
Abstract
We report a novel approach to synthesize chemical vapor deposition-grown three-dimensional graphene nano-networks (3D-GNs) that can be mass produced with large-area coverage. Annealing of a PVA/iron precursor under a hydrogen environment, infiltrated into 3D-assembled-colloidal silicas reduces iron ions and generates few-layer graphene by precipitation of carbon on the iron surface. The 3D-GN can be grown on any electronic device-compatible substrate, such as Al2O3, Si, GaN, or Quartz. The conductivity and surface area of a 3D-GN are 52 S/cm and 1,025 m2/g, respectively, which are much better than the previously reported values. Furthermore, electrochemical double-layer capacitors based on the 3D-GN have superior supercapacitor performance with a specific capacitance of 245 F/g and 96.5% retention after 6,000 cycles due to the outstanding conductivity and large surface area. The excellent performance of the 3D-GN as an electrode for supercapacitors suggests the great potential of interconnected graphene networks in nano-electronic devices and energy-related materials.
Similar content being viewed by others
Introduction
Three-dimensional (3D) bi-continuous structures with controlled symmetry and periodicity have found use in many applications in photonic crystals, phononic crystals and micro-electromechanical systems1,2. In addition, the large surface area and the availability of 3-dimensional responses to external stimuli provide further potential for using 3D structures in diverse areas of energy-related materials and tissue engineering3,4.
Graphene is a one-atom-thick planar sheet that is densely packed with sp2-bonded carbon atoms in a honeycomb crystal lattice with high optical transmittance, excellent electrical conductivity, high flexibility and mechanical stability, large theoretical specific surface area and unique transport properties5,6,7,8. Graphene grown on metal film by chemical vapor deposition (CVD) via carbon dissolution and a segregation mechanism on a metal catalyst surface has superior conductivity because it has fewer defects and a relatively large domain size9,10,11,12,13,14. Potential problems in CVD growth on a metal film are that the formation of single-crystalline graphene is limited to the 2D plane and production is limited to small quantities. The availability of more sophisticated 3D graphene nanonetworks (3D-GNs) on a bulk scale would enable important advances in energy-related materials, heat sinks and cell culture plates by taking advantage of large surface areas, outstanding high electrical/thermal conductivity and three dimensional environments. For example, it has recently been reported that 3D graphene networks grown on a 3D nickel frame have excellent mechanical strength, supercapacitance and thermal transport properties due to the nature of the 3D structures6,15,16. However, the dimensions of the structures fabricated by this approach are confined by the dimensions of the available nickel frame and are restricted to a scale of a few hundred micrometers, which might in turn hamper a variety of potential applications for the 3D graphene. Alternatively, a 3D graphene-frame on a micron scale has been fabricated by vacuum filtration of chemically modified graphene, although performance was rather low due to the use of reduced graphene oxide17,18. Exploring ways to synthesize graphene that allow for dimensional tunability down to the nanometer scale without loss of its original characteristics could play an important role in both fundamental study and the realization of future graphene applications. So far copper and nickel have been commonly used as metal catalysts for CVD growth of graphene with well established methods and minor progress has been recently achieved in growing multilayer graphene film on iron-based catalysts, which have been widely used for growing carbon nanotubes19,20,21. Utilizing iron as a catalyst will also broaden the applicability of graphene and be an attractive avenue of research, since iron is the fourth most common element in the earth's crust and it is nontoxic, inexpensive and easy to remove.
Supercapacitors are attractive as alternative energy storage devices to batteries and fuel cells due to their fast charging and discharging rates at high power densities, long life cycle, simple principle and low maintenance cost22,23,24,25. Among two different classes of supercapacitors classified by their energy storage mechanism, electrochemical double-layer capacitors (EDLCs) and pseudocapacitors (PCs), EDLCs use the adsorption/desorption of ions forming an electrical double-layer at the interface between an electrode and an electrolyte for storing charge. It is critical to guarantee effective surface area and surface properties of the electrode. In addition to high specific surface area, several other parameters, such as good electric conductivity, appropriate pore size and distribution and electrochemical, thermal and mechanical stability, are equally important to supercapacitor performance. In light of these requirements, porous carbon materials have been suggested as promising materials for EDLCs26,27. However, most porous carbon-based materials with high specific surface area suffer from rather low conductivity, which restricts their application in high performance supercapacitors. On the other hand, graphene provides an ideal alternative to existing EDLC materials, since it has very high electrical conductivity and excellent chemical and mechanical stability and extremely large surface area. However, its theoretical value of parameters such as conductivity (106 S/cm) and surface area (2,630 m2/g) still need to be realized, at least to some extent28,29.
Here, for the first time, we report a bi-continuous 3D-GN grown via a precursor-assisted CVD technique using solution processed iron precursors, which potentially allow for any arbitrary shape of graphene with dimensional scalability and mass-production capability. Graphene is grown on a 3D assembly of colloidal silica (CS) infiltrated with a PVA/iron precursor solution. Annealing PVA/FeCl3 in a hydrogen environment reduces the iron, which can serve as a catalyst for the dissolution of carbon during the subsequent graphene growth process. A 3D-GN has a substantially improved geometrical surface area of 1,025 m2/g and a conductivity of 52 S/cm, which greatly exceeds the values of traditional 3D graphene structures, e.g., graphite with 0.6 m2/g and tens of kΩ/□, respectively30,31,32,33. Moreover, as a result of employing solution-prepared metal precursors, high-quality 3D graphene can be grown on any inert substrate, such as Al2O3, quartz or GaN, etc., enabling the production of ready-to-use graphene for use in the semiconductor industry. As a proof of concept, we have demonstrated that 3D-GN-based EDLC exhibits an excellent specific capacitance of 245 F/g and 96.5% retention after 6,000 cycles. The superior performance of a 3D-GN as an electrode for supercapacitors is attributed to the realization of the effective synergies of the great conductivity and large surface area of graphene in the form of a 3-dimensional network with proper pore sizes. Artificially created 3D-GNs with controlled shape and spacing offer excellent potential for application in areas where the advantages of both graphene and nanoscale 3D structures are needed, such as 3D electrodes, energy conversion/storage devices and thermal management systems.
Results
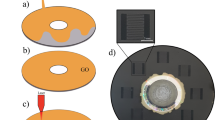
Figure 1 shows the four steps of the fabrication process for the 3D-GN. First, surface-activated colloidal silica (d = 30 nm, 150 nm and 220 nm, as shown in Figure S1) obtained by treatment with H2SO4 were self-assembled using Colvin's method34 into near single crystalline opals with a face-centered cubic (FCC) structure on a 300 nm thick SiO2/Si substrate with a thickness of 3 um. Then a solution of PVA-FeCl3 was infiltrated into the 3D-assembly of silica colloids. We chose iron chloride hexahydrate (FeCl3·6H2O) and water-soluble polyvinyl alcohol (PVA) as a precursor of the Fe catalyst and a solid carbon source for graphene growth, respectively. PVA was chosen because it can easily form a homogeneous dispersion with the iron precursor (FeCl3, in this case) due to the electrostatic interaction between the hydroxyl groups of PVA and iron ions in the FeCl3 solution35,36. In the third step, the composite film of 3D PVA-FeCl3/CS on the SiO2/Si substrate was heated to 1,000°C in a quartz tube. The detailed thermal treatment parameters and conditions are shown in Figure S2. Annealing the 3D PVA-FeCl3/CS composite in a hydrogen environment at high temperature reduced the iron ions (III) into iron (0) and subsequent growth of graphene using PVA as a carbon source on the surface of the iron metal (Figure S3). This process is supported by a recent report demonstrating CVD graphene growth on an iron film substrate21. By carefully controlling the ratio of FeCl3 to PVA, we determined the minimum amount of PVA that can be decomposed and diffused into reduced iron during high temperature annealing and segregated on the surface of the iron to form graphene upon cooling. In the final step, iron was removed with the HF/HCl solution leaving few-layer 3D-GN structures on the SiO2/Si substrate.

Schematic illustration of the fabrication process for a 3D-GN.
(I) Self-assembly of CS on SiO2/Si substrate. (II) Infiltration of PVA-FeCl3 into 3D assembly of CS. (III) Graphene growth by CVD on 3D PVA-FeCl3/CS assembly. (VI) Iron/CS etching, leaving the 3D-GN.
Figure 2 shows the morphological characteristics of a representative 3D-GN obtained by CVD growth followed by removing the iron and the assembly of 220 nm silica particles. Figure 2a is a scanning electron microscope (SEM) image of a 3D assembly of PVA-FeCl3/CS with a thickness of 3 um. Figure 2b is a SEM image of a low-density 3D-GN created from graphene growth and subsequent removal of iron/CS. The optimum ratio of PVA to FeCl3 was determined by careful control of the pressure and flow rate of the gas during the CVD process. The thickness of the 3D-GN generated from the removal of the CS template was reduced to approximately 80% of the original thickness due to the loss of carbon materials and a slight collapse of the 3D-GN near the substrate caused by the surface tension of aqueous solvent during the drying process. However, once the nanopore structures formed, they were rigid and stable enough to be used for further applications as expected from the high modulus (1,100 GPa) of graphene reported elsewhere6,28. Figures 2c and 2d are a TEM image of the 3D-GN near the edges clearly showing a nano-cavity of the 3D-GN and a close-up image of a single unit cell of the 3D-GN with a diameter of 220 nm, respectively. Additionally, we occasionally observed another set of nanopores with diameters of ∼40 nm on the surface of a single graphene ball. This is likely attributable to the removal of iron nanodomains formed due to the agglomeration of iron during the annealing process. More TEM images, confirming few-layer graphene are shown in Figure S4. Figure 2e is a selected area diffraction image of a single unit cell of graphene, revealing the single crystalline nature of graphene. Besides the formation of the network in the film, a bulk scale of the 3D-GN fabricated by the drop-casting of solution followed by CVD also yielded a comparable quality of graphene (Raman, conductivity, etc.). Figure S5 shows photographs of the 3D-GN in the form of a powder (a) and films (b,c) fabricated via drop-casting and spin-coating, respectively, indicating the large scale synthesis of 3D-GN (All the processes including silica templating for drop casting samples are the same as those for the film samples except the method of casting).

Morphological characterizations of 3D-GN.
(a) SEM image of 3D PVA-FeCl3/CS. (b) SEM image of 3D-GN created from graphene growth followed by removal of iron. (c) TEM image of 3D-GN taken near the edges. (d) Close-up image of a single graphene ball. (e) The selected area electron diffraction (SAED) pattern showing the nearly single crystalline nature of the graphene ball in the image of (d).
X-ray diffraction (XRD) measurements were made to confirm the reduction of Fe (III) to Fe (0). Figure 3a shows the XRD data of the PVA-FeCl3/CS (220 nm) film at a FeCl3 concentration of 350 phr before and immediately after annealing in a H2 gas environment. Compared to the data taken from the pristine PVA-FeCl3 film showing many broad and undefined peaks in red, two sharp peaks corresponding to the (200) and (110) planes of Fe (0) and another strong and sharp peak indexed to the (002) plane of graphene are evolved for the annealed sample in black (3D-GN containing iron)37. These peaks confirm that the iron ions are completely reduced to the nearly single crystalline iron metal with the preferred growth direction in the (110) plane, which shows the capability of successfully creating single crystalline graphene which appears at 2θ = 26°.

X-ray diffraction (XRD) and X-ray photoelectron (XPS) spectra before (all in red) and after (all in black) annealing of PVA-FeCl3/CS (220 nm) film at a FeCl3 concentration of 350 phr.
(a) The XRD of pristine iron chloride hexahydrate (FeCl3·6H2O) is shown in red and that of an annealed film of PVA-FeCl3/CS composite (3D-GN containing iron) is in black. (b–d) XPS spectra of Fe, Cl and C in the film at a FeCl3 concentration of 350 phr.
The transformation of FeCl3 to iron metal followed by CVD graphene growth can be further confirmed by analyzing the X-ray photoelectron spectroscopy (XPS) spectra of iron, chlorine and carbon in the same samples as the ones for XRD. Figures 3b–d show the XPS spectra of Fe2p, Cl2p and C1s in pristine PVA/FeCl3 (all in red) and converted 3D-GN/Fe (all in black) after the CVD growth procedure. Major differences between the iron ion species and metallic iron can be seen in the Fe2p region at around 700–730 eV, as shown Figure 3b. The Fe2p peak from the pristine PVA/FeCl3 sample is deconvoluted into two major peaks, at 711.5 eV and 724.54 eV, corresponding to the binding energy of Fe3+ in FeCl3 and Fe2O3, respectively, which suggests the coexistence of FeCl3 and oxidized Fe2O338,39. On the other hand, a peak at around 704 eV, corresponding to the binding energy of metal ions, appears for the annealed samples, clearly indicating the reduction of iron ions. The peak between binding energies of 195 eV and 204 eV in the XPS spectra (in Figure 3c) reveals the presence of chlorine in the samples. The two broad peaks in red at binding energies of 198.8 eV and 199.95 eV, corresponding to 2p3/2 and 2p1/2 electrons of chlorine ions (Cl−), respectively, in the pristine sample and the disappearance of the chlorine ion peak in the 3D-GN/Fe sample suggest that the chlorine is completely evaporated under the high temperature growth conditions of the 3D-GN. Finally, compared to the broad and significantly low C1s peak in the pristine samples in Figure 3d, the intense, sharp peak in the 3D-GN/Fe, centered at 284.18 eV, which originated from the C-C or C = C bond, indicates the conversion of amorphous carbon into high quality graphene. A full width at half maximum (FWHM) of 0.82 was obtained for the 3D-GN; this is comparable to the FWHM of a high quality single layer graphene40,41.
Discussion
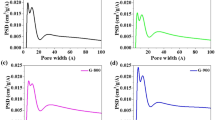
Figure 4 shows the Raman spectra of 3D-GNs synthesized by the diffusion and precipitation of carbon on the surface of 3D iron networks with various composition ratios of PVA to iron precursors. On the basis of previous reports that describe the growth of graphene on iron, one significant issue to be considered is that the relatively high solubility of carbon in iron could lead to the formation of multi-layer graphene with some defective areas21. We found that the initial amount of iron and carbon greatly affected the quality of graphene and the structural formation of the 3D-GN, since both elements are nearly used up during the growth process, as shown in the TGA data, Figure S3. Optimum growth conditions of graphene on PVA-FeCl3/3D-CS were determined by careful control of the pressure and the flow rate of the gas at 1,000°C in all the experiments during CVD until we acquired the best Raman peak (Figure S6). The typical Raman spectrum of the 3D-GN at various iron to PVA ratios characterized at more than 20 random locations are compared with that of pristine FeCl3/PVA with broad, strong D and G peaks as shown in the bottom of Figure 4a. The creation of graphene in the 3D-GN can be verified by the decrease in the intensity of the D band and the evolution of sharp G and 2D bands at the higher frequency region. The intensity of the G band steadily increased as the amount of FeCl3 increased (from bottom to top) implying the formation of a planar configuration of sp2 bonds. The dramatic increase in intensity of the 2D band of the 3D-GN at 350 phr indicates high-quality of graphene. The negligible D peak around 1,350 cm−1 for the 350 phr sample suggests that the 3D-GN at 350 phr has few defects or symmetry-broken sites. The maximum ratio of I2D/IG and the minimum ratio of ID/IG optimized at 350 phr are 1.74 and 0.15, respectively, as shown in Figure S6. Therefore, the Raman spectra indicate that amorphous parts of carbonized-C are nearly used up and transformed into few-layer 3D graphene networks when the ratio of PVA to iron was set at around 1∶3.5. Surprisingly, the average conductivity of the 3D-GN obtained from ∼20 samples was 52 S/cm, which is much greater than the previously reported value6. Moreover, the average conductivity of the 3D-GN before the removal of iron was ∼200 S/cm, implying potential for the direct use of 3D-GN/iron as a porous 3D electrode. More importantly, one of the significant advantages of the proposed method is that it allows for mass-production. Since the iron precursor can be homogenously dispersed with the carbon source to form the graphene frame, there is essentially no limit to the scale of production for this type of precursor-assisted CVD. For example, we could obtain 2 g of 3D-GN per batch of CVD, which is comparable to 2050 m2 of planar 2D graphene based on the surface area of our 3D-GN. Due to such large scale production, BET measurement is readily available. The surface area of the 3D-GN obtained by templating 220 nm uniform silica particles (in Figure 2) was measured to be 448 m2/g and the pore size of a 3D-GN range from approximately 210–230 nm in diameter, with some much smaller mesopores with a mean diameter of 14 nm, as shown in Figure S7. The surface area of 448 m2/g is much greater than that of same size CS templates (13 m2/g), which can be attributed to the presence of cavities as well as a few layers in graphene. We believe further improvement of the physical characteristics of 3D-GNs will be possible, i.e., much smaller pores by using a smaller size uniform silica template and better conductivity with more systematic control of composition ratios as well as growth conditions. In fact, we were able to obtain a 3D-GN with a specific surface area of 1,025 m2/g and conductivity of 5.4 S/cm by templating silica particles with a dimension of 20–30 nm in diameter on a bulk scale (Figures S7 and S8). Even though they had lower uniformity in particle size and shape and somewhat aggregated morphology compared to the larger size particles, comparable quality of Raman peaks and other characteristics to those of the uniform larger size and well assembled 3D-GNs were observed, as shown in the Supporting Information (Figure S5).

Raman spectra of 3D-GNs grown by CVD.
(a) Raman spectra with various composition ratios (phr: parts per hundred parts of resin) of PVA-FeCl3 by templaing 220 nm of CS. The Raman spectrum of pristine PVA after annealing at 1,000°C in a H2 environment is presented in black. (b) Raman spectra of 3D-GNs grown on various substrates by templating 220 nm of CS with PVA-FeCl3 solutions at a FeCl3 concentration of 350 phr. There is basically no restriction on the choice of substrate as long as it is stable and inert at high temperature.
Another very useful benefit of this technique compared to previously reported methods is that the graphene growth does not restrict the choice of substrate due to the use of metal precursor solutions, which essentially permits the dimensional scalability. Furthermore, this circumvents the need for a transfer process which can lead to the introduction of additional defects. Figures 4b shows the Raman spectra of a 3D-GN grown on any arbitrary substrate such as bare silicon, sapphire, GaN, or quartz. We used the same growth conditions here as employed for growth on a SiO2/Si substrate. The best Raman spectrum, with a more enhanced 2D band and a lower D band, was obtained from the 3D-GN grown on the sapphire substrate; this may be attributed to the close match of the sapphire (111) facet with the lattice of graphene, which provides an ideal surface for graphene epitaxy40,42. The direct growth of a 3D-GN on any electronic device-compatible substrate will open up the possibility of producing ready-to-use graphene with minimum defects in the semiconductor industry.
As a demonstration of the potential of 3D-GNs, the performance of the 3D-GN sample as an EDLC electrode was studied using cyclic voltammetry (CV) and galvanostatic charge/discharge measurements. Here we used a 3D-GN prepared with a specific surface area of 1,025 m2/g and conductivity of 5.4 S/cm fabricated on a bulk scale. The 3D-GN was p-doped by dipping it in a HNO3 solution to further enhance conductivity up to 6.9 S/cm. Figure 5a shows the CV results of 3 cell electrodes assembled with the 3D-GN as the active material at various scan rates in a range of −0.2 V to 0.8 V vs Ag/AgCl. Generally, for a supercapactior that uses a carbon-based electrode, the CV curve shape and the specific capacitance can significantly degrade as the voltage scan rate increases, showing distortion of the rectangular shape. The CV curves of the 3D-GN supercapacitor at all scan rates except the one at the highest scan rate of 500 mV/s were nearly rectangular, indicating that the 3D-GN is an ideal carbon electrode material with excellent capacitance behavior and low contact resistance. A deviation from an ideal double-layer capacitor at a very high scan rate of 500 mV/s could be related to the increased resistance. An outstanding specific capacitance of 245 F/g in a H2SO4 solution (1 M) was obtained at a scan rate of 5 mV/s, calculated by equation (1) (This is very encouraging since the capacitance value could be further improved by optimizing several parameters for cell fabrication).

The essentially greater specific capacitance of the 3D-GN relative to that of previously reported values for graphene electrode materials (101–205 F/g, as shown in Figure 5f) is attributable to the large accessible surface area achieved by proper-sized mesopores and 3-dimensionally interconnected conducting pathways of graphene. More specifically, compared to several previously reported supercapacitors with high specific surface area greater than 2000 m2/g, which show relatively low capacitance values due to the presence of predominant micro-pores less than 2 nm in diameter, our 3D-GNs have a reasonably great specific surface area of pores with controlled sizes of about 10 nm in diameter which can be effectively and fully used for electrochemical reaction sites43,44. The specific capacitance per unit surface area was as high as 23.9 μF/cm2, an outstanding value for double-layer capacitors. The galvanostatic charge/discharge method was applied to evaluate the capacitance performance of the 3D-GN. A well symmetric charge/discharge characteristic with a triangular shape distinctive for an ideal capacitor was observed even at a high current load of 100 A/g (Figure 5b). Importantly, the capacitance loss of graphene was less than 13%, with a current variation from 5 A/g to 100 A/g, as shown in Figure 5c.

Electrochemical performance of 3D-GN-based supercapacitor.
(a) Cyclic voltammogram curve at different scan rates. (b) Charging/discharging curve at different discharge current densities. (c) Capacitance change at various current densities. (d) Nyquist impedance plot. (e) Capacitance retention plot with cycle number. (f) Plot of recorded specific capacitances of EDLC electrodes acquired from various methods.
In order to obtain a comprehensive perspective on the capacitive response, an electrochemical impedance test was conducted. The Nyquist plot of the 3D-GN in Figure 5d exhibits a small semicircle at the high frequency region and a straight line in the medium frequency region. The small diameter of the semicircle at the high frequency region represents the low charge transfer resistance at the interface between the electrode and electrolyte. The nearly vertical second partial semicircle at the medium frequency region indicates the low charge transfer resistance for the adsorption process, which is greatly affected by the surface morphology of the electrode. The low resistance at both the high and medium frequency regions can be due to improved conductivity in the 3-dimensionally interconnected graphene networks through the nano-channels with proper diameters. The 3D-GN based supercapacitor retains ∼96.5% of its performance even after 6000 cycles, suggesting great life-cycle stability of the 3D-GN based electrode materials for their practical use in electrochemical capacitors as shown in Figure 5e.
In conclusion, we developed an easy and direct method for fabricating 3D networks of few-layer graphene via the CVD technique from a PVA/iron precursor on a bulk scale. Our substrate-free CVD method adds the dimensional tunability to high quality graphene, by enabling the creation of nanonetworks. A 3D-GN can be grown on any inert substrate such as Al2O3, quartz, or SiO2/Si wafer, etc. and can be further transferred onto any arbitrary substrate for flexible devices, cell culture plates, etc. The ratio of I2D/IG ( = 1.74) for the representative 3D-GN confirms the formation of few-layer graphene. A conductivity of ∼52 S/cm, high surface areas of 1,025 m2/g and a great porosity of around 3.4 cm3/g were recorded for the 3D-GN. Electrochemical measurements indeed proved that the high surface area 3D-GN based electrode, which provides easy contact and transportation of both charges and the electrolyte through the 3D conducting pathways, exhibited excellent specific supercapacitance. The outstanding performance of 3D-GNs fabricated with this easy and inexpensive method suggests a straightforward route to achieve nano-textured 3D graphene with strong potential for use in electronic devices and heat-dissipation systems, as well as for energy-related materials.
Methods
Preparation of CS/PVA-FeCl3·6H2O composite thin film
Poly vinyl alcohol (PVA, Mw = 31,000–50,000) and FeCl3·6H2O were obtained from Aldrich Chemical Company and used without any further purification. PVA (10 wt.%) was dissolved in DI-water at 90°C and then mixed with percents of 100, 200, 350 and 600 phr (phr: part per hundred parts of resin) by weight of FeCl3·6H2O. PVA/FeCl3 composite thin films were prepared by self-assembly of CS, as reported elsewhere. Any impurities in the solution were filtered through a 0.2 μm cellulose acetate syringe filter and the filtrated solution was spin-coated on a 300 nm SiO2/Si substrate. The prepared composite thin films were dried in a vacuum oven for one day. 3D-GNs was p-doped by dipping in 20 mol% of HNO3 solution for 30 min followed by washing in D.I. water in order to enhance the conductivity for supercapacitance measurements.
Fabrication of the 3D-GNs
The prepared composite films were placed in a quartz tube (Scientech Co.) with an outer diameter of 120 mm, heated to 1,000°C in a H2 (100 sccm)/Ar atmosphere at 4 torr with a heating rate of ∼20°C/min and then placed in isothermal conditions for 30 min. After annealing, the samples were cooled to an ambient temperature. The 3D-GN/Fe sample on a 300 nm SiO2/Si substrate was then immersed on BOE consisting of HF (5%) and HCl (3%) for 48 hrs to remove the SiO2 substrate and iron simultaneously.
Characterization of 3D-GNs
The structures of the samples were characterized by SEM (Nova Nano-SEM 230, 10 kV), TEM (JEM-2100, 200 kV) and Raman spectroscopy (WITec, alpha300R, excited by a 532nm laser). X-ray diffraction measurements were carried out with a Rigaku Co. High Power X-Ray Diffractometer D/MAZX 2500V/PC from 20° to 80°. Surface area determination was performed by Brunauer-Emmett-Teller (BET) methods using a BELSORP-max (BEL JAPAN INC.). The sheet resitances of 3D-GNs (both film and powder type) were characterized by 4 point-probe (Dasol Eng, FPP-RS8, pin-spacing 1 mm, pin-radius 100 μm). The conductivities were obtained with the equation of σ = 1/(R × d), where d is the thickness of the sample and R is the sheet resistance of the sample. To measure conductivities of powder type 3D-GN, the pellet with a diameter of 13 mm and thickness of 7 μm was prepared by mechanical milling and subsequent pressurizing (1,000 kg/cm2) of the 3D-GN. An energy dispersive X-ray analyser (EDX) was used to provide elemental identification and quantitative compositional information of samples, as shown in Figure S9.
Electrochemical measurement
The electrochemical properties of supercapacitor electrodes were measured in three-electrode systems by cyclic voltammetry using a computer controlled electrochemical interface (VMP3 biologic) from −0.2 to 0.8 V at room temperature. 3D-GNs, a graphite plate, Ag/AgCl and 1 M H2SO4 were used as the working electrode, counter electrode, reference electrode and electrolyte, respectively. To examine the electrochemical properties, the 0.5 mg of electro-active material (3D-GN) was mixed with acetylene black (10 wt.%) and polyvinylidenedifluoride (PVDF, 10 wt.%) as a binder and then the mixture was pasted onto a FTO electrode (2.56 cm2) and dried at 150°C for 20 min in an air atmosphere. The cyclic voltammetry was carried out at different scan rates ranging from 5 mV/s to 500 mV/s. Electrochemical impedance spectroscopy (EIS) was carried out in 1M LiClO4/acetonitrile solutions at a frequency range from 100 kHz to 0.1 Hz using a potentiostat (Versa STAT 3, AMETEK). The control sample for EIS measurement was fabricated with 10 wt.% of acetylene black and 10 wt.% of PVDF.
References
Jang, J.-H. et al. Three-Dimensionally-Patterned Submicrometer-Scale Hydrogel/Air Networks That Offer a New Platform for Biomedical Applications. Nano Lett. 8, 1456–1460 (2008).
Jang, J.-H., Ullal, C. K., Gorishnyy, T., Tsukruk, V. V. & Thomas, E. L. Mechanically Tunable Three-Dimensional Elastomeric Network/Air Structures via Interference Lithography. Nano Lett. 6, 740–743 (2006).
Jang, J.-H., Dendukuri, D., Hatton, T. A., Thomas, E. L. & Doyle, P. S. A Route to Three-Dimensional Structures in a Microfluidic Device: Stop-Flow Interference Lithography. Angew. Chem. 119, 9185–9189 (2007).
Zorlutuna, P. et al. Microfabricated Biomaterials for Engineering 3D Tissues. Adv. Mater. 24, 1782–1804 (2012).
Geim, A. K. & Novoselov, K. S. The rise of graphene. Nat. Mater. 6, 183–191 (2007).
Chen, Z. P. et al. Three-dimensional flexible and conductive interconnected graphene networks grown by chemical vapour deposition. Nat. Mater. 10, 424–428 (2011).
Park, S. & Ruoff, R. S. Chemical methods for the production of graphenes. Nat. Nanotech. 4, 217–224 (2009).
Dean, C. R. et al. Boron nitride substrates for high-quality graphene electronics. Nat. Nanotechnol. 5, 722–726 (2010).
Bae, S. et al. Roll-to-roll production of 30-inch graphene films for transparent electrodes. Nat. Nanotech. 5, 574–578 (2010).
Reina, A. et al. Large Area, Few-Layer Graphene Films on Arbitrary Substrates by Chemical Vapor Deposition. Nano Lett. 9, 30–35 (2009).
Bhaviripudi, S., Jia, X., Dresselhaus, M. S. & Kong, J. Role of Kinetic Factors in Chemical Vapor Deposition Synthesis of Uniform Large Area Graphene Using Copper Catalyst. Nano Lett. 10, 4128–4133 (2010).
Li, X. et al. Large-Area Synthesis of High-Quality and Uniform Graphene Films on Copper Foils. Science 324, 1312–1314 (2009).
Li, X., Cai, W., Colombo, L. & Ruoff, R. S. Evolution of Graphene Growth on Ni and Cu by Carbon Isotope Labeling. Nano Lett. 9, 4268–4272 (2009).
Kwak, J. et al. Near room-temperature synthesis of transfer-free graphene films. Nat. Commun. 3, 7 (2012).
Cao, X. H. et al. Preparation of Novel 3D Graphene Networks for Supercapacitor Applications. Small 7, 3163–3168 (2011).
Michael Thompson Pettes, H. J., Rodney, S. Ruoff & Li Shi. Thermal Transport in Three-Dimensional Foam Architectures of Few-Layer Graphene and Ultrathin Graphite. Nano Lett. 12, 2959–2964 (2012).
Jeong, H. M. et al. Nitrogen-Doped Graphene for High-Performance Ultracapacitors and the Importance of Nitrogen-Doped Sites at Basal Planes. Nano Lett. 11, 2472–2477 (2011).
Choi, B. G., Yang, M., Hong, W. H., Choi, J. W. & Huh, Y. S. 3D Macroporous Graphene Frameworks for Supercapacitors with High Energy and Power Densities. ACS Nano 6, 4020–4028 (2012).
Yao, Y. et al. Controlled Growth of Multilayer, Few-Layer and Single-Layer Graphene on Metal Substrates. J. Phy. Chem. C 115, 5232–5238 (2011).
He, Z. et al. Iron Catalysts for the Growth of Carbon Nanofibers: Fe, Fe3C or Both? Chem. Mater. 23, 5379–5387 (2011).
Xue, Y. Z. et al. Synthesis of large-area, few-layer graphene on iron foil by chemical vapor deposition. Nano Res. 4, 1208–1214 (2011).
Conway, B. E. Electrochemical Supercapacitor : Scientific Fundamentals and Technological Applications (Pleum; New York, 1999).
Kötz, R. & Carlen, M. Principles and applications of electrochemical capacitors. Electrochim. Acta 45, 2483–2498 (2000).
Wang, G., Zhang, L. & Zhang, J. A review of electrode materials for electrochemical supercapacitors. Chem. Soc. Rev. 41, 797–828 (2012).
Zhu, Y. et al. Carbon-Based Supercapacitors Produced by Activation of Graphene. Science 332, 1537–1541 (2011).
Bose, S. et al. Carbon-based nanostructured materials and their composites as supercapacitor electrodes. J. Mater. Chem. 22, 767–784 (2012).
Zhang, L. L. et al. Highly Conductive and Porous Activated Reduced Graphene Oxide Films for High-Power Supercapacitors. Nano Lett. 12, 1806–1812 (2012).
Lee, C., Wei, X., Kysar, J. W. & Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 321, 385–388 (2008).
Stoller, M. D., Park, S., Zhu, Y., An, J. & Ruoff, R. S. Graphene-Based Ultracapacitors. Nano Lett. 8, 3498–3502 (2008).
Shornikova, O. N., Kogan, E. V., Sorokina, N. E. & Avdeev, V. V. The Specific Surface Area and Porous Structure of Graphite Materials. Russian J. Phy. Chem. A 83, 1022–1025 (2009).
Ubbelohde, A. & Lewis, F. Graphite and Its Crystal Compounds. (Oxford, Clarendon Press, 1960).
Wassei, J. K. & Kaner, R. B. Graphene, a promising transparent conductor. Mater. Today 13, 52–59 (2010).
De, S. et al. Flexible, Transparent, Conducting Films of Randomly Stacked Graphene from Surfactant-Stabilized, Oxide-Free Graphene Dispersions. Small 6, 458–464 (2010).
Jiang, P., Bertone, J. F., Hwang, K. S. & Colvin, V. L. Single-Crystal Colloidal Multilayers of Controlled Thickness. Chem. Mater. 11, 2132–2140 (1999).
Babin, P., Gavrilov, A. & Trofimova, L. Absorption spectra and the nature of complexes in the polyvinyl Alcohol-Nickel (II) chloride system. J. Appl. Spectro. 73, 151–153 (2006).
Nouh, S. A., Radwan, M. M., Agami, W. R. & Morsy, M. Studies on the effect of laser radiation on the thermal stability of stabilized poly(vinyl chloride). J. Appl. Polym. Sci. 89, 2249–2255 (2003).
Kumar, S. & Chakarvarti, S. K. SEM morphology and XRD characterization of Ni microstructure arrays synthesized by dc electrodeposition in porous polycarbonate templates. J. Mater. Sci. 39, 3249–3251 (2004).
Grosvenor, A. P., Kobe, B. A., Biesinger, M. C. & McIntyre, N. S. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. and Interf. Anal. 36, 1564–1574 (2004).
Suzuki, S., Yanagihara, K. & Hirokawa, K. XPS study of oxides formed on the surface of high-purity iron exposed to air. Surf. and Interf. Anal. 30, 372–376 (2000).
Miyasaka, Y. N. Atsushi; Temmyo, Jiro. Graphite Thin Films Consisting of Nanograins of Multilayer Graphene on Sapphire Substrates Directly Grown by Alcohol Chemical Vapor Deposition. JPN J. Appl. Phys. 50, 04DH12–04DH12-04 (2011).
Mérel, P., Tabbal, M., Chaker, M., Moisa, S. & Margot, J. Direct evaluation of the sp3 content in diamond-like-carbon films by XPS. Appl. Surf. Sci. 136, 105–110 (1998).
Song, H. J. et al. Large scale metal-free synthesis of graphene on sapphire and transfer-free device fabrication. Nanoscale 4, 3050–3054 (2012).
Qu, D. Y. Studies of the activated carbons used in double-layer supercapacitors. J. Power Sources 109, 403–411 (2002).
Shi, H. Activated carbons and double layer capacitance. Electrochim. Acta 41, 1633–1639 (1996).
Acknowledgements
This work is supported by NRF with the contract no. NRF-20120019408, NRF-R1A1A2043076 and NRF-2012-0002413.
Author information
Authors and Affiliations
Contributions
J.Y. and J.L. contributed equally to this work. The experiments were designed by J.Y., J.L. and S.K. with suggestions from J.J.; J.Y. prepared 3D-GNs samples. J.L. and K.K. performed electron microscopy studies. J.Y. performed XPS analysis. S.K. performed supercapacitor measurements. J.J. analyzed the data and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
supporting information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Yoon, JC., Lee, JS., Kim, SI. et al. Three-Dimensional Graphene Nano-Networks with High Quality and Mass Production Capability via Precursor-Assisted Chemical Vapor Deposition. Sci Rep 3, 1788 (2013). https://doi.org/10.1038/srep01788
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01788
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
