed through a neutral Al2O3 column with dichloromethane as the eluent to remove the catalyst. After concentration by solvent evaporation under reduced pressure, the polymer was precipitated from cold ether (dry ice bath) for 3 times. And then it was dried in a vacuum oven at 30°C for 2 days. Yield: 75%. The composition of PEO43-b-PTMA60 was determined from the 1H NMR spectrum (see Fig. S4B in the supplementary information) by comparing the integrals of the resonance peaks of PEO (δ = 3.63) and PTMA (δ = 5.10).
1H NMR (400 MHz, CDCl3): δ 5.10 (broad, 60H, OCHO), 4.07 (broad, 123H, OCH2CH2OCH), 3.87 − 3.78 (broad, 245H, OCH2CH2OCH, CH2OCHO), 3.63 (broad, 170H, OCH2CH2O), 1.91 − 1.82 (broad, 371H, CHCH2CH2CH2, CH2CCH3), 1.07 − 0.92 (broad, 190H, CH3O, CH2CCH3).
Syntheses of PEO-b-P(DEA-stat-TMA) block copolymers by ATRP
DEA (0.500 g, 2.67 mmol), TMA (0.540 g, 2.67 mmol) and bpy (18.5 mg, 0.120 mmol) were added to a solution of PEO43-Br (0.120 g, 0.0600 mmol) in methanol (3.00 mL). The mixture was degassed by passing argon through for 30 min at room temperature. Cu(I)Br (8.60 mg, 0.0600 mmol) was quickly added into the flask before it was immersed into a preheated oil bath. The reaction was carried out for 48 h at 50°C with magnetic stirring. The crude product was passed through a neutral Al2O3 column with dichloromethane as the eluent to remove the catalyst. After concentration of the solution by solvent evaporation under reduced pressure, then it was dried in a vacuum oven at 30°C for 2 days. Yield: 88%. The composition of PEO43-b-P(DEA33-stat-TMA47) was determined from the 1H NMR spectrum (see Fig. S4C, SI) by comparing the integrals of the resonance peaks of PEO (δ = 3.65), PTMA (δ = 5.10) and PDEA (δ = 2.69).
1H NMR (400 MHz, CDCl3): δ 5.10 (broad, 47H, OCHO), 4.12 − 4.07 (broad, 163H, OCH2CH2N, OCH2CH2OCH), 3.90 − 3.81 (broad, 192H, OCH2CH2OCH, CH2OCHO), 3.65 (broad, 170H, OCH2CH2O), 2.69 (broad, 201H, (CH2)3N), 1.91 − 1.82 (broad, 354H, CHCH2CH2CH2, CH2CCH3), 1.21 − 0.92 (broad, 453H, CH3O, CH2CCH3, NCH2CH3).
The same method was adopted to synthesize PEO43-b-P(DEA24-stat-TMA24) except for a shorter reaction time of 24 h and different ingredient ratios, yield: 91%. 1H NMR spectrum: see Fig. S5 in the supplementary information. 1H NMR (400 MHz, CDCl3): δ 5.10 (broad, 24H, OCHO), 4.12 − 4.07 (broad, 98H, OCH2CH2N, OCH2CH2OCH), 3.85 − 3.76 (broad, 94H, OCH2CH2OCH, CH2OCHO), 3.69 (broad, 170H, OCH2CH2O), 2.70 − 2.55 (broad, 145H, (CH2)3N), 1.96 − 1.78 (broad, 199H, CHCH2CH2CH2, CH2CCH3), 1.17 − 0.92 (broad, 297H, CH3O, CH2CCH3, NCH2CH3).
Self-assembly of block copolymers into vesicles
The self-assembly of PEO43-b-PTMA60, PEO43-b-PDEA41, PEO43-b-P(DEA33-stat-TMA47) and PEO43-b-P(DEA24-stat-TMA24) copolymers were based on solvent-switch method. In a typical protocol, PEO43-b-PTMA60 (8.0 mg) was dissolved in THF (4.0 mL) and then double volume of distilled water was added dropwise with syringe into the solution at a rate of about one drop every 3 seconds with magnetic stirring. The light blue solution of PEO43-b-PTMA60 polymer vesicles was stirred for 30 min to reach equilibrium after all the water was added. Subsequently, the solution was dialyzed against pure water to remove THF in a dialysis tubing with a molecular weight cutoff from 8000 to 14000. The solution was diluted to a concentration of 0.15 mg/mL for DLS measurement. The same self-assembly method was adopted for PEO43-b-P(DEA33-stat-TMA47), PEO43-b-P(DEA24-stat-TMA24) and PEO43-b-PDEA41 copolymers except for the quantity of copolymers used.
Ultrasound effect study
The ultrasound radiation experiment was carried out as following: a bottle of 15 mL of diluted solution (0.15 mg/mL) was immersed into the middle of ultrasonic cleaner bath and then exposed to ultrasound irradiation with the power of 180 W for 10, 30, 60, 90, 120 and 150 min, intermittently. In addition, 5 mL of the DOX loaded polymer 3 vesicle solution was exposed to ultrasound irradiation with the power of 180 W for 3, 5, 7, 10, 15, 20, 25, 30, 40, 50 and 60 min, intermittently. The hydrodynamic diameters and size distribution of copolymer vesicles were determined by DLS at each interval.
Haemolysis test for PEO43-b-P(DEA33-stat-TMA47) vesicles
Fresh blood from rats, collected in heparinized tubes, was centrifuged at 8°C for 10 min and washed several times with phosphate buffer solution (PBS) until the supernatant was colorless. 500 μL of serial dilutions of PEO43-b-P(DEA33-stat-TMA47) vesicle solution in PBS were mixed with 500 μL of 2.5% (v/v) suspensions of erythrocytes in 2 mL centrifuge tubes. After incubation of 60 min at 37°C in an incubator, the blood cells were removed by centrifugation and the supernatants were investigated spectroscopically at 540 nm for the release of hemoglobin. As controls, PBS (negative, 0%) and 0.2% Triton X-100 solution (positive, 100%) were used27.
Cytotoxicity test
Cellular viability was determined using the Cell Counting Kit-8 assay (CCK-8, Dojindo, Japan). Human L02 liver cells or HCCLM3 liver cancer cells were seeded with equal density in each well of 96-well plates (4000 cells/well) in 100 μL of Dulbecco's Modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) for 24 h at 37°C in a humidified 5% CO2-containing atmosphere. Then 20 μL of polymer 3 vesicle solution with final concentrations of 62.5, 125, 250, 500 and 1000 μg/mL, respectively, was added and incubated with cells for additional 48 h. In addition, 20 μL of polymer 3 vesicles (loaded with DOX·HCl) solution with final concentrations of loaded DOX of 0.1 μg/mL, 0.5, 2.5, 5.0 and 10 μg/mL, respectively, was added and incubated with HCCLM3 liver cancer cells for additional 48 h. Untreated cells served as a control group. At the end of the treatment, CCK-8 dye was added to each well and the plates were incubated for additional 1 h at 37°C. Subsequently, the absorbance was measured by dual wave length spectrophotometry at 450 nm and 630 nm using a microplate reader. Each treatment was repeated five times. The relative cell viability (%) was determined by comparing the absorbance at 450 nm with control wells containing only cell culture medium.
Drug loading and in vitro release of block copolymer vesicles
The encapsulation and controlled release of anti-cancer drug experiments were carried out according to the literature protocol28,29 and modified as following. Copolymers (80.0 mg) and DOX·HCl (16.0 mg) were dissolved in THF (40.0 mL) to make a polymer/DOX·HCl mixture solution. Then pure water (80.0 mL) was dropwise added to the solution within 2.5 h to form DOX-loaded polymer vesicles and the mixture was stirred overnight to reach equilibration. Then the organic solvent (THF) was removed by rotary evaporation. The unloaded free drug and residual THF were removed by dialysis using a dialysis tube (cutoff Mn = 8000–14000) against 1000 mL pure water at 25°C with 300 r/min of stirring. Pure water was renewed for 6 times within 3 h (every 0.5 h). The final volume of vesicle/DOX·HCl aqueous solution was 85.8 mL and the drug loading efficiency of polymer vesicles was measured by fluorescence spectroscopy after dialysis (the dialyzed solution was diluted to 20 times by adding 0.01 M pH 7.4 tris buffer before fluorescence measurement). The final drug release process was carried out by dialyzing 5.0 mL of DOX-loaded vesicles in the dialysis tube (cutoff Mn = 8000–14000) against 100 mL tris buffer (0.01 M; pH 7.4 or pH 5.5 or pH 4.0) in a beaker (100 mL) at 37°C and 190 r/min of stirring. Two of the each copolymer samples (pH 7.4) were irradiated by ultrasound for 20 and 60 min (t = 20, 60 min) at the beginning of drug release. Three parallel experiments were carried out for all the release experiments at different conditions. At different time intervals, the concentration of solution in the beaker was measured by fluorescence spectroscopy (excitation at 461 nm and emission at 591 nm) and the cumulative release curve of DOX was obtained. The calibration curve was shown in Fig. S15, SI. The drug loading efficiency (DLE) and drug loading content (DLC) were calculated according to the following equations29.


Where Me is the weight of drug encapsulated in vesicles, Mf is the weight of drug in feed and Mp is the weight of polymer used. Mf = 16 mg, Mp = 80 mg.
According to the release results, a control solution of pure DOX·HCl was prepared by simply adding 1.8 mg of DOX·HCl to 50 mL water. The concentration was the same as DOX·HCl loaded in polymer vesicles. Then the control experiment was carried out under the same conditions as mentioned above.
References
Du, J. Z. & O'Reilly, R. K. Advances and challenges in smart and functional polymer vesicles. Soft Matter 5, 3544–3561 (2009).
Sanson, C. et al. Doxorubicin loaded magnetic polymersomes: Theranostic nanocarriers for MR imaging and magneto-chemotherapy. Acs Nano 5, 1122–1140 (2011).
Du, J. Z., Tang, Y. Q., Lewis, A. L. & Armes, S. P. pH-sensitive vesicles based on a biocompatible zwitterionic diblock copolymer. J. Am. Chem. Soc. 127, 17982–17983 (2005).
Napoli, A., Valentini, M., Tirelli, N., Muller, M. & Hubbell, J. A. Oxidation-responsive polymeric vesicles. Nat. Mater. 3, 183–189 (2004).
Roy, D., Cambre, J. N. & Sumerlin, B. S. Future perspectives and recent advances in stimuli-responsive materials. Prog. Polym. Sci. 35, 278–301 (2010).
Qin, S. H., Geng, Y., Discher, D. E. & Yang, S. Temperature-controlled assembly and release from polymer vesicles of poly(ethylene oxide)-block-poly(n-isopropylacrylamide). Adv. Mater. 18, 2905–2909 (2006).
Coleman, A. C. et al. Light-induced disassembly of self-assembled vesicle-capped nanotubes observed in real time. Nat. Nanotech. 6, 547–552 (2011).
Yan, Q. et al. Voltage-responsive vesicles based on orthogonal assembly of two homopolymers. J. Am. Chem. Soc. 132, 9268–9270 (2010).
Lin, C.-W., Chen, Y.-H. & Chen, W.-S. Application of ultrasound and ultrasound-guided intervention for evaluating elbow joint pathologies. J. Med. Ultras. 20, 87–95 (2012).
Kudo, M., Hatanaka, K. & Maekawa, K. Defect reperfusion imaging, a newly developed novel technology using sonazoid in the treatment of hepatocellular carcinoma. J. Med. Ultras. 16, 169–176 (2008).
Kudo, M., Hatanaka, K. & Maekawa, K. Sonazoid-enhanced ultrasound in the diagnosis and treatment of hepatic tumors. J. Med. Ultras. 16, 130–139 (2008).
Lee, M.-H., Lin, H.-Y., Chen, H.-C. & Thomas, J. L. Ultrasound mediates the release of curcumin from microemulsions. Langmuir 24, 1707–1713 (2008).
Marin, A. et al. Drug delivery in pluronic micelles: Effect of high-frequency ultrasound on drug release from micelles and intracellular uptake. J. Controlled Release 84, 39–47 (2002).
Lin, H. Y. & Thomas, J. L. Peg-lipids and oligo(ethylene glycol) surfactants enhance the ultrasonic permeabilizability of liposomes. Langmuir 19, 1098–1105 (2003).
Skirtach, A. G. et al. Ultrasound stimulated release and catalysis using polyelectrolyte multilayer capsules. J. Mater. Chem. 17, 1050–1054 (2007).
Wang, J., Pelletier, M., Zhang, H. J., Xia, H. S. & Zhao, Y. High-frequency ultrasound-responsive block copolymer micelle. Langmuir 25, 13201–13205 (2009).
Xuan, J. A., Pelletier, M., Xia, H. S. & Zhao, Y. Ultrasound-induced disruption of amphiphilic block copolymer micelles. Macromol. Chem. Phys. 212, 498–506 (2011).
Discher, D. E. & Eisenberg, A. Polymer vesicles. Science 297, 967–973 (2002).
Du, J. Z. et al. Organic/inorganic hybrid vesicles based on a reactive block copolymer. J. Am. Chem. Soc. 125, 14710–14711 (2003).
Lomas, H. et al. Non-cytotoxic polymer vesicles for rapid and efficient intracellular delivery. Faraday Discuss. 139, 143–159 (2008).
Blana, A., Walter, B., Rogenhofer, S. & Wieland, W. F. High-intensity focused ultrasound for the treatment of localized prostate cancer: 5-year experience. Urology 63, 297–300 (2004).
Du, J. Z. & Armes, S. P. pH-responsive vesicles based on a hydrolytically self-cross-linkable copolymer. J. Am. Chem. Soc. 127, 12800–12801 (2005).
Yu, S. Y., Azzam, T., Rouiller, I. & Eisenberg, A. "Breathing" vesicles. J. Am. Chem. Soc. 131, 10557–10566 (2009).
Ng, K. H. International guidelines and regulations for the safe use of diagnostic ultrasound in medicine. J. Med. Ultras. 10, 5–9 (2002).
Petersen, H., Fechner, P. M., Fischer, D. & Kissel, T. Synthesis, characterization and biocompatibility of polyethylenimine-graft-poly(ethylene glycol) block copolymers. Macromolecules 35, 6867–6874 (2002).
Sun, L. & Du, J. Z. Revisiting the time for removing the unloaded drug by dialysis method based on a biocompatible and biodegradable polymer vesicle. Polymer 53, 2068–2073 (2012).
Marmottant, P. & Hilgenfeldt, S. Controlled vesicle deformation and lysis by single oscillating bubbles. Nature 423, 153–156 (2003).
Ren, T. B. et al. Multifunctional polymer vesicles for ultrasensitive magnetic resonance imaging and drug delivery. J. Mater. Chem. 22, 12329–12338 (2012).
Du, J. Z., Fan, L. & Liu, Q. M. pH-sensitive block copolymer vesicles with variable trigger points for drug delivery. Macromolecules 45, 8275–8283 (2012).
Acknowledgements
J.D. is supported by Shanghai 1000 Plan (SH01068), the program for professor of special appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (P2009011), National Natural Science Foundation of China (21074095 and 21174107), New Century Excellent Talents in Universities of Ministry of Education (NCET-10-0627), Ph.D. program Foundation of Ministry of Education (20110072110048), Fok Ying Tong Education Foundation (132018), Shanghai Pujiang program (10PJ1409900) and the fundamental research funds for the Central Universities. Dr Jing Chen was thanked for performing the cell viability experiments.
Author information
Authors and Affiliations
Contributions
J.D. led the project, designed the experiments, analyzed the data, supervised W.C. and wrote the paper with assistance from W.C. W.C. also performed the experiments and analyzed the data. Both authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
supporting information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Chen, W., Du, J. Ultrasound and pH Dually Responsive Polymer Vesicles for Anticancer Drug Delivery. Sci Rep 3, 2162 (2013). https://doi.org/10.1038/srep02162
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02162
This article is cited by
-
Injectable Fiber Electronics for Tumor Treatment
Advanced Fiber Materials (2022)
-
Ultrasound-responsive polymer-based drug delivery systems
Drug Delivery and Translational Research (2021)
-
On the origin and regulation of ultrasound responsiveness of block copolymer nanoparticles
Science China Chemistry (2020)
-
Emerging era of “somes”: polymersomes as versatile drug delivery carrier for cancer diagnostics and therapy
Drug Delivery and Translational Research (2020)
-
Ultrasound-responsive Homopolymer Nanoparticles
Chinese Journal of Polymer Science (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
