
Abstract
In vivo two-photon microscopy has revealed vital information on neural activity for brain function, even in light of its limitation in imaging events at depths greater than several hundred micrometers from the brain surface. We developed a novel semiconductor-laser-based light source with a wavelength of 1030 nm that can generate pulses of 5-picosecond duration with 2-W output power and a 20-MHz repetition rate. We also developed a system to secure the head of the mouse under an upright microscope stage that has a horizontal adjustment mechanism. We examined the penetration depth while imaging the H-Line mouse brain and demonstrated that our newly developed laser successfully images not only cortex pyramidal neurons spreading to all cortex layers at a superior signal-to-background ratio, but also images hippocampal CA1 neurons in a young adult mouse.
Similar content being viewed by others


Introduction
It is essential for brain research to elucidate how a single neuron projects its dendrites and axon fiber into the layers of the brain. Indeed, neuron morphology has provided insight into understanding brain function. Typically, dendrites of cortical pyramidal neurons spread widely across several layers of the cortex and the length of these dendrites can vary from ∼20 micrometers to 1 mm. The width and branching of the dendrites depend on the distance from the soma, suggesting that such spatial differences in morphology relate to the functionality of the neuron, including the electrophysiological characteristics of the dendrite and synaptic efficiency1,2. There is often discordance between results obtained from an acute brain slice preparation and results from the intact brain, likely because severing the connectivity among neurons is unavoidable. Thus, a method is needed to visualize dendrites and all of the projections in the brain with high spatial resolution.
In comparison to confocal or other optical microscopy systems, two-photon microscopy offers a striking advantage in visualizing intact neurons within deeper layers of the living mouse brain3,4,5,6,7. Indeed, the penetration depth for in vivo two-photon microscopy is superior compared to confocal microscopy in the same preparation among various organs, likely because the excitation wavelength for two-photon excitation is in the range of near infrared, where the absorption coefficient is lowest in biological specimens. We have already visualized cell bodies and basal dendrites of layer V pyramidal neurons at ∼700 μm from the brain surface in the live H-line mouse brain8. However, we were not able to detect enhanced yellow fluorescent protein (eYFP) signals at layers deeper than layer V with a sufficient signal-to-background ratio required for visualizing neurons. In addition, in these deeper regions, the spatial resolution tends to degrade. Therefore, to observe neurons with a superior signal-to-background ratio, we sought a novel light source for eYFP excitation, of which the intensity is stronger and the wavelength is longer compared to a commonly used laser—Ti:Sapphire laser.
For conducting in vivo observation over an extended period of time, a mechanically stable apparatus for securing the mouse head is necessary. Open-skull surgery (or cranial window) is usually performed on anesthetized mice for two-photon microscopic observation of their brains in vivo9,10,11. In open-skull surgery, a part of the skull is replaced with a piece of cover slip. This technique is thought to be suitable for observing the deeper regions of the brain because it avoids light scattering in the bone under the objective lens. When the cover slip is not perpendicular to the axial direction of the objective lens, optical aberrations can occur and result in degradation in focusing the excitation light in the specimen12. Thus, this complication necessitates an apparatus to secure the mouse head during surgery and observation that would allow for accurate adjustment of the cover slip at an angle perpendicular to the axial direction of the objective lens.
In this study, we developed a novel near-infrared semiconductor-laser-based picosecond 1030-nm optical pulse source, which is directly led to a laser-scanning microscope, without a negative chirp system. The specification of our laser has 2-W maximum average output, 20-MHz repetition rate and 5-picosecond duration at individual pulse. The peak-power of the pulse at the output of the optical main amplifier is 10 kW, because the gain of the main amplifier was achieved over a thousand times compared with our previously reported laser13,14,15. In addition, the pulse width of 5 picoseconds did not broaden at the specimen and thus did not require a negative chirp system. Furthermore, we also report a method to maintain the mouse head on an adapter stage with a horizontal adjustment mechanism, which will compensate for variations in the head position of the cover slip for each surgical operation. Here, by in vivo deep imaging of an H-line mouse (Thy1-eYFP) brains8, we demonstrate the advantages of our newly developed two-photon microscopy technique, such as the increased penetration depth and successful in vivo imaging of hippocampal CA1 neurons.
Results
Novel near-infrared semiconductor-laser-based light source with an upright two-photon microscope
Our newly developed laser system, composed of a seed laser and amplifier, generated a 1030-nm picosecond laser pulse. The duration of individual pulses of the picosecond laser beam was approximately 5 picoseconds (Figure 1a, inset). The M2 value (laser-beam-quality indication parameter) of the output laser beam was measured to be 1.2; this indicates that the beam profile is close to the ideal Gaussian beam (M2 = 1). The output power of the 1030-nm picosecond laser was manually controlled by adjusting the current in the main amplifier. The laser light was led directly to the scanner port of an upright two-photon microscope (BX61/FV1000, Olympus) (Figure 1a). We placed a chirper in the light path from the Ti:Sapphire laser; however, a chirper was not necessary with the new laser, since the femto-second pulse broadened its width at the focal plane due to group velocity dispersion in the microscopy16. We placed a changing mirror switch (**) in the middle of the optical pathway to compare the performance with the 910-nm femto-second pulse (∼100 femto-second) laser light generated by a Ti:Sapphire laser (Tsunami, Spectra-Physics). Shared by both lasers, a pair of lenses, i.e., Kepler's optics, was placed after the changing mirror in order to adjust the diameter in the laser beam to be suitable for the objective lens (XLPLN 25X WMP, Olympus) in the microscope.

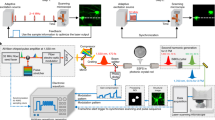
Schematic diagrams for in vivo two-photon microscopy using a novel picosecond pulse laser and a system to secure the head of mouse under standard upright microscope stage.
(a) Optical arrangement for the 1030-nm picosecond (newly developed) and 910-nm femto-second (Ti:Sapphire) lasers to be introduced into the microscope. The optical pathway from each laser is shown in green and red lines, respectively. The duration of an individual pulse of the 1030-nm laser beam was about 5 picoseconds (left). (b) The newly developed apparatus, with a horizontal adjustment mechanism, to secure the head of the mouse under an upright microscope stage. Three adjuster bolts are located on the adapter stage and a 35-mm disposable dish was mounted on the head of the mouse. The mouse head was suspended from the adapter stage by the 35-mm dish and the body was held under the stage by soft cloth. The stage angle could be manually controlled by adjuster bolt to ensure that the cover slip and objective lens were parallel, which would reduce optical aberrations.
Compatible adapter stage under the microscope
We developed an apparatus, with a horizontal adjustment mechanism, to secure a mouse head under an upright microscope stage (Figure 1b). To secure the mouse head onto the adapter plate, a 35-mm plastic culture dish with a hole was cemented onto the skull bone around the cranial window via dental cement. The body of the mouse was underneath the adapter stage (see Experimental procedure). The angles of the plate in relation to the axial direction of the objective lens could be adjusted by turning three screws, resulting in the reduction of aberrations that occur when the objective lens and the cover slip on the skull are not parallel (data not shown).
Deep imaging of an H-Line mouse brain by in vivo two-photon microscopy with 910-nm femtosecond and 1030-nm pico-second lasers
To compare the penetration depth of the newly developed 1030-nm picosecond laser to that of the 910-nm femto-second laser, we examined eYFP expressing pyramidal neurons in 4-week-old H-line mice (Figure 2). We captured three-dimensional stacks as z-series of cross-sectional xy-images of eYFP fluorescence and adjusted the power of the excitation laser lights according to the depth of the focal plane from the brain surface. When we obtained the xy-images at layers deeper than 500 μm from the surface of the brain, the power of the 910-nm femto-second or 1030-nm picosecond laser was 35 or 174 mW after the objective lens, respectively. Those values were the maximum we could apply for each laser. Figure 2a shows that apical dendrites of pyramidal neurons spreading from layer V to layer I in the cerebral cortex were visualized with the 910-nm femto-second laser, while almost no signals were detected in layers deeper than 800 μm, except for several fiber-like structures in the white matter region at ∼900 μm depth. On the other hand, the 1030-nm picosecond laser revealed not only a panoramic view of cortical pyramidal neurons, it also revealed hippocampal pyramidal neurons beneath the cortex in a young adult mouse (Figure 2b). Between the depths of 600 and 900 μm, we identified several somata in cerebral cortex pyramidal neurons, likely residing in layer V and VI. We also observed the basal dendrites of cerebral cortex pyramidal neurons (Figure 3b). At the border of the cortical area, white matter and alveus hippocampi were clearly observed. Surprisingly, the somata of CA1 pyramidal neurons and apical dendrites were visualized in the hippocampal area. These results indicate that the penetration depth of the 1030-nm picosecond laser was ∼1.4 mm.

Two-photon fluorescence imaging of cortical and hippocampal CA1 pyramidal neurons with 910-nm femto-second and 1030-nm picosecond excitation in a young adult H-line mouse brain.
(a) Maximum intensity projections of three-dimensional stacks were obtained with 910-nm and 1030-nm lasers (b). Four normalized xy frames from the z-stack at various depths are shown, including the dura matter, apical dendrite, soma, white matter and hippocampus, respectively (left panel). The arrowhead shows a part of a basal dendritic process at cortex layer V.

Two-photon fluorescence imaging of cortical pyramidal neurons with 910-nm and 1030-nm excitation in an adult H-line mouse brain.
(a) Maximum intensity projections of three-dimensional stacks were obtained with 910-nm and 1030-nm lasers (b). Four normalized xy frames from the z-stack at various depths including the dura matter, apical dendrite, soma and white matter, respectively (left panels). The arrowhead shows a part of a basal dendritic process at cortex layer V.
Next, we compared the three-dimensional stacks from the young-adult mouse (4-weeks-old) to those obtained from a live adult H-line mouse (over 8 weeks-old; Figure 3). With the 910-nm femto-second laser, we acquired fluorescence images of apical dendrites spreading from layer V to layer I. In the cerebral cortex, however, we found somata of cerebral cortex pyramidal neurons lying horizontally only at the top of layer V (∼700 μm); this suggests that the 910-nm femto-second laser failed to visualize cerebral cortex pyramidal neurons occupying (vertically) the thick area just below the top region of layer V (Figure 3a; see also Figures 2a and 2b)17. It is possible that we could not obtain sufficient signals due to the tendency for the optical transparency of the organ to degrade with aging18. The 1030-nm picosecond laser had superior penetration depth in the live adult mouse brain compared to the 910-nm femto-second laser: eYFP signals were detected in all layers of the cortex and white matter region (Figure 3b). In addition, the 1030-nm picosecond laser enabled the visualization of basal dendrites at ∼900 μm (Figure 3b; see also Figure 2b).
Finally, we analyzed the three dimensional-stacks to evaluate the signal-to-background ratio and penetration depth of the 1030-nm picosecond laser. To distinguish a neural process in the xy-plane, the ratio of the fluorescence intensity to the background is critical, especially in the deeper regions of dim fluorescence. To avoid variation in the eYFP intensity as a result of morphological structure or fluctuation in the fluorescence image, we measured two average intensities: (1) in circles containing a section of an identical neural process of the single neuron for the “signal” and (2) in a neighboring circle for the “background” (Figure 4a; see also Experimental procedure). We calculated signal-to-background ratios (R) among seven individual pyramidal neurons and plotted them in Figures 4b and 4c. These figures show that, both in young-adult and adult mice, R decreased rapidly in layers deeper than 700 μm in the case of the 910-nm femto-second laser. In contrast, R was sufficiently higher even at similar depths in the case of the 1030-nm picosecond laser. Notably, for in vivo images of hippocampal CA1 neurons, we calculated higher R's with the 1030-nm picosecond laser in layers deeper than 1 mm in the young-adult mouse (Figure 4b).

Quantitative analysis of signal-to-background ratios for in vivo imaging of neurons in the live mouse brain.
(a) A cross-sectional- (xy-) image obtained at 600-μm depth in a young adult mouse. The left white or left yellow box indicates the “soma” (upper right) or “apical dendrite” (lower right) region, respectively. In the upper and lower right panels, the averages of “signal” and “background” were calculated from fluorescence intensities in a circle (20 μm diameter) around an individual neural process (“signal”: red circle) and the background area (“background”: green circle), respectively. Ratio R at the each process was obtained from the average signal divided by the background (see Experimental procedure). Depth dependence of the average and SEM of signal-to-background ratios (R) obtained in the young adult (b) and the adult mouse (c). Red and black circles indicate averages with the 1030-nm picosecond and 910-nm femto-second lasers, respectively (n = 7 processes, ±: SEM).
Discussion
In this study, we described a novel in vivo two-photon microscopy technique that employs a newly developed 1030-nm high-power picosecond pulse laser enabling improvement in penetration depth up to ∼1.4 mm (Figures 3 and 4). Apical and basal dendrites and somata of cortical pyramidal neurons in live H-line mouse brains were successfully visualized by this novel laser (Figure. 2, 3). We were also able to observe hippocampal CA1 pyramidal neurons in a young adult H-line mouse in vivo (Figure 2).
With a 5-picosecond duration and 20-MHz repetition rate, this 1030-nm pulse laser has sufficiently induced two-photon excitation and revealed the morphology of neurons within deeper layers of the live mouse brain in vivo. We found that the laser wavelength of 1030 nm is suitable for the two-photon excitation cross-section of eYFP as described previously19. In contrast, the Ti:Sapphire laser could not excite eYFP at maximum efficiency, because it was not able to generate sufficient power at a wavelength over 1000 nm. Furthermore, a longer pulse duration of 5-picoseconds is suitable for in vivo two-photon microscopy as shown in this study and our previous study14. It is possible that the inefficiency of the two-photon excitation to a longer pulse duration may be compensated by a repetition rate of 20 MHz, a higher laser power of 2 W, or both. In addition, the 1030-nm picosecond laser light was generated by a semi-conductor diode laser system, while the 910-nm femto-second laser light was generated by a solid-state laser, the Ti:Sapphire laser. The laser intensity of the 1030-nm picosecond laser light was thus extremely stable, leading to a high signal-to-background ratio and was easily handled compared to Ti:Sapphire lasers. These features might enable the visualization of hippocampal CA1 pyramidal neurons at ∼1.4 mm depth. This in vivo microscopy technique will facilitate the examination of spatial features of a single intact neuron in more detail, including the branching of dendrites and distribution of various types of spines in a live brain, which could reveal the underlying mechanisms of how the function of neural circuits emerges from the activity of neural cells. Indeed, two-photon microscopy has a wide range of applications; clinical applications, in particular, could benefit from this novel technique.
It has been reported that light near infrared is relatively harmless to living organs, which may be why two-photon microscopy with a near infrared laser is suitable for in vivo observation. For example, photo-bleaching or photo-damage did not occur during imaging of the hippocampal region, even when the maximum output of the 1030-nm picosecond laser (174 mW) was applied. Immediately after the mouse died, however, damage to brain tissue at the focal point was observed when the maximum power of the laser was applied (data not shown). This observation is consistent with the phenomenon that the live brain continually removes heat generated by the circulation of cerebrospinal fluid and/or blood flow. Such cooling action of circulation might permit in vivo two-photon imaging in live brains.
For live imaging over a long time period, it is critical to suppress vibrations produced by a beating heart or breathing. In our preparation, it took over 90 min to get a z-series of xy-images for a single stack (Figures 2 and 3). Various setups for securing the mouse head under the upright microscope have been proposed for in vivo two-photon microscopy9,11,20. However, here we proposed a stable stage with a horizontal adjustment mechanism. This procedure has an advantage in that there is no need to exchange a normal microscopy stage for a special or custom-built stage for in vivo observation. Our adapter stages were composed of a wide plastic dish containing emersion liquid (water or PBS) and were cemented tightly to the skull bone. With these conditions, we were able to conduct observations over a long time. Furthermore, our stage compensated for optical aberrations due to the angle of the cover slip against the surface of the objective12. Indeed, the angle was easily adjusted during observation by manipulating a bolt on the stage.
As discussed, our novel laser enabled the observation of whole cortical neuron processes, even in the adult mouse brain. However, it failed to visualize the synapses of hippocampal pyramidal neurons in a young adult mouse. Several researchers have reported the visualization of hippocampal pyramidal neuron synapses under in vivo-like conditions via the insertion of a GRIN lens directly into the cortex from the skull hole or the removal of a part of the cortex by local aspiration21,22,23. For elucidating brain function, these invasive methods of in vivo imaging are not ideal because of the potential effects of lesioning in the cortex. The imaging technique using our novel laser, on the other hand, was essentially non-invasive and could provide accurate information about brain function.
Recently, in vivo two-photon imaging of blood vessels using a longer-wavelength laser (1280 nm) was reported and included imaging of vasculature within the layers up to a depth of ∼1.6 mm in the live brain24,25. In this study, a red polar fluorescent dye, i.e., Texas Red, was injected for visualizing blood. Simultaneous multicolor imaging with different fluorescent colors in the visible range would be difficult under that condition. In contrast, the 1030-nm picosecond laser light sufficiently excited eYFP fluorescence for visualization, suggesting that the wavelength is suitable for multicolor fluorescence imaging using eYFP and other shorter fluorescent dyes.
The present microscopy technique failed to visualize any structures in the St. Radiatum region of the hippocampal CA1 area deeper than 1.4 mm (Figure 2b). This limitation in the penetration depth is likely due to the working distance of the objective lens used here (2.0 mm; XLPLN 25X WMP), since the fluorescence seemed to be substantially strong in the area lying vertically above St. Radiatum (1.1–1.4 mm; Figure 4b). The difference between the working distance and the penetration depth corresponded to dead space, consisting of the cover slip and the cerebral spinal fluid, between the surface of the objective lens and the brain surface. In the future, an objective lens with a longer working distance is warranted for the evaluation of the penetration depth limit, leading to improvement in the penetration depth for in vivo two-photon microscopy. In summary, this novel in vivo two-photon microscopy technique can reveal the functional properties of cortical and hippocampal neurons using a functional fluorescent indicator26; this technique could provide insight into episodic memory4,27 or other functions of neural circuits involving the hippocampus.
Methods
Animals
Young-adult (4 weeks-old) and adult (6 to 9 weeks-old) transgenic mice expressing yellow fluorescent protein (YFP) (YFP-H line8) for in vivo imaging were used in this study. All mice were housed with food and water ad libitum on a 12∶12 light-dark cycle (lights on from 8:00 to 20:00), with controlled temperature and humidity. The study was carried out in accordance with the recommendations in the Guidelines for the Care and Use of Laboratory Animals of the Animal Research Committee of Hokkaido University. The protocol was approved by the Committee on the Ethics of Animal Experiments in the Hokkaido University (No. 10-0119)
Cranial window for in vivo two-photon imaging
For high-resolution imaging of synaptic structures in the cortex of living mice, the overlying opaque skull bone should be partially removed to make a cranial window, namely, the “open-skull” glass window3,7,28. In the open-skull preparation a piece of the cranial bone is removed (about 3 mm in diameter), leaving the dura intact and the exposed brain is covered with a thin glass cover slip (for detailed methods, see published protocols20,29,30,31).
Image acquisition
The image stacks consisted of over 200 optical sections with 5-μm Z-steps and were taken from the brain surface, an area covering 500 × 500 μm (512 × 512 pixels, 0.994 μm/pixel). These were obtained using a two-photon laser microscopy customized for in vivo imaging (FV1000 confocal microscope, Olympus; tsunami, Spectra-Physics Inc.) with a 25x water objective lens (NA 1.05). All of the fluorescence signals under 690-nm wavelengths were detected via the Non-Descanned Detector.
Data analysis
To calculate the signal-to-background ratio, all images were analyzed by NIS element Ver.4.00 (NIKON). The average signal and background intensity of each individual image from all depths were measured from two 20 μm–diameter areas (one around pyramidal neuron process and the other a non-signal area, n = 7 each).
The signal-to-background ratio (R) was defined as follows:

Here,  and
and  are the fluorescence signal intensity in a single pixel and the number of the pixels in the region of interest (ROI) in the “signal” or “background” region, respectively (see Figure 4).
are the fluorescence signal intensity in a single pixel and the number of the pixels in the region of interest (ROI) in the “signal” or “background” region, respectively (see Figure 4).
References
Matsuzaki, M., Honkura, N., Ellis-Davies, G. C. & Kasai, H. Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766 (2004).
Nicholson, D. A. et al. Distance-dependent differences in synapse number and AMPA receptor expression in hippocampal CA1 pyramidal neurons. Neuron 50, 431–442 (2006).
Grutzendler, J., Kasthuri, N. & Gan, W. B. Long-term dendritic spine stability in the adult cortex. Nature 420, 812–816 (2002).
Nakashiba, T. et al. Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell 149, 188–201 (2012).
Nemoto, T. Living cell functions and morphology revealed by two-photon microscopy in intact neural and secretory organs. Mol Cells 26, 113–120 (2008).
Pan, F. & Gan, W. B. Two-photon imaging of dendritic spine development in the mouse cortex. Developmental neurobiology 68, 771–778 (2008).
Trachtenberg, J. T. et al. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420, 788–794 (2002).
Feng, G. et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51 (2000).
Jia, H., Rochefort, N. L., Chen, X. & Konnerth, A. In vivo two-photon imaging of sensory-evoked dendritic calcium signals in cortical neurons. Nat Protoc 6, 28–35 (2011).
Kim, S. K., Eto, K. & Nabekura, J. Synaptic structure and function in the mouse somatosensory cortex during chronic pain: in vivo two-photon imaging. Neural Plast 2012, 640259 (2012).
Noguchi, J. et al. In vivo two-photon uncaging of glutamate revealing the structure-function relationships of dendritic spines in the neocortex of adult mice. J Physiol 589, 2447–2457 (2011).
Pawley, J. B. Handbook of biological confocal microscopy. 2nd edn, (Plenum Press, 1995).
Kuramoto, M. et al. Two-photon fluorescence bioimaging with an all-semiconductor laser picosecond pulse source. Opt Lett 32, 2726–2728 (2007).
Yokoyama, H. et al. Nonlinear-microscopy optical-pulse sources based on mode-locked semiconductor lasers. Opt Express 16, 17752–17758 (2008).
Yokoyama, H. et al. Two-photon bioimaging utilizing supercontinuum light generated by a high-peak-power picosecond semiconductor laser source. J Biomed Opt 12, 054019 (2007).
Rullier, C. Femtosecond Laser Pulses. Second edn, (Springer, 2005).
Helmchen, F. & Waters, J. Ca2+ imaging in the mammalian brain in vivo. European journal of pharmacology 447, 119–129 (2002).
Oheim, M., Beaurepaire, E., Chaigneau, E., Mertz, J. & Charpak, S. Two-photon microscopy in brain tissue: parameters influencing the imaging depth. J Neurosci Methods 111, 29–37 (2001).
Blab, G. A., Lommerse, P. H. M., Cognet, L., Harms, G. S. & Schmidt, T. Two-photon excitation action cross-sections of the autofluorescent proteins. Chem Phys Lett 350, 71–77 (2001).
Yang, G., Pan, F., Parkhurst, C. N., Grutzendler, J. & Gan, W. B. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat Protoc 5, 201–208 (2010).
Barretto, R. P. et al. Time-lapse imaging of disease progression in deep brain areas using fluorescence microendoscopy. Nature medicine 17, 223–228 (2011).
Jung, J. C., Mehta, A. D., Aksay, E., Stepnoski, R. & Schnitzer, M. J. In vivo mammalian brain imaging using one- and two-photon fluorescence microendoscopy. Journal of neurophysiology 92, 3121–3133 (2004).
Mizrahi, A., Crowley, J. C., Shtoyerman, E. & Katz, L. C. High-resolution in vivo imaging of hippocampal dendrites and spines. The Journal of neuroscience : the official journal of the Society for Neuroscience 24, 3147–3151 (2004).
Kobat, D. et al. Deep tissue multiphoton microscopy using longer wavelength excitation. Opt Express 17, 13354–13364 (2009).
Kobat, D., Horton, N. G. & Xu, C. In vivo two-photon microscopy to 1.6-mm depth in mouse cortex. J Biomed Opt 16, 106014 (2011).
Zhao, Y. et al. An expanded palette of genetically encoded Ca(2)(+) indicators. Science 333, 1888–1891 (2011).
Liu, X. et al. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature 484, 381–385 (2012).
Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S. & Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 3974–3980 (2009).
Holtmaat, A. et al. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc 4, 1128–1144 (2009).
Kelly, E. A. & Majewska, A. K. Chronic imaging of mouse visual cortex using a thinned-skull preparation. J Vis Exp (2010).
Mostany, R. & Portera-Cailliau, C. A craniotomy surgery procedure for chronic brain imaging. J Vis Exp (2008).
Acknowledgements
We thank A. Koizumi and M. Kikuchi of Hokkaido University for technical assistance. This research was supported by Grant-in-Aid for Scientific Research (KAKENHI) from the Ministry of Education, Science, Sports and Culture of Japan.
Author information
Authors and Affiliations
Contributions
R.K. and T.N. designed and performed the imaging experiments. K.S. and H.T. assisted with the imaging experiments. H.Y., A.S., S.S. and Y.K. developed the picosecond laser light source. R.K., H.Y., S.S., Y.K. and T.N. designed and wrote manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kawakami, R., Sawada, K., Sato, A. et al. Visualizing hippocampal neurons with in vivo two-photon microscopy using a 1030 nm picosecond pulse laser. Sci Rep 3, 1014 (2013). https://doi.org/10.1038/srep01014
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01014
This article is cited by
-
Large-scale cranial window for in vivo mouse brain imaging utilizing fluoropolymer nanosheet and light-curable resin
Communications Biology (2024)
-
Advances in adaptive optics–based two-photon fluorescence microscopy for brain imaging
Lasers in Medical Science (2020)
-
Optical second harmonic generation microscopy: application to the sensitive detection of cell membrane damage
Biophysical Reviews (2019)
-
Two-photon imaging of neuronal activity in motor cortex of marmosets during upper-limb movement tasks
Nature Communications (2018)
-
Aberration correction considering curved sample surface shape for non-contact two-photon excitation microscopy with spatial light modulator
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
