
Abstract
Time-resolved fluorescence measurements of graphene oxide in water show multiexponential decay kinetics ranging from 1 ps to 2 ns. Electron-hole recombination from the bottom of the conduction band and nearby localized states to wide-range valance band is suggested as origin of the fluorescence. Excitation wavelength dependence of the fluorescence was caused by relative intensity changes of few emission species. By introducing the molecular orbital concept, the dominant fluorescence was found to originate from the electronic transitions among/between the non-oxidized carbon regions and the boundary of oxidized carbon atom regions, where all three kinds of functionalized groups C-O, C = O and O = C-OH were participating. In the visible spectral range, the ultrafast fluorescence of graphene oxide was observed for the first time.
Similar content being viewed by others
Introduction
Following the exciting research of graphene1,2,3, chemically derived graphene oxide (GO) and reduced GO (rGO) nowadays have attracted great interest due to the candidate status instead of graphene in some aspects, facile synthesis and promising applications4,5,6,7. GO and rGO supercapacitor composites8, permeation and conducting thin films7,9 have been realized and the tunable optical characteristics have also raised much attention from light-emitting, biosensing and nonlinear optics communities10. For the as-prepared GO with the typical C:O ratio (2–4)6, the widespread fluorescence has been reported from visible to near infrared (NIR) range and the maximum intensity is located between 500 and 800 nm6,10,11,12,13,14,15. For rGO, blueshift11,16,17,18 and redshift12 of the fluorescence from UV to NIR have been observed. Luo et al12 proposed that the bond distortions may contribute to the fluorescence of GO and rGO. Chien and Eda et al11,16 have pointed out the disorder-induced states and the newly formed graphitic domains of sp2 clusters during reduction are responsible for GO photoluminescence (PL) and rGO blue PL, respectively. Galande et al19 have studied the pH-dependent fluorescence of GO and suggested the emission of quasi-molecular fluorophores accounted for this behavior. Besides, Gokus et al20 have observed the similar PL features in oxygen plasma treated graphene compared to that from GO and assigned it to CO-related localized electronic states of oxidation sites. Till now, although much progress has been achieved in GO-related studies, the origin of the fluorescence of GO is still a controversial issue.
In this work, we focus on the fluorescence of as prepared GO in water. Steady state and time-resolved fluorescence (TRF) measurements have been conducted in order to interpret the origin of GO emission. For the first time, TRF measurements with femtosecond resolution were performed. The GO model was built according to experimental data; the related electronic structures and properties were calculated based on density functional theory (DFT). Both experimental data and theoretical analysis suggest that the GO fluorescence is due to electron-hole recombination from conduction band (CB) bottom and nearby localized electronic states to wide-range valance band (VB). In view of atomic structure, the GO emission is predominantly from the electron transitions among/between the non-oxidized carbon region (-C = C-) and the boundary of oxidized carbon atom region (C-O, C = O and O = C-OH).
Results
XPS, XRD and Raman spectra
Figure 1a shows the C1s signal of the prepared graphite oxide powder obtained by x-ray photoelectron spectrometer (XPS). This signal was fitted by four components: C = C&C-C (46.8%, 284.6 eV), C-O (43.1%, 286.7 eV), C = O (5.0%, 288.0 eV) and O = C-OH (5.1%, 288.9 eV). The estimated C/O ratio is ~ 2.4. These features are consistent with the previous studies5,6,21,22. Figure 1b presents the x-ray diffraction (XRD) spectrum, where the main diffraction peak appears at 12.4° with a full width at half maximum (FWHM) of ~1.3°, being similar to other reports23,24. Correspondingly, the interlayer distance in graphite oxide is ~0.71 nm. The typical Raman spectrum of graphite oxide is shown in Figure 1c, where the D and G bands were observed clearly at ~1350 and ~1598 cm−1 with the intensity ratio (or area ratio) ID/IG of ~1 (or ~1.76), comparable with the previous observations23,25,26. The other three weak Raman bands, 2D, D+G and 2G appear at ~2700, ~2925 and ~3156 cm−1, respectively.

(a) XPS, (b) XRD and (c) Raman spectra of graphite oxide.
Steady-state and transient absorption
Figure 2a shows the absorption spectrum of GO suspension in 2 mm cuvette ranging from 200 to 900 nm. One main peak at ~231 nm comes from the π-π* transition of C-C and C = C bonds in sp2 hybrid regions and a shoulder peak at ~303 nm is due to n-π* transition of the C = O bond in sp3 hybrid regions14,27. In addition, there is no obvious band edge absorption feature found up to 900 nm. Figure 2b presents the transient absorption spectra in the visible range from 530 to 770 nm. The positive absorbance change indicates the excited state absorption is dominant in the probing region. The small valley around 740 nm is caused by induced absorption of water since there is a water absorption band in this range28. Transient absorption signal at 560 to 760 nm decays multiexponentially (Figure S1); global fit gives four time constants: 2.1 ± 0.2, 13 ± 2, 90 ± 10 and 2000 ± 200 ps. The first three components correspond to the reported values in the previous pump-probe studies on GO29,30. The observed longest component is assigned to the carrier lifetime at the bottom of CB, which is further confirmed by TRF measurements.

(a) Steady state and (b) transient absorption spectra of GO in water.
In Ref. 29, an ultrafast dynamics was reported by use of degenerate pump-probe (800 nm) spectroscopy. The turnover of kinetic curves within the first picosecond was observed with the increase of pump intensity. According to their proposed mechanism, the negative and positive transient absorption signals are due to the saturable absorption in sp2 regions and the two-photon absorption in sp3 regions, respectively. In particular, they mentioned that the optical transition for 800 nm in the sp3 domains is forbidden due to the large energy gap of sp3 domains. In other words, the electrons in the ground state of sp3 domains could not be effectively excited at low pump intensity. With the increase of pump intensity, the positive and negative transient absorption becomes comparable, thus, the turnover of kinetic curves was observed. However, the turnover of kinetic curves was not observed in our case and Ref 30. The main reason is that we have used non-degenerate pump-probe technique with λexc = 480 nm (Ref. 30 used λexc = 400 nm). Both sp2 and sp3 domains are excited monophotonically with low pump intensity (comparable with case C on Fig. 3 of Ref.29). In the present work, the observed positive transient absorption spectra and kinetics are similar to those in Ref 30, where only positive transient absorption kinetics of GO and rGO were observed. Particularly, the longest lifetime component (τ3+τ4) is larger than 300 ps for both GO and rGO30, which is consistent with our observations.

Fluorescence excitation (a) and emission (b) spectra of GO in water; (c) fluorescence excitation-emission map of GO; two trains of strong signals indicated by dashed white lines are due to scattering of excitation light and its second order; (d) fluorescence emission energy/wavelength maxima versus excitation energy/wavelength.
Fluorescence
Figure 3a shows the fluorescence excitation spectra for the emission wavelengths (λem) from 350 nm to 650 nm. It is clear that the excitation spectrum broadens with λem and there is no obvious sharp absorption peak. The excitation light between 280 nm and λem all can be absorbed, which indicates the continuous absorption band exists in our sample. Moreover, our observation at λem = 600 nm is well consistent with that measured at λem = 605 nm by others11. The clear structured fluorescence excitation spectra of GO at λem = 511 and 540 nm were observed at various pH values in Ref.19, which was not seen here. The diversity probably results from the different dielectric environment of GO. However, some of our broad excitation spectra show resemblance to those observed at λem = 665 nm19 and may contain similar spectral components. Figure 3b shows the fluorescence emission spectra with excitation wavelength (λexc = 280 – 650 nm). The broad emission band appears between 400 and 800 nm analogous to previous reports on fluorescence spectra of GO6,10,11,12,13,14,17 and oxygen-plasma treated graphene20. The maximum of the emission spectrum red-shifts versus λexc (See Figure 3c), most pronounced in the excitation region II as shown in Figure 3d. In Region I (350–525 nm), the emission maximum is constant. On the whole, there is obvious dependence of both, excitation spectra versus λem and emission spectra versus λexc. Such a behavior is indicative for multiple chromophore/fluorophore system31 involved with aromatic and oxidation groups.
Figures 4a–c present the TRF decay curves of GO at λexc = 400 nm by time-correlated single photon counting (TCSPC) system. Similar measurements were also executed at λexc = 480 nm. The kinetic curves at λem = 500–780 nm show a multiexponential decay feature. Four time constants are necessary to fit these curves and the corresponding fractional amplitudes are shown in Figure 4d, respectively. For λexc = 400 or 480 nm, the lifetimes are τ1 = 30 ± 15 ps (or 30±15 ps); τ2 = 140 ± 10 ps (or 145±10 ps); τ3 = 510 ± 20 ps (or 540 ± 20 ps); τ4 = 1680 ± 50 ps (or 1780 ± 60 ps). There is no obvious dependence of lifetime on λexc found for these two excitation cases. Recently, three emission lifetimes for GO: 200, 500, 1400 ps have been extracted11, which well agree with obtained τ2, τ3 and τ4 in the present study. In addition, similar multiexponential decay (40, 250 and 1000 ps) was also found in the oxygen-plasma treated graphene20. Note that, the longest component from fluorescence decay is comparable to the longest carrier lifetime (2000 ps) obtained from transient absorption data, which supports that emission corresponding to τ4 originates from the bottom of CB. As shown in Figure 4d, with the increase of λem from 500 to 700 nm, the amplitude of τ1 decreases while the other three increase gradually. At λem > 700 nm, the amplitudes of all components maintain. The small fluctuation around 740 nm is related to the absorption of water, as indicated above.

(a–c) TCSPC measurements of GO in water at λexc = 400 nm; (d) fractional amplitudes of four time components obtained by TCSPC measurements at λexc = 400 and 480 nm.
Furthermore, by use of the emission spectra at λexc of 400 and 475 nm (Figure 3b) and the fractional amplitudes obtained from TRF measurements (Figure 4d), the contribution of four components was extracted, as shown in Figure 5. As expected, the peaks from τ2, τ3 and τ4 are dominant for λem> 600 nm and the peak from τ1 plays important role in the shorter wavelength region (500–600 nm). For both cases, the peak positions of τ2, τ3 and τ4 are comparable. It is also noted that the emission peaks blue shift with the decrease in lifetime. In other words, these observations indicate that the emission lifetime decreases from the lower to higher excited states. Observed rapid red-shift of λem maximum versus λexc (region II in Figure 3d) can be explained in terms of stronger contribution (fractional intensities) of longer time components of emission to the steady-state fluorescence (Figure 3b).

Fluorescence emission spectra at (a) λexc = 400 nm and (c) λexc = 480 nm and the corresponding spectra (b) and (d) of four time components as denoted by square (τ1), circle(τ2), upward(τ3) and downward(τ4) triangles, respectively.
Figures 6a–f show the femtosecond fluorescence upconversion data for GO suspensions at λexc = 400 nm and λem = 540–660 nm. In the first 50 ps, there are mainly two decay components: τ0 = 0.9 ± 0.2 ps; τ1 = 26 ± 3 ps. The relative fractional amplitudes vary with λem (Figure 6g) and the contribution of τ1 increases with λem. Decay lifetime τ1 corresponds well to the shortest component measured with TCSPC. Our up-conversion measurements indicate that another ultrafast emission state exists in the GO sample, which is responsible for τ0 decay. The origin of the ultrafast component is attributed to the higher excited state emission (see below).

(a–f) Fluorescence upconversion kinetics at λexc = 400 nm and λem = 540–660 nm; (g) relative fractional amplitudes of two lifetime components (τ0 = 0.9 ± 0.2 ps; τ1 = 26 ± 3 ps) versus wavelength.
DFT calculations
Figure 7a shows top (upper panel) and side (lower panel) views of the theoretical atomic structural model of GO cell, which is built based on the experimental data, mainly XPS and absorption spectra. In the following calculations, the periodic structure made of large amount of GO cells was introduced, which is closer to the real structure compared with the single cell. By adjusting the relative positions of these bonds, the absorption spectra (Figure S2) and absorption wavelengths (Figure S3) were calculated, which are in agreement with the experimental data (Figure 2a). It indicates the positions of C-C, C-O, C = O and O = C-OH in sample are well reproduced in the model.

(a) Top view and side view of GO model; (b) electronic band structure and density of states (DOS) of GO; (c) schematic of fluorescence excitation and emission of GO; (d) quasi-molecular orbital energy levels of GO; (e) fluorescence related orbitals of GO.
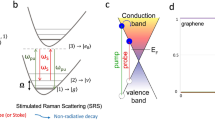
Previously, the theoretical calculations32,33 on oxidized graphene suggested a band gap ranging from 0 to few eV, which depends on the ratio of carbon bonds to oxidation functional groups and their different arrangements. By estimating the typical sp2 cluster size (2.5~6 nm)16,26, the predicted band gap in GO is from 0.58 to 0.24 eV6. Experimentally, a 0.25 eV band gap of oxidized graphene was determined by use of scanning probe microscopy34. Bansal et al have directly observed the density of states near Fermi level of GO by capacitive photocurrent spectroscopy35. In our case, the electronic band structure (left panel) and density of states (DOS: right panel) of GO were calculated as shown in Figure 7b. The band gap of this GO structure is ~0.6 eV, which implies that the band edge-related absorption peak may appear in the infrared range. Meanwhile, the calculation shows a band edge transition at 1987 nm (Figure S3). Indeed, an absorption band of GO film on quartz was observed at ~1914 nm in the near IR region as shown in Figure S4, which further supports the validity of the used model. Moreover, it is noted that there are some relative flat bands nearby the bottom of CB, where the localized electronic states exist. For the dominant excited state absorption (Figure 2b), the corresponding transitions are from the CB bottom and nearby localized states to the higher excited states. For the observed fluorescence of λem = 400–800 nm, the origin is attributed to the electronic transitions from the bottom of CB and the nearby localized states to the VB as shown in Figure 7c. The multiple exponential photoluminescence decay was caused by different emission states of band edge (τ4) and localized electronic states (τ0- τ3) in CB. Furthermore, as indicated by red dots in Figure 7b, the lowest three bands of CB contain five localized states. Therefore, observed five fluorescence decay times at λem = 540–660 nm will originate from these states. Moreover, the lowest two bands in CB mainly contribute to the steady-state emission at λem> 600 nm (Figure 5).
Physically, these states originate from the Anderson localization36,37,38 of electronic wavefunctions in the heterogeneous and disordered sp2/sp3 complex structure. As shown in Figure 7b, the energy bands near theses localized states are flat, which indicates the electron effective masses at these regions are close to infinity. Thus, the excited electrons in these flat bands are confined in the certain momentum space. These electrons will relax radiatively (interband transition) rather than non-radiatively (intraband relaxation). Previously, the localization of electronic wavefunctions in disordered graphene39, chemically modified graphene40 and graphene nanoribbons41 has been studied theoretically. The existence of localized states in disordered graphene nanoribbons was also proved experimentally42. Structurally, these localized states are probably from few kinds of chromophone/fluorophore structures consist of aromatic and oxidation groups in GO and not the states caused by random disorder in GO. It is supported by measurements of other GO samples with different proportions of functional groups (Figure S7–S15).
Molecular-like features (the pH-dependent visible fluorescence) of GO have recently been revealed by the steady state fluorescence measurements19, where the emission is assigned to quasi-molecular fluorophores consisting of O = C-OH groups and neighboring carbon atoms. In order to check this viewpoint, the GO has been treated as a large-size two dimensional molecule. The orbital energy levels of this structure were calculated (Figure 7d) and the corresponding electronic orbitals are presented in Figure 7e. The energy differences between HOMO and LUMO are about 0.6 eV and the non-oxidized C-C& C = C regions take main responsibility for their formation. Obviously, the observed fluorescence of GO between 400 and 800 nm is not from LUMO to HOMO electronic transition. Recently, Itoh has reviewed the fluorescence from higher excited states of organic molecules43, such as azulenes, aromatic acenes and aromatic carbonyl compounds. In GO sample, large amount of analogues of aromatic acenes, o-hydroxybenzaldehyde and other fluorescent molecules exist. Therefore, the fluorescence from higher excited states should be taken into account. For the emission near 640 nm, LUMO, L+1, L+2, HOMO and H-1 to H-5 could be responsible, where the corresponding atom groups include -C = C- with -C-O, -C = O and O = C-OH. As seen in Figure 7e, L+1, L+2 and H-1 to H-5 mainly come from the regions containing both -C = C- and oxidized groups. Particularly, for the dominant yellow (500-800 nm) fluorescence of GO, the allowed electronic transitions are from LUMO to H1-5 and from L+1, L+2 to HOMO. Moreover, the oxidized regions connected to non-oxidized -C = C- more strongly contribute to the electronic orbitals than that from the inner part of oxidized regions. In other words, mainly the boundary of the oxidized regions that consists of oxidized carbon atoms and functionalized groups participates into the fluorescence emission rather than inner atoms from oxidized regions. Thus, the observed fluorescence mainly resulted from the electronic transitions among/between the non-oxidized -C = C- and the boundary of oxidized regions. Even though there is no direct experimental evidence for the precise role of three functional groups, we have observed that the fluorescence features of GO vary with different proportions of oxidized groups (Figures S10–S15). Our data indicates that the functionalized groups together with oxidized carbon atoms and non-oxidized -C = C- form the dominant emission species in GO.
Discussion
Previously, Chien and Eda et al suggested that the fluorescence of as prepared GO may originate from the optical transitions from structural disorder-induced localized states in the π-π* gap of sp2 sites11,16. Luo et al also proposed that the fluorescence may be caused by bond disorder induced energy gaps rather than quantum confinement effect12. The influence of functionalized groups on the fluorescence has not caused enough attention in these two groups11,12,16. Our studies show that the functionalized groups and some oxidized sp3 sites have important contribution to the fluorescence. Broad yellow fluorescence was also observed in O2 plasma-treated graphene by Gukus et al20. By analyzing the fluorescence band broadening and time-resolved data, they suggested CO-related localized electronic state at oxidation sites being the fluorophores. It was noted that not sp2 sites emit fluorescence20. In our case, the sp2 sites are critical for the emission and the fluorescence band was found to consist of few components (emission species), as shown in Figure 5. Recently, Galande et al proposed that the GO fluorescence is from quasi-molecular fluorophores formed by COOH groups and nearby sp2 carbon atoms according to steady state studies of pH-dependent fluorescence19. We found all three kinds of functionalized groups C-O, C = O and O = C-OH are related to the emission. In addition, the tunable fluorescence of rGO11,17,18 and graphene quantum dots44,45,46 has started to attract interest and the emission shows some similarities with that of GO, such as excitation wavelength and pH dependent emission. The current results of GO fluorescence could be linked to understand those emissions from rGO and graphene quantum dots. In contrast to the previous studies11,12,16,19,20, the present mechanism considers roles of three functionalized groups, sp2 and oxidized sp3 sites; the λexc dependent fluorescence and its multiexponential decays were interpreted in terms of both viewpoints of semiconductor electronic band structure and molecular orbital, which agree well with the detailed steady-state and TRF data.
In conclusion, fluorescence of GO in water was studied by use of steady state and time-resolved spectroscopic techniques. We found the excitation wavelength dependent fluorescence and the multiexponential fluorescence decay kinetics at λem = 500–800 nm. An ultrafast fluorescence (~1 ps) of GO in water was observed at λem = 540–660 nm for the first time. The origin of fluorescence is attributed to the electron-hole recombination from the bottom of CB and neighbor localized states to wide-range VB. The carrier lifetime at the bottom is about 2 ns, which is confirmed by both transient absorption and TRF measurements. Moreover, based on molecular orbital concept, the emission species of predominant visible fluorescence are demonstrated being non-oxidized carbon atom regions and the boundary of oxidized carbon atoms. All three kinds of functionalized groups C-O, C = O and O = C-OH are involved in the fluorescence of GO.
Methods
Preparation of GO
GO was prepared according to a modified Hummer's strategy47,48,49. First, with stirring, graphite (1 g) was put into 50 ml of high concentration H2SO4 (98%) in an ice bath. And then, KMnO4 (6.0 g) was gently introduced to avoid a rapid increase in the temperature of the suspension. At 30°C, the obtained mixture was stirred for 1 hour as following. After that, 80 ml deionized water was gradually added under strong stirring. Meanwhile, the temperature was increased to 90°C and the stirring maintained at this temperature for 30 min. Deionized (200 ml) water and H2O2 (6 ml, concentration 30%) were further injected and the color of the suspension changed from dark brown to yellow. Subsequently, centrifugation, washing and freeze drying processes were carried out to get the graphite oxide. The aqueous suspension of GO (concentration 0.5 mg/ml) was prepared by ultrasonic dispersion of graphite oxide in water. The GO film on quartz was obtained by naturally drying GO solution. Optical images of GO samples can be found in Figure S5. Besides, another four supplementary GO samples (S1–S4) were also prepared by the modified Hummer's method with different graphite/H2SO4 mass ratios, oxidation times and temperatures.
Sample characterization and optical measurements
X-ray photoelectron spectroscopy (Thermo Fisher Scientific, UK) was used to determine the element contents. X-ray powder diffraction measurements were carried out by use of a Bruker D8 Advance diffractometer with Cu Kα radiation. A Raman system (CRM 200, WITec) with a 532 nm excitation source of diode-pumped laser was employed to measure Raman spectra. Steady state absorption spectra between 200 and 900 nm were detected by a UV-Vis spectrophotometer (Cary 100 Bio, Varian) with 1.0 nm spectral resolution. The NIR absorption spectrum from 900 to 2450 nm was taken by a UV/Vis/NIR Spectrometer (PerkimElmer Lambda 950). A spectrofluorometer (Fluorolog-3, HORIBA Jobin Yvon) was used to record the steady state fluorescence excitation and emission spectra.
Transient absorption spectra were taken by ultrafast pump-probe system, which has been described in the previous publications50,51. The pump wavelength of 480 nm was chosen and the white light continuum was used as the probe beam. Time-resolved fluorescence (TRF) spectra were measured at room temperature by use of time-correlated single photon counting (TCSPC) technique with resolution of 10 ps (PicoQuant PicoHarp 300). For TRF, the second harmonic laser beams of Titanium sapphire laser (Chameleon, Coherent Inc.) at 400 and 480 nm (100 fs, 80 MHz) were utilized as the excitation source52. For TCSPC measurements, the instrument response function is about 30 ps. After deconvolution fitting, the temporal resolution is ~10 ps. A femtosecond fluorescence up-conversion Spectrometer (FOG100, CDP) was used to measure the ultrafast fluorescence decay signals with 400 nm excitation femtosecond laser source (100 fs, 80 MHz). Absorption/TCPSC and upconversion/pump-probe measurements were performed in 1 cm and 2 mm quartz cuvettes, respectively. For the fluorescence upconversion case, a flow cuvette was used.
DFT calculations
The model of GO was built according to the experimental XPS data, which gave C:O = 2.4:1 and where 5*6 super-cell (including 60 carbon atoms) was used. Density functional theory (DFT/GGA-PBE) was implemented to calculate the molecular orbital, band structure, density of states in Material studio 5.5 Dmol3 module with the DNP 4.4 basis including TS correction53. Core electron was treated by All Electron. All the parameters are fine. Beside, the absorption spectrum was calculated in Castep module using the same method, where the pseudopotentials are norm-conserving and the energy cut off is 750 eV.
References
Novoselov, K. S. et al. Two-dimensional gas of massless Dirac fermions in graphene. Nature 438, 197–200 (2005).
Geim, A. K. Graphene: Status and prospects. Science 324(5934), 1530–1534 (2009).
Novoselov, K. S. Graphene: Materials in the flatland. Rev. Mod. Phys. 83, 837–849 (2011)
Dikin, D. A. et al. Preparation and characterization of graphene oxide paper. Nature 448, 457–460 (2007).
Dreyer, D. R., Park, S., Bielawski, C. W. & Ruoff, R. S. The chemistry of graphene oxide. Chem. Soc. Rev. 39, 228–240 (2010).
Eda, G. & Chhowalla, M. Chemically derived graphene oxide: towards large-area thin-film electronics and optoelectronics. Adv. Mater. 22, 2392–2415 (2010).
Nair, R. R., Wu, H. A., Jayaram, P. N., Grigorieva, I. V. & Geim, A. K. Unimpeded permeation of water through helium-leak–tight graphene-based membranes. Science 335, 442–444 (2012).
Stoller, M. D., Park, S., Zhu, Y., An, J. & Ruoff, R. S. Graphene-based ultracapacitors. Nano Lett. 8(10), 3498–3502 (2008).
Eda, G., Fanchini, G. & Chhowalla, M. Large-area ultrathin films of reduced graphene oxide as a transparent and flexible electronic material. Nat. Nanotechnol. 3, 270–274 (2008).
Loh, K. P., Bao, Q., Eda, G. & Chhowalla, M. Graphene oxide as a chemically tunable platform for optical applications. Nat. Chem. 2, 1015–1024 (2010).
Chien, C.-T. et al. Tunable photoluminescence from graphene oxide. Angew. Chem. Int. Ed. 51, 1–6 (2012).
Luo, Z., Vora, P. M., Mele, E. J., Johnson, A. T. C. & Kikkawa, J. M. Photoluminescence and band gap modulation in graphene oxide. Appl. Phys. Lett. 94, 111909 (2009).
Sun, X. et al. Nano-graphene oxide for cellular imaging and drug delivery. Nano Res 1, 203–212 (2008).
Cuong, T. V. et al. Photoluminescence and Raman studies of graphene thin films prepared by reduction of graphene oxide. Mater. Lett. 64, 399–401 (2010).
Shukla, S. & Saxena, S. Spectroscopic investigation of confinement effects on optical properties of graphene oxide. Appl. Phys. Lett. 98, 073104 (2011).
Eda, G. et al. Blue photoluminescence from chemically derived graphene oxide. Adv. Mater. 22, 505–509 (2010).
Subrahmanyam, K. S., Kumar, P., Nag, A. & Rao, C. N. R. Blue light emitting graphene-based materials and their use in generating white light. Solid State Commun. 150, 1774–1777 (2010).
Xin, G. et al. Tunable photoluminescence of graphene oxide from near-ultraviolet to blue. Mater. Lett. 74, 71–73 (2012).
Galande, C. et al. Quasi-molecular fluorescence from graphene oxide. Sci. Rep. 1, 85 (2011).
Gokus, T. et al. Making graphene luminescent by oxygen plasma treatment. ACS Nano 3(12), 3963–3968 (2009).
Paredes, J. I., Villar-Rodil, S., Martinez-Alonso, A. & Tascon, J. M. D. Graphene oxide dispersions in organic solvents. Langmuir 24, 10560–10564 (2008).
Kolmakov, A. et al. Graphene oxide windows for in situ environmental cell photoelectron spectroscopy. Nat. Nanotechnol. 6, 651–657 (2011).
Marcano, D. C. et al. Improved synthesis of graphene oxide. ACS Nano 4(8), 4806–4814 (2010).
Jeong, H. Y. et al. Graphene Oxide Thin films for flexible nonvolatile memory applications. Nano Lett. 10, 4381–4386 (2010).
Kudin, K. N. et al. Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett. 8(1), 36–41 (2008).
Mattevi, C. et al. Evolution of electrical, chemical and structural properties of transparent and conducting chemically derived graphene thin films. Adv. Funct. Mater. 19, 2577–2583 (2009).
Luo, Z., Lu, Y., Somers, L. A. & Johnson, A. T. C. High yield preparation of macroscopic graphene oxide membranes. J. Am. Chem. Soc. 131, 898–899 (2009).
Langford, V. S., McKinley, A. J. & Quickenden, T. I. Temperature dependence of the visible-near-infrared absorption spectrum of liquid water. J. Phys. Chem. A 105, 8916–8921 (2001).
Liu, Z.-B. et al. Ultrafast dynamics and nonlinear optical responses from sp2- and sp3-hybridized domains in graphene oxide. J. Phys. Chem. Lett. 2, 1972–1977 (2011).
Kaniyankandy, S., Achary, S. N., Rawalekar, S. & Ghosh, H. N. Ultrafast relaxation dynamics in graphene oxide: Evidence of electron trapping. J. Phys. Chem. C 115, 19110–19116 (2011).
Subach, F. V. & Verkhusha, V. V. Chromophore transformations in red fluorescent proteins. Chem. Rev. 10.1021/cr2001965, in press (2012).
Yan, J.-A., Xian, L. & Chou, M. Y. Structural and electronic properties of oxidized graphene. Phys. Rev. Lett. 103, 086802 (2009).
Yan, J. A. & Chou, M. Y. Oxidation functional groups on graphene: Structural and electronic properties. Phys. Rev. B 82, 125403 (2010).
Pandey, D., Reifenberger, R. & Piner, R. Scanning probe microscopy study of exfoliated oxidized graphene sheets. Surf. Sci. 602, 1607–1613 (2008).
Bansal, T. et al. New insights into the density of states of graphene oxide using capacitive photocurrent spectroscopy. Carbon 50, 808–814 (2012).
Anderson, P. W. Absence of diffusion in certain random lattices. Phys. Rev. 109, 1492–1505 (1958).
Evers, F. & Mirlin, A. D. Anderson transitions. Rev. Mod. Phys. 80, 1355–1417 (2008).
Das Sarma, S., Adam, S., Hwang, E. H. & Rossi, E. Electronic transport in two-dimensional graphene. Rev. Mod. Phys. 83, 407–470 (2011).
Pereira, V. M., Guinea, F., Lopes dos Santos, J. M. B., Peres, N. M. R. & Neto, A. H. C. Disorder induced localized states in graphene. Phys. Rev. Lett. 96, 036801 (2006).
Leconte, N. et al. Quantum transport in chemically modified two-dimensional graphene: From minimal conductivity to Anderson localization. Phys. Rev. B 84, 235420 (2011).
Martin, I. & Blanter, Y. M. Transport in disordered graphene nanoribbons. Phys. Rev. B 79, 235132 (2009).
Schubert, G. & Fehske, H. Metal-to-insulator transition and electron-hole puddle formation in disordered graphene nanoribbons. Phys. Rev. Lett. 108, 066402 (2012).
Itoh, T. Fluorescence and phosphorescence from higher excited states of organic molecules. Chem. Rev. 10.1021/cr200166m, in press, (2012).
Pan, D., Zhang, J., Li, Z. & Wu, M. Hydrothermal route for cutting graphene sheets into blue-luminescent graphene quantum dots. Adv. Mater. 22, 734–738 (2010).
Peng, J. et al. Graphene quantum dots derived from carbon fibers. Nano Lett. 12, 844−849 (2012).
Zhu, S. et al. Graphene quantum dots with controllable surface oxidation, tunable fluorescence and up-conversion emission. RSC Advances 2, 2717–2720 (2012).
Hummers, Jr, W. S. & Offeman, R. E. Preparation of graphitic oxide. J. Am. Chem. Soc. 80(6), 1339–1339 (1958).
Cote, L. J., Kim, F. & Huang, J. Langmuir-blodgett assembly of graphite oxide single layers. J. Am. Chem. Soc. 131, 1043–1049 (2009).
Zhou, X. & Liu, Z. A scalable, solution-phase processing route to graphene oxide and graphene ultralarge sheets. Chem. Commun. 46, 2611–2613 (2010).
Shang, J., Yu, T., Lin, J. & Gurzadyan, G. G. Ultrafast electron-optical phonon scattering and quasiparticle lifetime in CVD-grown graphene. ACS Nano 5(4), 3278–3283 (2011).
Shang, J., Yu, T. & Gurzadyan, G. G. Femtosecond energy relaxation in suspended graphene: Phonon-assisted spreading of quasiparticle distribution. Appl. Phys. B 107, 131–136 (2012).
Ma, L. et al. Singlet fission in rubrene single crystal: direct observation by femtosecond pump–probe spectroscopy. Phys. Chem. Chem. Phys. 14, 8307–8312 (2012).
Tkatchenko, A. & Scheffler, M. Accurate molecular Van Der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 102, 073005 (2009).
Acknowledgements
We are grateful to Professor Maria-Elisabeth Michel-Beyerle for continuous support. We thank Prof. Qihua Xiong and Mr. Dehui Li for near infrared absorption measurements. Shang thanks Dr. Zhiqiang Luo, Dr. Rui Chen, Mr. Jiaxu Yan, Mr. Mingjie Li, Mr. Jun Yin, Mr. Jilei Liu, Ms. Linfei Lai and Dr. Xiangyang Wu for their discussions. Yu thanks the support of the Singapore National Research Foundation under NRF Award No. NRF-RF2010-07 and MOE Tier 2 MOE2009-T2-1-037.
Author information
Authors and Affiliations
Contributions
JS, LM, TY and GGG conceived the experiments; JS, LM and GGG performed the experiments; JS and WA were involved in the sample preparation; JL performed the DFT calculations; JS, TY and GGG prepared the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Shang, J., Ma, L., Li, J. et al. The Origin of Fluorescence from Graphene Oxide. Sci Rep 2, 792 (2012). https://doi.org/10.1038/srep00792
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00792
This article is cited by
-
Evaluation of Absorption and Photoluminescence in Graphene Oxide Obtained Through a Simple Route
Journal of The Institution of Engineers (India): Series D (2023)
-
SERS monitoring of photoinduced-enhanced oxidative stress amplifier on Au@carbon dots for tumor catalytic therapy
Light: Science & Applications (2022)
-
DNA-aptamer-nanographene oxide as a targeted bio-theragnostic system in antimicrobial photodynamic therapy against Porphyromonas gingivalis
Scientific Reports (2022)
-
A Comprehensive Review on Ion Beam-Reduced Graphene Oxide: Tailoring the Reduction with Optical, Electrical and Electronic Structural Properties
Journal of Electronic Materials (2022)
-
Development of a highly sensitive fluorescence method for tartrazine determination in food matrices based on carbon dots
Analytical and Bioanalytical Chemistry (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
