
Abstract
The aggregation and deposition of amyloid-β(1–42) (Aβ42) in the brain is implicated in the aetiology of Alzheimer's disease (AD). While the mechanism underlying its deposition in vivo is unknown its precipitation in vitro is influenced by metal ions. For example, Aβ42 is known to bind copper, Cu(II), in vitro and binding results in aggregation of the peptide. The biophysical properties of Cu(II)-Aβ42 aggregates are of significant importance to their putative involvement in the amyloid cascade hypothesis of AD and are currently the subject of strong debate. In particular the question has been raised if sub- and super-stoichiometric concentrations of Cu(II) act in opposing ways in respectively accelerating and preventing amyloid fibril formation by Aβ42. Herein we have used fluorimetry and transmission electron microscopy to provide unequivocal evidence that under near-physiological conditions both sub- and super-stoichiometric concentrations of Cu(II) prevented the assembly of Aβ42 into ThT-positive β-sheet rich amyloid fibrils.
Similar content being viewed by others

Introduction
A central tenet of the amyloid cascade hypothesis (ACH) of Alzheimer's disease (AD) is that the extracellular deposition in the brain of amyloid-β1–42 (Aβ42) as β sheets is integral to disease aetiology1,2. While it is unknown why Aβ42 forms deposits of β sheets in vivo there is much speculation as to the potential role of metal ions in driving the conformational changes which underlie the process3. A number of metal ions have been shown to associate with monomeric Aβ42 in vitro and while each of these interactions may ultimately result in the precipitation of the peptide the detailed structures of the deposits that are formed are varied4,5,6. For example, the evidence to-date suggests that Aβ42 binds Cu(II) with the greatest avidity5,7 though this interaction results in the precipitation of amorphous deposits of the peptide and not the β sheets which are the signatures of Aβ42 deposits in vivo, for example, at the core of neuritic or senile plaques in AD4. The propensity for Cu(II) to prevent Aβ42 from adopting a β sheet conformation is well known4,8,9 and widely accepted and, additionally this property of Cu(II) has also been demonstrated for other amyloidogenic peptides10,11,12. However, certain aspects of the structural relationship between Aβ42 and Cu(II) have recently been challenged and suggestions have been made that the propensity for Cu(II) to precipitate Aβ42 as amorphous deposits is only valid at ‘high’ concentrations of peptide and at stoichiometric ratios of Cu(II) to peptide ≥ 113. Outside of these specific conditions it is suggested that Cu(II) accelerates β sheet formation by Aβ42. While there are actually very few published data to support these contentions they are widely advertised14,15,16 and it is important that they are resolved as they could have important implications for a putative role for Cu(II) in AD. Recent evidence has indicated that not only is the copper content of human brain tissue lower in AD17,18 and in congophilic amyloid angiopathy (CAA)19 but that there is a negative relationship between brain copper content and amyloid pathologies in AD and CAA20. Lower copper leads to more extensive and more aggressive amyloid pathology. Herein we have thoroughly investigated the affect of Cu(II) on the propensity for Aβ42 to form ThT-positive β sheet rich amyloid fibrils and we have found no evidence under near-physiological conditions to support an accelerant effect of Cu(II) on fibril formation by Aβ42.
Results
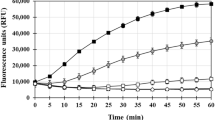
We first examined how the ratio of Cu(II) to Aβ42 affected β sheet-rich fibril formation as measured using ThT fluorescence and as confirmed by TEM. At a total peptide concentration of 8 μM and in the absence of added Cu(II) Aβ42 formed ThT-positive β sheets instantaneously and ThT fluorescence peaked at approximately 200 AU after about 1 h incubation at 37°C. ThT fluorescence remained high at 3 h and fell after 24 h incubation (Table 1). In the presence of added Cu(II) and at each incubation time interval, 1,3 and 24 h, ThT fluorescence were significantly lower at metal:peptide ratios of 0.5, 1.0, 2.0, 5.0 and 10.0. The influence of Cu(II) at each metal:peptide ratio was effectively complete within 1 h incubation at 37°C as there were no statistically significant differences in data relating to its effects after 1, 3 and 24 h incubation at 37°C (Fig. 1a). Cu(II) reduced ThT fluorescence by at least 75% when it was equimolar or greater and at a metal:peptide ratio of 10 the ThT fluorescence was zero at each time point. Both the presence of characteristic negatively-stained amyloid fibrils in the absence of added Cu(II) and the total lack of such fibrils at a 10-fold molar excess of Cu(II) were confirmed in these treatments by TEM (Fig. 2a,b).

Copper affects the propensity for Aβ42 to form ThT-positive β sheets.
(a) The influence of the metal:peptide ratio on the ThT fluorescence of Aβ42 (8.0 μM) after incubation at 37°C for 1, 3 and 24 h. Mean and SD are plotted, n = 4. (b) The influence of sub-, equimolar and super-stoichiometric concentrations of Cu(II) on the ThT fluorescence of Aβ42 (5.0 μM) after incubation at 37°C for 24 h. Mean and SD are plotted, n = 3. (c) Abolition by 100 μM EDTA of the effects of sub- to equimolar concentrations of Cu(II) on the ThT fluorescence of Aβ42 (5.0 μM) after incubation at 37°C for 24 h. Mean and SD are plotted, n = 3. (d) The influence of the concentration of Aβ42 on the propensity for Cu(II) at a metal:peptide ratio of 0.5 to form ThT-positive β sheets. Mean and SD are plotted, n = 3.

Copper abolishes the negatively-stained fibrillar structure of Aβ42 as viewed by TEM.
(a) An 8.0 μM preparation of Aβ42 incubated for 3 h at 37°C under near-physiological conditions assembles into negatively stained amyloid single and double fibrils. (b) An 8.0 μM preparation of Aβ42 in the presence of 80 μM Cu(II) incubated for 3 h at 37°C under near-physiological conditions forms amorphous positively stained deposits showing remnants of a fibril-like morphology. Scale bar 200 nm for both TEM images.
When the metal:peptide ratio was widened to include more points which were sub-stoichiometric with respect to Cu(II) the ThT fluorescence of a 5 μM Αβ42 treatment incubated at 37°C for 24 h was significantly reduced at each ratio ≥ 0.25 and was effectively zero at a ratio of 2.0 and 5.0 (Fig. 1b). We found no significant difference in ThT fluorescence between treatments with no added Cu(II) and those with a Cu(II):peptide ratio of 0.1. TEM showed the presence of negatively stained amyloid fibrils at Cu(II):peptide ratios ≤ 0.5 and was unable to confirm the presence of any negatively-stained fibrillar materials at Cu(II):peptide ratios ≥ 1.0.
A further even more detailed examination of how sub-stoichiometric concentrations of Cu(II) influenced the formation of β sheet-rich amyloid fibrils of a 5 μM preparation of Aβ42 in the absence and presence of 100 μM EDTA showed dose-dependent reductions in ThT fluorescence which were almost wholly abolished in media which additionally included EDTA (Fig. 1c). TEM confirmed the presence of negatively-stained fibrillar material in preparations which additionally included 100 μM EDTA (Fig. 3). The presence of a significant molar excess of EDTA effectively prevented the effects of Cu(II) on β sheet formation by Aβ42. TEM confirmed that in the absence of EDTA incremental increases in the metal:peptide ratio from ca 0.2 through to ≥ 1.0 produced similarly incremental changes in the visible structure of the precipitated amyloid-like materials, from distinct negatively-stained fibrils of diameter ca 10 nm to amorphous ribbon-like deposits with little characteristic negative staining (Fig. 4).

The copper chelator EDTA abolishes the effect of Cu(II) on the fibrillar structure of Aβ42.
(a) A 5.0 μM preparation of Aβ42 in the presence of 4.0 μM Cu(II) incubated for 24 h at 37°C under near-physiological conditions forms fibril-like deposits showing both negative and positive staining. (b) The identical preparation in the presence of 100 μM EDTA forms negatively-stained fibrils showing characteristic amyloid morphology. Scale bar 200 nm for both TEM images.

Incremental increases in Cu(II):Aβ42 ratio results in incremental changes to the structure of amyloid deposits.
(a) 5.0 μM Aβ42 in the presence of 1.0 μM Cu(II) results in classic negatively-stained fibrillar morphologies which are not distinct from peptide preparations in the absence of added Cu(II). (b) 5.0 μM Aβ42 in the presence of 2.0 μM Cu(II) results in fewer negatively-stained fibrils which were generally shorter in length than peptide preparations in the absence of added Cu(II). (c) 5.0 μM Aβ42 in the presence of 3.0 μM Cu(II) results in fibril-like morphologies which are not obviously negatively-stained and are amorphous in appearance as compared to peptide preparations in the absence of added Cu(II). (d,e) 5.0 μM Aβ42 in the presence of 4.0/5.0 μM Cu(II) results in primarily positively-stained fibril-like deposits which are distinctly amorphous in comparison to peptide preparations in the absence of added Cu(II). (f) 5.0 μM Aβ42 in the presence of 10.0 μM Cu(II) results in primarily positively-stained amorphous deposits showing no semblance of fibrillar structure. Scale bars on all TEM images are 200 nm.
A final experiment investigated how the concentration of Aβ42 influenced Cu(II) at a metal:peptide ratio of 0.5 to affect the formation of ThT-positive β sheet-rich amyloid fibrils. At each concentration of Aβ42 the affect of Cu(II) was to significantly reduce the ThT fluorescence though the effect was significantly lower proportionately at a peptide concentration of 1 μM than 10 μM (Fig. 1d).
Discussion
We have investigated how Cu(II) affects the propensity for Aβ42 to form ThT-positive β sheet-rich amyloid fibrils under near-physiological conditions. We define the latter in terms of the composition of extracellular milieu which are characteristic of environments, such as brain interstitial fluid, where amyloid might be expected to form and precipitate as, for example, senile plaques. The inorganic composition of these environments will be similar to serum21 and, importantly, will include ca 1 mM of both Ca2+ and Mg2+. The pH is likely to be 7.40 and the temperature 37°C and we believe that these chemical and physical properties are essential minimum criteria for studying amyloidogenesis under in vitro conditions4. The physiological concentration of Cu(II) in brain extracellular fluids can vary between ca 0.5 μM in CSF to 100–250 μM in the synaptic cleft22 and, as such, the metal:peptide ratios investigated herein can be considered as near-physiological. We have identified the presence of β sheet-rich amyloid fibrils using a combination of ThT fluorimetry and TEM. These are the optimal methods for studying the formation of β sheets in amyloid preparations containing near-physiological concentrations of peptide4. A range of concentrations of Aβ42 from sub-saturation (1.0 μM) to super-saturated (10.0 μM) were studied in association with metal:peptide ratios representing sub-stoichiometric (e.g. 0.1) to super-stoichiometric (e.g. 10.0) conditions and under no combination of the aforementioned conditions did the presence of added Cu(II) accelerate or augment ThT-positive β sheet-rich fibril formation relative to the equivalent peptide-only preparations. To the contrary, the consistent effect of Cu(II) addition was to reduce the rate of formation or completely abolish the formation of Aβ42 structures in β sheet conformations (Fig. 1; Fig. 4). The formation of ThT-reactive amyloid was practically instantaneous, with or without added Cu(II) in media and fluorescence maxima were generally achieved within hours and showed a tendency to fall over subsequent days incubation at 37°C. The presence of Cu(II) resulted in incremental falls in ThT fluorescence for increasing sub-stoichiometric concentrations of Cu(II) and effectively abolished ThT-positive β sheets of Aβ42 once Cu(II) was present to molar excess. These affects upon ThT fluorescence were mirrored by structural changes observed by TEM in the deposits of Αβ42 from characteristic negatively-stained fibrils at a metal:peptide ratio of 0.2 to amorphous ribbon-like materials at equimolar concentrations and finally ill-defined positively-stained precipitates when Cu(II) was present to excess (Fig. 4). It was of interest that under conditions where the rate of formation of ThT-positive β sheets were favoured, e.g. 10 μM Aβ42, the propensity for Cu(II) to prevent this process was proportionately higher than at lower concentrations of peptide, e.g. 1 μM Aβ42, for a constant metal:peptide ratio (Fig. 1d). In treatments which included 100 μM EDTA the addition of Cu(II) neither accelerated, augmented nor reduced ThT fluorescence relative to treatments without added Cu(II) (Fig. 1c; Fig. 3). Since EDTA was expected to bind Cu(II) up to 4 orders of magnitude more strongly than Aβ4223 and EDTA was present to significant excess then the presence of EDTA was expected to abolish any effect of Cu(II) on the aggregation of Aβ42 (Fig. 3). Clearly we have found no evidence to support Cu(II) as an accelerant of amyloid fibril formation by Aβ42 and our data are in full agreement with the majority of the published literature, as is discussed further below.
The first indication that Cu(II) prevented Aβ42 from aggregating as β sheets was by Zou et al8. where it was shown using a combination of ThT fluorescence and atomic force microscopy (AFM) that a 5-fold molar excess of Cu(II) prevented 100 μM Aβ42 from forming β sheets at pH 5, 6, 7, 8 and 9 and 37°C. This research was quickly followed by that of Yoshiike et al9. where it was shown using ThT fluorescence that a 2-fold molar excess of Cu(II) prevented 10 μM Aβ42 from forming β sheets while at 1 μM Aβ42 sub-stoichiometric levels of added Cu(II) produced incremental reductions in ThT fluorescence which were very similar to those reported herein (Fig. 1). We followed up these seminal studies4 by demonstrating using ThT fluorescence and TEM that under near-physiological conditions a 10-fold molar excess of Cu(II) prevented a 1 μM preparation of Aβ42 from forming β sheet structures even after incubation at 37°C for 32 weeks. We additionally showed that this effect of Cu(II) was completely abolished in the presence of 1 mM EDTA. These three seminal studies together already negate the very recent suggestion that experiments had not previously accounted for Cu(II) affects at low concentrations of Aβ42 and sub-stoichiometric metal:peptide ratios13. However, these studies combined with more recent research24,25,26,27,28,29 and notably that of Smith et al.30, should leave little doubt as to how the addition of Cu(II) affects the propensity for Aβ42 to form β sheets under near-physiological conditions and yet many investigators recently reviewing this chemistry remain confused13,14,15,16. The origin of this confusion would appear to be a solitary paper by Sarell et al31. where it was reported that sub-stoichiometric concentrations of Cu(II) accelerated amyloid fibril formation by Aβ42. Data were shown in one supplementary figure for ThT fluorescence of preparations of 1, 3 and 5 μM Aβ42 in the presence of 0.5 mole equivalents of Cu(II) incubated at 30°C for up to 250+ hours. The figure purports to show that Cu(II) accelerated fibril formation though only after a lag period of approximately 50 hours up until which neither Aβ42 only nor Aβ42 + Cu(II) produced any significant ThT fluorescence. These results are clearly at odds with all other data in the aforementioned literature and specifically with the data reported herein. How might these differences be reconciled? While there are no clear explanations for the anomalous results of Sarell et al31. there are significant differences in the way in which their experiments were carried out relative to those herein and elsewhere. Specifically, their media did not include Ca2+ or Mg2+ and the incubations were at 30 not 37°C. While no information was given on the concentration of ThT in their fibril assays or their volumes it is known that ThT was coincubated with peptide in well plates, working volume ca 250–350 μL, for up to 250+ hours with 30 seconds of agitation every 30 minutes and without any convincing effective precautions against effects due to evaporation. Our assays were carried out in closed, low volume (100 μL) PE fluorescence cuvettes with ThT being added to the preparation only after a background reading (in the absence of ThT) was taken and thereafter only incubating the ThT and peptide for a sufficient period to achieve a fluorescence plateau, usually less than 600 seconds4. It is well known that co-incubation of ThT with peptide preparations which are undergoing amyloidogenesis will, for any set period of time, result in higher readings of ThT fluorescence than if the peptide was incubated in the absence of the fluor and ThT was only added immediately prior to a measurement being taken. This effect might be explained by ThT having access to more binding sites during the assembly of β sheets than when β sheet fibrils were preformed prior to the addition of the fluor. It might also be explained by a propensity for ThT to actually instigate amyloid fibril formation as has recently been demonstrated for ThT and Aβ4032. A final difference in methodology between Sarell et al31. and the research herein relates to the preparation of the stock solution of Aβ42. We dissolve the peptide in 0.01 M NaOH at a concentration of ca 1 mg/mL and we have shown that the peptide is fully dissolved in this highly alkaline, ca pH 12, preparation and can be used immediately thereafter4. Sarell et al31. dissolved their peptide at the same concentration but in water at pH 10. They then incubated the stock at 5°C for 72 h before using it. Over many years of practice we have learned that our Aβ42 stock remains stable for up to 24 h at 4°C and thereafter its propensity to produce ThT-reactive amyloid when diluted into media is gradually diminished over time. Whether these differences in preparation of Aβ42 stocks combined with the distinctly different assay media (e.g. presence of Ca2+ and Mg2+ in our media) are sufficient to account for the extreme lag phase observed by Sarell et al31. is unknown. However, neither of these factors would necessarily explain the Cu(II) effects observed by Sarell et al. since other research groups have used different ways of preparing their Aβ42 stocks and invariably they have not added divalent metals to their assay media and, similar to herein, they have not observed any Cu(II)-induced acceleration of Aβ42 fibril formation8,9,24,25,26,27,28,29,30. It is hard to conclude anything other than the results of Sarell et al31. are anomalous and are not representative of the consensus of scientific opinion on this matter.
When all of the data which together define how Cu(II) affects how Aβ42 forms amyloid fibrils with β sheet structure are considered in the context of everything that is known about these reactions they raise an interesting conundrum. The question is raised whether in these in vitro assays where there is always a saturated concentration of Aβ42 the kinetically-favoured and hence dominant reaction is the binding of Cu(II) by monomeric Aβ42 or is it the reaction of Aβ42 with itself and the formation of a β sheet conformer? We are inclined to believe that it is the latter which is the kinetically favoured reaction and that Cu(II) is only bound by Aβ42 under such conditions once it has self-aggregated and adopted a fibrillar β sheet structure. Where is the evidence to support such an heretical suggestion? To begin, it is absolutely the case that the binding affinity of Aβ42 for Cu(II) has never actually been measured under any conditions where self-aggregation of the peptide was not favoured and on-going. We emphasise the full length peptide here as there is little evidence that shorter fragments of this peptide, including Aβ40, are reliable mimics of Cu(II) binding by Aβ4233. In addition where any attempt has been made to compare the binding affinities of monomeric and fibrillar Aβ42 for Cu(II) the result was identical both quantitatively as a binding constant and qualitatively in respect of the ligands responsible for binding Cu(II)7. It is also true that Cu(II) can both prevent the formation of ThT-positive β sheets of Aβ42 and, importantly, abolish the β sheet conformation of preformed fibrillar material34. Coincidentally the TEM images of Aβ42 aggregates formed under both of these conditions appear identical. In addition we have noted herein that incremental increases in the Cu(II):Aβ42 ratio produced similarly incremental changes in the visible structures of the precipitated ‘amyloid’ materials. From distinct negatively-stained fibrils at low ratios to flattened, ribbon-like and less obviously negatively-stained ‘amyloid’ materials as equimolar levels of metal and peptide were approached (Fig. 4). These images certainly offer the suggestion that the changes in fibrillar structures occurred post-fibril formation as opposed to being the result of assembly from copper-Aβ42 monomers. The data presented herein that the efficacy with which Cu(II) prevented Aβ42 from forming ThT-positive β sheets was significantly proportionately greater at higher concentrations of peptide (Fig. 1d) also supports the notion that β sheet aggregates are required to form before their structure can be disrupted by Cu(II) binding. Finally and perhaps most convincingly, our recent observation that serum amyloid P component (SAP) (100 nM) catalysed and stabilised the formation of amyloid from an under saturated preparation of Aβ42 (500 nM) and that this effect was independent of the presence of a 10-fold metal:peptide molar excess of Cu(II) supported the suggestion that the formation of amyloid fibrils are prerequisites to their β sheet structure being disrupted by Cu(II)35. The effect of SAP was achieved through it being bound by preformed fibrils of Aβ42 and it was speculated that the population of the fibrils by many molecules of SAP prevented Cu(II) also being bound and so prevented Cu(II)-induced disruption of the β sheet structure. If the first and kinetically-favoured interaction under these conditions was Cu(II) being bound by monomeric Aβ42 then there would not be any amyloid fibrils in a β sheet conformation to bind and be stabilised by SAP. Regardless of the precise mechanism whereby Cu(II) disrupts the β sheet structure of Aβ42 a lower concentration of biologically available Cu(II) in the extracellular milieu of the brain could contribute towards a more extensive and more aggressive manifestation of the amyloid cascade hypothesis20 and therefore, possibly, to the aetiology of AD.
Methods
Amyloid β-protein (1–42) was bought from Bachem and used directly to prepare 200 μM stocks in 0.01 M NaOH. The peptide is fully dissolved under these highly alkaline conditions and exists only as monomers4. Peptide stocks are stable in this form for 24 h at 4°C. All treatments were prepared by diluting the appropriate volume of peptide stock into a physiologically-significant medium containing the requisite concentrations of Cu(II) and/or 100 μM ethylenediaminetetraacetic acid (EDTA). The physiologically-significant medium is a modified Krebs-Henseliet buffer including 118.5 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 1.4 mM CaCl2, 11.0 mM glucose and it is buffered at pH 7.40 ± 0.05 with 100 mM PIPES4. The buffer also includes 0.05% w/v sodium azide as an antimicrobial. Cu(II) is added to the required concentration using a certified copper standard provided by PE Life Sciences. All treatments are prepared and maintained at 37°C to accurately reflect physiological milieu.
The formation of β sheets was measured using ThT fluorescence as decribed previously4 and briefly herein. ThT was prepared freshly as required in ultra-pure water (conductivity < 0.067 μS/cm) as a 100 μM stock which when diluted into assays was used at a concentration of 10 μM. ThT fluorescence was measured using a PE LS50B fluorimeter with temperature controller (set at 37°C) and using a 100 μL low volume cuvette with lid to prevent evaporation. All treatments were measured (Ex. 450 nm; Em. 482 nm) before and after the addition of ThT and after either 300 s or the establishment of a plateau if this occurred within this timeframe. Background fluorescence (AU) was subtracted from ThT fluorescence to give a final value.
The occurrence of amyloid fibrils and other structures were confirmed using transmission electron microscopy (TEM)34. Pre-coated S162 200 mesh formvar/carbon coated copper grids (Agar Scientific), were inserted into 20 or 50 μL of the beaded treatment for 2 mins, then wicked, passed through ultra-pure water, re-wicked and placed into 30 μL 2% uranyl acetate (in 70% ethanol), for 30 s. Following staining with uranyl acetate, grids were removed, wicked, passed through ultra-pure water, re-wicked and placed into 30 μL of 30% ethanol for 30 s. Grids were finally re-wicked following this step, covered and allowed to dry for up to 24 h, prior to analysis via TEM. Samples, 3 full grids from each sample, were viewed on a JEOL 1230 transmission electron microscope operated between 100.0 – 120.0 kV, equipped with a Megaview III digital camera from Soft Imaging Systems (SIS). Images were obtained on the iTEM universal TEM imaging platform software.
References
Hardy, J. A. & Higgins, G. A. Alzheimer's-disease – the amyloid cascade hypothesis. Science 256, 184–185 (1992).
Checler, F. & Turner, A. J. Journal of neurochemistry special issue on Alzheimer's disease: amyloid cascade hypothesis-20 years on preface. J. Neurochem. 120, CP9–CP10 (2012).
Roberts, B. R., Ryan, T. M., Bush, A. I., Masters, C. L. & Duce, J. A. The role of metallobiology and amyloid peptides in Alzheimer's disease. J. Neurochem. 120, 149–166 (2012).
House, E. et al. Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Abeta42 in a manner which may have consequences for metal chelation therapy in Alzheimer's disease. J. Alzh. Dis. 6, 291–301 (2004).
Atwood, C. S. et al. Characterisation of copper interactions with Alzheimer amyloid beta peptides: Identification of an attomolar-affinity copper binding site on amyloid beta 1–42. J. Neurochem. 75, 1219–1233 (2000).
Garzon-Rodriguez, W., Yatsimirsky, A. K. & Glabe, C. G. Binding of Zn(II), Cu(II) and Fe(II) ions to Alzheimer's Aβ peptide studied by fluorescence. Bioorg. Med. Chem. Lett. 9, 2243–2248 (1999).
Sarell, C. J., Syme, C. D., Rigby, S. E. J. & Viles, J. H. Copper(II) binding to amyloid-β fibrils of Alzheimer's disease reveals a picomolay affinity: Stoichiometry and coordination geometry are independent of Aβ oligomeric form. Biochemistry 48, 4388–4402 (2009).
Zou, J., Kajita, K. & Sugimoto, N. Cu2+ inhibits the aggregation of amyloid β-peptide(1–42) in vitro. Angew. Chem. Int. Ed. 40, 2274–2277 (2001).
Yoshiike, Y. et al. New insights on how metals disrupt amyloid β-aggregation and their effects on amyloid-β cytotoxicity. J. Biol. Chem. 276, 32293–32299 (2001).
Khan, A., Ashcroft, A. E., Korchazhkina, O. V. & Exley, C. Metal-mediated formation of fibrillar ABri peptide. J. Inorg. Biochem. 98, 2006–2010 (2004).
Ward, B., Walker, K. & Exley, C. Copper(II) inhibits the formation of amylin amyloid in vitro. J. Inorg. Biochem. 102, 371–375 (2008).
Exley, C. et al. Copper is a potent inhibitor of the propensity for human ProIAPP1–48 to form amyloid fibrils in vitro. J. Diabet. Res. Clin. Med. 1, 3 (2012).
Viles, J. H. Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer's, Parkinson's and prion diseases. Coord. Chem. Rev. 256, 2271–2284 (2012).
Hureau, C. Coordination of redox active metal ions to the amyloid precursor protein and to amyloid-β peptides involved in Alzheimer's disease. Part 1: An overview. Coord. Chem. Rev. 256, 2164–2174 (2012) .
Leal, S. S., Botelho, H. M. & Gomes, C. M. Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coord. Chem. Rev. 256, 2253–2270 (2012).
Miller, Y., Ma, B. & Nussinov, R. Metal binding sites in amyloid oligomers: Complexes and mechanisms. Coord. Chem. Rev. 256, 2245–2252 (2012).
Zecca, L. et al. Iron, copper and their proteins in substantia nigra of human brain during aging. J. Radioanal. Nucl. Chem. 263, 733–737 (2005).
Schrag, M., Mueller, C., Oyoyo, C., Smith, M. A. & Kirsch, W. M. Iron, zinc and copper in the Alzheimer's disease brain: A quantitative meta analysis. Some insight on the influence of citation bias on scientific opinion. Prog. Neurobiol. 94, 296–306 (2011).
Schrag, M. et al. Effect of cerebral amyloid angiopathy on brain iron, copper and zinc in Alzheimer's disease. J. Alzh. Dis. 24, 137–149 (2011).
Exley, C., House, E., Polwart, A. & Esiri, M. M. Brain burdens of aluminium, iron and copper and their relationships with amyloid-β pathology in 60 human brains. J. Alzh. Dis. 31, 725–730 (2012).
Strange, K. Regulation of solute and water balance and cell volume in the central nervous system. J. Am. Soc. Nephrol. 3, 12–27 (1992).
Gaier, E. D., Eipper, B. A. & Mains, R. E. Copper signalling in the mammalian nervous system:Synaptic effects. J. Neurosci. Res. 91, 2–19 (2013).
Sillén, L. G. & Martell, A. E. Stability Constants. Chem. Soc. Special Publ. No. 17 634–641 (The Chemical Society, London, 1964).
Khan, A., Dobson, J. P. & Exley, C. Redox cycling of iron by Aβ42 . Free Rad. Biol. Med. 40, 557–569 (2006).
Ha, C., Ryu, J. & Park, C. B. Metal ions differentially influence the aggregation and deposition of Alzheimer's beta-amyloid on a solid template. Biochemistry 46, 6118–6125 (2007).
Olofsson, A., Lindhagen-Persson, M., Vestling, M., Sauer-Eriksson, A. E. & Ohman, A. Quenched hydrogen/deuterium exchange NMR characterisation of amyloid-beta aggregates formed in the presence of Cu2+ and Zn2+. FEBS J. 276, 4051–4060 (2009).
Innocenti, M. et al. Trace Cu(II) or Zn(II) ions drastically modify the aggregation behaviour of amyloid-beta (1–42): An AFM study. J. Alzh. Dis. 19, 1323–1329 (2010).
Bolognin, S. et al. Aluminium, copper, iron and zinc differentially alter amyloid-A beta (1–42) aggregation and toxicity. Int. J. Biochem. Cell Biol. 43, 877–885 (2011).
Jiang, D., Rauda, I., Han, S., Chen, S. & Zhou, F. Aggregation pathways of the amyloid-β(1–42) peptide depend upon its colloidal stability and ordered β-sheet stacking. Langmuir 28, 12711–12721(2012).
Smith, D. P. et al. Concentration dependent Cu2+ induced aggregation and dityrosine formation of the Alzheimer's disease amyloid-β peptide. Biochemistry 46, 2881–2891 (2007).
Sarell, C. J., Wilkinson, S. R. & Viles, J. H. Substoichiometric levels of Cu2+ ions accelerate the kinetics of fiber formation and promote cell toxicity of amyloid-β from Alzheimer's disease. J. Biol. Chem. 285, 41533–41540 (2010).
D'Amico, M. et al. Thioflavin T promotes Aβ(1–40) amyloid fibrils formation. J. Phys. Chem. Lett. 3, 1596–1601 (2012).
Mantri, Y., Fioroni, M. & Baik, M-H. Computational study of the binding of Cu(II) to Alzheimer's amyloid-β peptide: Do Aβ42 and Aβ40 bind copper ion identical fashion? J. Biol. Inorg. Chem. 13, 1197–1204 (2008).
House, E. et al. Copper abolishes the β-sheet secondary structure of pre-formed amyloid fibrils of Aβ42 . J. Alzh. Dis. 18, 811–817 (2009).
Mold, M., Shrive, A. K. & Exley, C. Serum amyloid P component accelerates the formation and enhances the stability of amyloid fibrils in a physiologically significant under-saturated solution of Aβ42 . J. Alzh. Dis. 29, 875–881 (2012).
Acknowledgements
MM was supported by an EPSRC Doctoral Training Grant and a Keele Acorn Studentship.
Author information
Authors and Affiliations
Contributions
M.M., L.O.-G., B.W. & C.E. performed the experiments. C.E. and M.M. wrote the manuscript and prepared the figures. All authors read and commented upon the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Mold, M., Ouro-Gnao, L., Wieckowski, B. et al. Copper prevents amyloid-β1–42 from forming amyloid fibrils under near-physiological conditions in vitro. Sci Rep 3, 1256 (2013). https://doi.org/10.1038/srep01256
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01256
This article is cited by
-
CERTL reduces C16 ceramide, amyloid-β levels, and inflammation in a model of Alzheimer’s disease
Alzheimer's Research & Therapy (2021)
-
Effects of Cu(II) on the aggregation of amyloid-β
JBIC Journal of Biological Inorganic Chemistry (2019)
-
Copper, dityrosine cross-links and amyloid-β aggregation
JBIC Journal of Biological Inorganic Chemistry (2019)
-
Interaction of Amyloid Aβ(9–16) Peptide Fragment with Metal Ions: CD, FT-IR, and Fluorescence Spectroscopic Studies
International Journal of Peptide Research and Therapeutics (2019)
-
Intracellular tracing of amyloid vaccines through direct fluorescent labelling
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
