
Abstract
The coagulation/fibrinolysis system is essential for wound healing after vascular injury. According to the standard paradigm, the synthesis of most coagulation factors is restricted to liver, platelets and endothelium. We challenged this interpretation by measuring coagulation factors in nine human primary cell types. FX mRNA was expressed by fibroblasts, visceral preadipocytes/adipocytes and hepatocytes, but not in macrophages or other cells. All cells expressed FVIII except endothelial cells. Fibroblasts, endothelial cells and macrophages produced thrombomodulin but not FV. Interestingly, vascular-related cells (platelets/monocytes) that expressed FV did not express FX and vice versa. Monocytes expressed FV, FVIII and FXIIIA, which are positive regulators of clot formation, but these cells also contained thrombomodulin, a negative regulator of coagulation. Our data show that the expression of coagulation factors is much more complex than previously thought and we speculate that this intricate regulation of coagulation factor expression is necessary for correct fine-tuning of fibrinogenesis versus fibrinolysis.
Similar content being viewed by others

Introduction
Atherothrombosis is a precursor of atherosclerotic plaque formation and is thus involved in a major cause of death worldwide1. The capacity of the coagulation system to repair blood vessel damage provoked by loss of vascular integrity (e.g., trauma or oxidized LDL2, as shown in Figure 1) is critically dependent on the presence of coagulation factors. This dependence is emphasized by the serious health problems observed in patients suffering from the congenital absence of such factors and modulators (e.g., thrombomodulin (TM) or protein C), which normally ensure rapid and appropriate fibrin clot formation or lysis. Upon vascular injury, tissue factor (TF) expressed from the damaged vasculature and TF-bearing cells, such as monocytes, activate the initiation phase3 of the coagulation pathway by forming a complex with circulating factor VIIa (FVIIa)4, resulting in the activation of factor X (FX) (See Figure 1 for an illustration of the vascular endothelial cell layers, including coagulation factors and cells and a summary of the results of this paper). In addition, FVIII is an important cofactor that plays an unambiguous role in the activation of FX in the tenase complex (FIXa, FVIII and FX)5,6,7. In addition, the prothrombinase complex, which mediates coagulation through fibrinogenesis, requires FV as a cofactor. Knowledge of the mechanisms governing coagulation factor synthesis would be valuable for understanding atherosclerotic disease and other disorders involving aberrant coagulation.

General structure of blood vessels and the factors involved in coagulation.
The layers above the lumen show an intact blood vessel, while those below the lumen show a blood vessel after injury. The blood vessel is divided into four major sections: 1) tunica adventitia, 2) tunica media, 3) tunica intima and 4) lumen. The tunica adventitia is composed of fibroblasts, connective tissue and collagen and is separated from the tunica media by the external elastic lamina. The tunica media contains fibroblasts, smooth muscle cells (SMCs), connective tissue and collagen and is isolated from the tunica intima by the internal elastic lamina. The tunica intima, which is bordered on one side by the tunica media and on the other side by the lumen, contains endothelial cells and in the case of new intima, the internal elastic lamina is damaged and therefore their contents leaks into the tunica media. In a normal vascular system, blood cells circulate in a regulated way (including monocytes, platelets and others). After a vascular injury, the wound healing and repair process will be immediately activated. However, OX-LDL particles are one of the major causes of chronic vascular injury. After a vascular injury, TF is released by the wound and forms a complex with the activated FVII (FVIIa) present in circulation, as shown by the black arrow at the site of injury (indicated in the middle of the lumen). At the same time, monocytes and platelets arrive quickly to the site of the injury to stop bleeding and repair the injury via the direct activation of coagulation cascade components (FV/FVIII). These two cofactors are involved in the optimal activation of FXa. Both platelets and monocytes ensure the stability of the fibrin network via cross-linking with FXIIIA, which is delivered by these two cell types. Thrombomodulin (TM) is also expressed by monocytes, macrophages, endothelial cells and fibroblasts. When monocytes enter the tissue at the site of injury, they are converted to macrophages (Mφ). Macrophages do not express FV or FX, while fibroblasts express FX but not FV.
The liver and platelets are two major sites of FV synthesis and the liver is a major site of FX production1,4,7,8,9. However, FX and FV have been detected in other cells, suggesting that apart from the liver, other cell types contribute to its expression10,11. This possibility has not been explored systematically. Moreover, although numerous studies show that FVIII forms a complex with von Willebrand factor (vWF) released from endothelial cells, no studies have been conducted to determine whether other circulating cells, such as platelets and monocytes, express FVIII. The absence of such studies seriously limits our ability to obtain a comprehensive understanding of how coagulation factor production is regulated.
During the inflammatory reaction following bacterial infection or tissue damage, the activation of coagulation and the formation of fibrin, which is the final event in the coagulation cascade, represent a defense mechanism that walls off infection and permits repair of the vascular wound8,9. Although the nature and the origin of inflammation and of fibrin formation are completely different, these processes are closely linked: inflammation induces the coagulation cascade, while coagulation modifies the inflammation state or restrains inflammation after completion of wound healing. Coagulation is thus required after inflammation to eliminate the infection, as was postulated by Mackman4. Thus, because defects in coagulation lead to complications in many inflammatory conditions, knowledge of the regulatory mechanisms of coagulation factor production is of major importance for our understanding of vascular disease, cancer and other pathologies.
In general, the prevailing view is that coagulation factor production is restricted to three compartments: the liver, where most coagulation factor production takes place; the endothelium, which is characterized, for example, by high levels of vWF; and the platelets, which, despite being devoid of a nucleus, seem to be capable of substantial coagulation factor production12. However, there is reason to doubt this view of coagulation factor synthesis. For instance, in the lung, alveolar macrophages express FVIIa13, while epithelial cells express TF14, FVIIa15 and Xa10 and fibroblasts express tissue factor pathway inhibitor16. Similarly, in atherosclerosis, coagulation factors are synthesized in the atheroma. No systematic investigation of the capacity of other cell types to produce coagulation factors has been reported. Hence, we decided to apply ultrapure human primary cell cultures to assess coagulation factor production. We opted for vascular related cells like endothelial cells (ECs), fibroblasts, macrophages, monocytes, bone marrow derived hematopoietic stem cells and platelets as these cells are important for thrombus formation under pathophysiological conditions. Moreover, we studied preadipocytes and adipocytes as these cells are tightly linked with obesitas, which in turn correlates with an increased risk of venous thrombosis17,18. Finally, hepatocytes were included as these cells are the best characterized cells for coagulation factor synthesis.
Results
To investigate whether human cells besides the major sites of coagulation factor production express clotting factors(s), FV mRNA expression was measured. As depicted in Figure 2A , FV mRNA was expressed at high levels in human monocytes and hepatocytes, with hepatocytes, the traditional major site of FV expression, serving as a positive control. Remarkably, FV was not detected in other cells, including human macrophages, which are derived from monocytes ( Figure 2A ). Thus, despite its detection in monocytes and hepatocytes, the gene is subject to stringent transcriptional regulation.

(A–C) Expression of FV (A), FX (B) and FVIII (C) by eight human primary cell types: stem cells, fibroblasts, endothelial cells, macrophages, preadipocytes and adipocytes, hepatocytes and monocytes, with and without exposure to LPS.
The level of each mRNA was measured in triplicate and error bars show standard deviations of means for triplicate cultures per condition and is represented as the mean ± SEM. The p-value obtained from a paired t-test with a two-tailed test samples (T-test). P < 0.05 is considered statistically significant. (A) shows the expression of the FV gene by human monocytes (dark-gray bar) and hepatocytes (light-gray bar). Monocytes and hepatocytes were not exposed to LPS. Hepatocytes were used as a positive control for coagulation factor expression. Human primary hepatocytes die upon exposure to LPS and monocytes were not exposed to LPS because LPS treatment results in the differentiation of monocytes into macrophages. mRNA expression is expressed as 2log (logarithm base 2) values on the y-axis. Relative mRNA intensity was obtained from three measurements and is represented as the mean ± SEM. (B) shows the expression of the FX gene by human fibroblasts, preadipocytes, adipocytes and hepatocytes. Gene expression is expressed as 2log values on the y-axis. Relative mRNA intensity was obtained from triplicate measurements and is represented as the mean ± SEM. Hepatocytes served as a positive control for coagulation factor expression. (C) shows the expression of the FVIII gene by human primary cells. FVIII mRNA expression was expressed as2log values on the y-axis. FVIII mRNA expression was observed in all cells tested except endothelial cells. Relative mRNA intensity was obtained from three measurements and is represented as the mean ± SEM. Hepatocytes served as a positive control for coagulation factor expression.
Because FV is a cofactor for FX proteolytic activity, we were interested in investigating whether FX expression would resemble that of FV. As shown in Figure 2B , FX, like FV, was subject to stringent transcriptional regulation, as it was only expressed in a subset of cell types. Its expression, however, was markedly different from FV and was limited to fibroblasts, visceral preadipocytes and adipocytes, where its transcription was as strong as in hepatocytes, the standard site of its synthesis. Thus, despite being parts of the same enzymatic complex and despite both FV and FX showing expression outside the traditional locales of their expression, the regulation of these genes is markedly different.
In addition to FV, FVIII also acts as cofactor to FX. The partially mutually exclusive expression of FV and FX raises questions as to the regulation of FVIII. Interestingly, while FV and FX showed restricted expression, FVIII was more or less ubiquitously expressed. All cell types examined expressed the gene to a high level, except endothelial cells, which only contained trace amounts of FVIII ( Figure 2C ).
vWF forms a complex with FVIII, which stabilizes FVIII. As FVIII is also a member of the tenase complex, the expression of vWF was investigated in all nine human primary cell populations. As shown in Figure 3A , vWF expression was high in endothelial cells. In addition, mRNA analysis showed that vWF was expressed by fibroblasts, macrophages, preadipocytes, hepatocytes and monocytes. However, the expression of vWF was inhibited in preadipocytes upon exposure to lipopolysaccharide (LPS) ( Figure 3A ), suggesting that inflammatory stimuli inhibit its production in this cell type.

(A-C). mRNA expression of vWF, FXIIIA and thrombomodulin in eight human primary cell types treated either with or without LPS.
The level of each mRNA was measured in triplicate and error bars show standard deviations of means for triplicate cultures per condition and is represented as the mean ± SEM. The p-value obtained from a paired t-test with a two-tailed test samples (T-test). P < 0.05 is considered statistically significant. (A) mRNA expression of vWF in eight different human primary cell types treated with or without LPS. vWF mRNA expression is expressed as 2log values on the y-axis. vWF mRNA expression was detected in all cells tested, except preadipocytes and adipocytes. The highest level of expression was in endothelial cells. Relative mRNA intensity was obtained from three measurements. Hepatocytes served as a positive control for coagulation factor expression. (B) mRNA expression of FXIIIA in eight human primary cell types treated with or without LPS. FXIIIA mRNA expression was synthesized only by macrophages and monocytes. LPS inhibited mRNA expression in macrophages. FXIIIA mRNA expression is expressed as 2log values on the y-axis. Relative mRNA intensity was obtained from three measurements. Hepatocytes served as a positive control for coagulation factor expression. (C) mRNA expression of thrombomodulin (TM) in eight different human primary cells treated with or without LPS. TM mRNA expression was observed in all cells except hepatocytes, preadipocytes and adipocytes (2log lower than or close to 1). TM mRNA expression was inhibited by LPS. TM mRNA expression is expressed as 2log values on the y-axis. Hepatocytes served as a positive control for coagulation factor expression.
After analyzing these coagulation factors that activate prothrombin (the inactive form of thrombin), the mRNA expression of FXIIIA, a fibrin clot stabilizer and that of thrombomodulin, an important and potent thrombin cofactor in the anticoagulant pathway, were studied. As illustrated in Figure 3B , FXIIIA was produced at a high level by monocyte-derived macrophages, as well as by monocytes and was only weakly expressed by hepatocytes. FXIIIA expression was not detected in other cells ( Figure 3B ) and is thus subject to rigorous transcriptional control. The mRNA expression of thrombomodulin was high in bone marrow-derived stem cells ( Figure 3C ). In addition, thrombomodulin was strongly expressed in endothelial cells, macrophages, monocytes and, to a lesser extent, fibroblasts; however, similar to FXIIIA expression in macrophages, thrombomodulin expression was inhibited after exposure of macrophages to LPS. No thrombomodulin expression was detected in visceral preadipocytes, adipocytes, or hepatocytes, again highlighting the intricate transcriptional control to which the members of the coagulation cascade are subject.
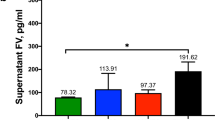
Finally, we focused on human platelets, which have obvious relevance to the coagulation process but do not lend themselves well to common technology for expression analysis. Thus, platelets were screened for the presence or absence of coagulation factors using mass spectrometry. Platelets were isolated by iodixanol-gradient centrifugation and the production of coagulation factors was subsequently assessed by LC-MS/MS. Orbitrap LTQ-XL (LC-MS/MS) analysis detected FV, FXIIIA and vWF in the platelet fraction ( Figure 4 , 5 and 6 , respectively), further completing the complicated picture of the sites of coagulation factor expression.

Proteomic analysis of FV production by platelets.
After one-step iodixanol-gradient centrifugation, purified platelet fractions were collected and subjected to LC-MS/MS (LC-tandem mass spectrometry). The results are displayed graphically with the intensity expressed as the ion count (y-axis) and plotted against the m/z ratio of the ions (x-axis). The precursor ion-scan mass spectrum of the FV tryptic digest resulted in an m/z of 830.62, as shown in green. The MS/MS product ion spectrum obtained from the fragmentation of this precursor ion at m/z 832·62 with b-ions (red peaks) and y-ions (blue peaks) corresponds to the sequence MPMGLSTGIISDSQI from the tryptic FV peptide MPMGLSTGIISDSQIK, as shown in the table (inset). Five peptides matched the FV protein sequence. Each sample was subjected twice to LC-MS/MS.

Proteomic analysis of FXIIIA production by platelets.
After one-step iodixanol-gradient centrifugation, purified platelet fractions were collected and subjected to LC-MS/MS (LC-tandem mass spectrometry). The results are displayed graphically with the intensity expressed as the ion count (y-axis) and plotted against the m/z ratio of the ions (x-axis). The precursor ion-scan mass spectrum of the FXIIIA tryptic digest resulted in an m/z of 631.79, as shown in green. The MS/MS product ion spectrum obtained from the fragmentation of this precursor ion at m/z 931·79 with b-ions (red peaks) and y-ions (blue peaks) corresponds to the sequence LALETALMYGA from the tryptic FXIIIA peptide LALETALMYGAK, as shown in the table (inset). Five peptides matched the protein sequence of FXIIIA. Each sample was subjected twice to LC-MS/MS.

Proteomic analysis of vWF production by platelets.
After one-step iodixanol-gradient centrifugation, purified platelet fractions were collected and subjected to LC-MS/MS (LC-tandem mass spectrometry). The results are displayed graphically with the intensity expressed as the ion count (y-axis) and plotted against the m/z ratio of the ions (x-axis). The precursor ion-scan mass spectrum of the vWF tryptic digest resulted in an m/z of 573.96, as shown in green. The MS/MS product ion spectrum obtained from the fragmentation of this precursor ion at m/z 573·96 with b-ions (red peaks) and y-ions (blue peaks) corresponds to the sequence LSEAEFEVL from the tryptic vWF peptide LSEAEFEVLK, as shown in the table (inset). Six peptides matched the sequence of vWF. Each sample was subjected twice to LC-MS/MS.
Discussion
The standard view of coagulation factor production holds that the present of these factors is restricted to three compartments, listed as follows, in order of importance: the liver, the endothelium and platelets. To investigate whether other cells are also sites of expression of FV, FVIII, vWF and FX, the mRNA levels of these factors were investigated in nine human primary cell types before and after exposure to LPS, with the exception of monocytes and hepatocytes, which were only studied in the absence of LPS exposure. Human hepatocytes cannot survive exposure to LPS and the exposure of monocytes to LPS results in their differentiation into macrophages. LPS was used to monitor whether the expression of coagulation factors in these cells is biologically active. Human primary hepatocytes, the standard site of coagulation factor synthesis, served as a positive control in our experiments. LC-MS/MS-based proteomic analysis of a highly purified human platelet population, isolated by iodixanol-gradient centrifugation, was also performed. We observed a complicated pattern of expression of these factors, which seems to contrast with the relatively simple scheme depicted in the literature, in which coagulation factor production is largely restricted to the liver, with contributions from the platelet fraction and vWF synthesis in the endothelium.
FV is the most important FX cofactor and is expressed only by monocytes and hepatocytes. Mutations resulting in the absence or dysfunction of activated FV (FVa) lead to hemorrhagic disease; conversely, mutations resulting in excessive activity of FV are associated with thrombosis. In particular, the R506Q mutation (also known as FV Leiden) is associated with thrombophilia and APC resistance and is a significant clinical problem19. Here, we observed that monocytes express FV. Hence, a prediction from our observations is that allogeneic bone marrow transplantation (from which monocytes originate) may be a rational treatment for FV deficiency20.
Although the formation of fibrin is crucial during vascular injury, a rapid response and acute fibrin clot formation are essential for the prevention of bleeding and for vascular wound healing. Because we showed that coagulation factor FV is expressed by monocytes, monocytes can no longer be considered merely a membrane surface for activated coagulation factors to be primed by tissue factor: monocytes may play a much more active role and may also be directly involved in FX activation (FXa) via cell-autonomous FV delivery. FV can be activated by elastase and cathepsin G, which are present in monocytes21, further stimulating FX activity through FV-dependent mechanisms.
Interestingly, almost all components of the coagulation cascade except FV act in plaques to form the prothrombinase complex. Notably, monocytes and platelets are only circulating cells, which supply the necessary FV to activate FX and cannot be considered tissue-resident cells. When monocytes extravasate by crossing the endothelium, they differentiate into macrophages (tissue cells) and lose FV expression ( Figure 1 ). Because coagulation factors need to be released into the blood, the expression of FV by circulating monocytes suggests that an important function of monocytes may be to maintain FV plasma levels. The possibility that monocytes are a main source of FV in plasma may explain why previous attempts at rectifying genetically aberrant FV expression with liver transplantation have failed20. This theory suggests that co-transplantation of bone marrow (which produces monocytes) could constitute a viable therapeutic approach.
Lipids, collagens, calcium and inflammatory cells are well-established components of atherosclerotic plaques22. In addition, fibroblasts play an important role in wound healing by synthesizing collagen and extracellular matrix23. It is intriguing that the expression of FX is absent in cells that express FV and vice versa (hepatocytes are not in direct contact with blood vessels). Proteomic analysis of highly purified platelets from human plasma reveals that FV and vWF are present in platelets but not FVIII or FX. Intriguingly, both monocytes and platelets produce FV and vWF but not FX. The previously established expression of FV and vWF by platelets4,21,24,25,26 was confirmed in our study. Monocytes are different from fibroblasts and macrophages with respect to the expression of FV and FX and it is tempting to suggest that the different localization of monocytes (circulating) versus fibroblasts and macrophages (present in tissue and matrix) is functionally relevant, but obviously further experimentation is necessary to test this notion.
Because FXIIIA27,28,29 plays an unambiguous role in the stabilization of the fibrin clot during wound healing and because thrombomodulin, an anticoagulant, functions in a complex with thrombin and is important for the degradation and lysis of fibrin after vascular repair, cells were also screened for the expression of FXIIIA and thrombomodulin. FXIIIA mRNA was expressed by monocytes and proteomic analysis of highly purified platelets revealed that these cells also produce FXIIIA protein. The expression of FXIIIA by these two cell types also suggests that monocytes and platelets are involved not only in priming coagulation but are also involved in the final event of fibrin stabilization. Indeed, we observed that monocytes and macrophages, but not platelets, express thrombomodulin. Thrombomodulin has been observed on the surface of endothelial cells and monocytes30,31,32,33, which is in agreement with our observations. Based on these data, we suggest that both platelets and monocytes arrive at the site of injury and synergistically initiate primary hemostasis and, as a final event, stabilize the fibrin clot to prevent bleeding and aid repair of the wound.
Our data show that coagulation factor production is complex and that the role of local production in vascular injury of such factors may be larger than previously thought (see Figure 1 ). Monocytes have a direct and dual role in coagulation by producing important cofactors for FX (FV, FVIII and FXIIIA), but additional regulation is provided through the production of thrombomodulin, which destabilizes fibrin clots30, impairs platelet function27,28,29,30,31,32,33 and activates the anticoagulant protein C pathway. Furthermore, the observation that LPS can regulate coagulation factor production by monocytes shows that the system is dynamic and responds to local cues, further supporting the hypothesis that regulation of coagulation factor production is highly context dependent and much more complex than thought. A full understanding of the mechanisms involved could potentially advance our understanding of the atherothrombotic process and open novel rational avenues for treatment.
Methods
Nine human primary cell types were used in this study, including human primary mesenchymal stem cells from bone marrow (hMSC-BM) (PromoCell, Germany), human primary dermal fibroblasts (PromoCell, Germany), human primary umbilical vein endothelial cells (HUVECs) (University Medical Center Groningen (UMCG) endothelial Facility), human primary peripheral blood CD14+ monocytes (LONZA, USA/Belgium), macrophages (monocytes differentiated to macrophages by phorbol-12-myristate-13-acetate (PMA), Poietics™ human primary visceral preadipocytes (LONZA, USA/Belgium), adipocytes (preadipocytes differentiated to adipocytes) and hepatocytes (Becton & Dickinson (BD) USA/Germany). All cells were kept in culture until reaching confluence (4-5 days). Then, cells were washed 5 times with FCS-free medium. The cells were cultured in the presence or absence of LPS (200 ng/ml). Cells were exposed to LPS for 24 h. Subsequently, total RNA was collected. The mRNA expression of the prothrombinase complex (FV and FX), tenase complex (FVIII), FVIII-stabilized protein (vWF), fibrin clot stabilizer (FXIIIA) and thrombomodulin: thrombin (THR: TM) complex, which activates protein C, was measured in triplicate for the 16 cell types. Platelet-associated proteins, collected from a highly pure platelet population isolated by iodixanol-gradient centrifugation, were analyzed by LC-MS/MS as described by Birschmann et al.34 (two independent isolations and two independent LC-MS/MS analyses were performed). Blood was collected from healthy volunteers after fasting by venipuncture into tubes containing the anticoagulant EDTA. All healthy volunteers provided informed consent. Subsequently, different iodixanol concentrations were prepared and layered within 3-ml tubes. The gradients were then centrifuged. Highly purified platelet fractions were isolated from the tubes by aspiration through a needle. Subsequently, platelet-associated proteins were trypsinized at 37°C for 24 h to convert proteins to peptides. After 12 h of incubation, 100 ng of trypsin was added to the fraction to ensure the complete conversion of proteins into peptides. After washing the peptide fraction through a C18 column, 5 μl of the purified platelet-associated protein (peptide) fraction was assayed by liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS). LTQ ORBITRAP-XL (Thermo Fisher, USA/The Netherlands) was used for MS.
Characterization of cells
Human mesenchymal stem cells from bone marrow
Rigid quality control tests were performed for each lot of mesenchymal stem cells. They were tested for cell morphology, adherence rate and viability. Furthermore, they were characterized by flow-cytometric analysis of a comprehensive panel of markers, e.g., PECAM HCAM, CD45 and endoglin. Differentiation performance was guaranteed up to 10 population doublings (PDs). In addition, all cells were tested for the absence of HIV-1, HBV, HCV and microbial contaminants (e.g., fungi, bacteria and mycoplasma).
Human primary dermal fibroblasts (HPDFs)
Rigid quality control tests were performed for each lot of human primary dermal fibroblasts. They were tested for cell morphology, adherence rate and cell viability. Immunohistochemical tests for the fibroblast-specific antigen CD90 were performed for each lot. Growth performance was tested through multiple passages up to 10 or 15 PDs under culture conditions without antibiotics and antimycotics. All cells were tested for the absence of HIV-1, HBV, HCV and microbial contaminants (e.g., fungi, bacteria and mycoplasma).
Human primary peripheral blood CD14+ monocytes
Monocytes are floating cells. Lonza human primary CD14+ monocytes were isolated from the peripheral blood of screened, healthy donors. Peripheral blood was collected from the donors using apheresis. Monocytes were then isolated using positive immunomagnetic selection directed against CD14. CD14 was expressed on most monocytes but not on other mononuclear cells in the blood. The purity of the population of cells was confirmed by flow cytometry detecting CD14 and CD11b. Each donor was tested and found non-reactive by an FDA-approved method for the presence of HIV-I, hepatitis B virus and hepatitis C virus. Where these tests were not possible, cell products were tested for the presence of viral nucleic acid from HIV, hepatitis B virus and hepatitis C virus. Cells were then cryopreserved.
Human primary macrophages
In contrast to monocytes, macrophages are attached cells. PMA is routinely applied to differentiate monocytes into macrophages35,36,37. After the confluence of normal human peripheral blood CD14+ monocytes were differentiated to macrophages by stimulation with 100 ng/ml PMA for 48 h. Cell quality was confirmed by their attachment to tissue-culture plastic. After two days, the medium with PMA was replaced with FCS-free medium with and without containing LPS for 24 h. Then, the cells were lysed to isolate total RNA.
Human primary visceral preadipocytes
Human primary visceral preadipocytes are precursor cells isolated from abdominal tissue that develop into adipocytes when fully differentiated and are characterized by accumulation of intracellular triglycerides in lipid droplets. The visceral preadipocyte cell system contains human visceral preadipocytes. Cell viability, morphology and proliferative capacity were measured after recovery from cryopreservation. Visceral pre-adipocytes were characterized morphologically at day 10 using AdipoRed™. After removing medium and washing the cells, the cells were incubated with the differentiation medium without serum and incubated either with or without LPS for 24 h. Then, the cells were lysed to isolate total RNA.
Human primary visceral adipocytes
Human visceral preadipocytes were differentiated into adipocytes according to instructions provided by PromoCell. Briefly, human primary preadipocytes were cultured to confluence in growth medium provided by PromoCell in 5% CO2 at 37°C. When the cells reached confluence, the growth medium was removed and replaced with preadipocyte differentiation FCS-free medium supplemented with 0.5 mM 3-isobutyl-1-methyxanthine, 400 ng/ml dexamethasone, 0.5 μg/ml bovine insulin, 9 ng/ml L-thyroxine, 3 μg/ml ciglitazone and 8 μg/ml D-biotin (all from PromoCell) for 72 hours. After 3 days, the differentiation medium was removed and replaced with adipocyte nutrition medium containing 3% FCS, 8 μg/ml D-biotin, 0.5 μg/ml bovine insulin and 400 ng/ml dexamethasone for 16 days (mature adipocytes). After maturation of adipocytes, the nutrition medium was removed and the cells were washed 5 times with the FCS-free differentiation medium without supplement. The cells were then incubated with the differentiation medium without FCS and supplement and incubated either with or without LPS for 24 h. Then, the cells were washed with PBS 5 times. Finally, the cells were lysed to isolate total RNA.
Human primary fresh hepatocytes
Human primary fresh hepatocytes maintain their morphology and function for long-term assays, such as enzyme induction, toxicity, metabolism of stable compounds and drug transporter assays. These fresh human hepatocytes were characterized for phase I and II enzyme activity. All cells were tested for CYP3A4 activity with a 6β-testosterone hydroxylase assay. BD human primary fresh hepatocytes were prepared from fresh liver tissue to maintain cell health, morphology and viability. A BD Matrigel overlay created a more physiological-like environment.
Human umbilical vein endothelial cells (HUVECs)
University Medical Center Groningen has had an Endothelial Cell Facility since 1995. HUVECs obtained from the Endothelial Cell Facility of UMCG (The Netherlands)38,39,40,41 were isolated from umbilical cords. The endothelial cells were characterized morphologically by growth contact inhibition and their typical cobblestone appearance at confluence and by immunologically using indirect immunofluorescence for intracellular vWF. Cells were phenotyped by flow cytometry for the presence of the endothelial cell marker CD31. All cells were cultured at 37°C in a humidified 5% (v/v) CO2/95% air atmosphere.
mRNA expression analysis
Total RNA was collected from human primary cell lysates before and after LPS treatments using a NucleoSpin RNA II kit (BIOKE) according to manufacturer's instructions, including DNase I treatment (BIOKE). RNA quality and concentration were assessed in a Bioanalyzer (Experion), (Bio-Rad, USA/The Netherlands) using the Agilent RNA 6000 Nano kit (Agilent, The Netherlands). A Total RNA Amplification Kit was applied to amplify and label the RNA (Applied Biosystems, The Netherlands). Total RNA was reverse-transcribed into cDNA and the concentrations were determined in NanoDrop as above. cRNA was biotinylated with an RNA Amplification Kit (Ambion, USA) according to the manufacturer's instructions, starting with 200 ng total RNA. Samples were purified using the RNeasy kit (Qiagen, The Netherlands). Hybridization to the Sentrix Human Expression BeadChip (Illumina, USA), washing and scanning were performed according to the BeadStation 500 manual (revision C). One BeadChip with 12 samples was used. Each slide was scanned immediately. After the scanning, the following steps were performed: 1: quality check; 2: background correction; 3: normalization to a housekeeping gene; and 4: utile normalization of the data using Beadstudio Expression module v 3.2.7.After these 4 steps, a relative mRNA intensity of 4 was considered cut-off and designed as the threshold level.
This study was approved by the Medical Ethics Committee of the University Medical Center Groningen with the reference number METc2007.081.
References
Rauch, U. et al. Thrombus formation on atherosclerotic plaques: pathogenesis and clinical consequences. Ann. Intern. Med. 134, 224–238 (2001).
Li, D. & Mehta, J. L. Oxidized LDL, a critical factor in atherogenesis. Cardiovasc. Res. 68, 353–354 (2005).
Monroe, D. M., Hoffman, M. & Roberts, H. R. Platelets and thrombin generation. Arterioscler. Thromb. Vasc. Biol. 22, 1381–1389 (2002).
Mackman, N. The role of tissue factor and factor VIIa in hemostasis. Anesth. Analg. 108, 1447–1452 (2009).
Fay, P. J. Activation of factor VIII and mechanisms of cofactor action. Blood Rev. 18, 1–15 (2004).
Saenko, E. L. & Scandella, D. The acidic region of the factor VIII light chain and the C2 domain together form the high affinity binding site for von willebrand factor. J. Biol. Chem. 272, 18007–18014 (1997).
Webster, W. P. et al. Factor VIII synthesis: hepatic and renal allografts in swine with von Willebrand disease. Am. J. Physiol. 230, 1342–1348 (1976).
Bouchard, B. A. & Tracy, P. B. Platelets, leukocytes and coagulation. Curr. Opin. Hematol. 8, 263–269 (2001).
Nicolaes, G. A. & Dahlback, B. Factor V and thrombotic disease: description of a janus-faced protein. Arterioscler. Thromb. Vasc. Biol. 22, 530–538 (2002).
Scotton, C. J. et al. Increased local expression of coagulation factor X contributes to the fibrotic response in human and murine lung injury. J. Clin. Invest. 119, 2550–2563 (2009).
Shantsila, E. & Lip, G. Y. H., The role of monocytes in thrombotic disorders Insights from tissue factor, monocyte-platelet aggregates and novel mechanisms. Thromb. Haemost. 102, 916–924 (2009).
Barile, C. J. et al. Inhibiting platelet-stimulated blood coagulation by inhibition of mitochondrial respiration. Proc. Natl. Acad. Sci. U. S. A. 109, 2539–2543 (2012).
Wilcox, J. N., Noguchi, S. & Casanova, J. Extrahepatic synthesis of factor VII in human atherosclerotic vessels. Arterioscler. Thromb. Vasc. Biol. 23, 136–41 (2003).
Shetty, S. et al. Induction of Tissue Factor by Urokinase in Lung Epithelial Cells and in the Lungs. Am. J. Respir. Crit. Care Med. 181, 1355–1366 (2010).
Shinagava, K., Ploplis, V. A. & Castellino, F. J. A severe deficiency of coagulation factor VIIa results in attenuation of the asthmatic response in mice. Am. J. Physiol. Lung Physiol. 296, L763–L770 (2009).
Bajaj, M. S., Steer, S., Kuppuswamy, M. N., Kisiel, W. & Bajaj, S. P. Synthesis and expression of tissue factor pathway inhibitor by serum-stimulated fibroblasts, vascular smooth muscle cells and cardiac myocytes. Thromb. Haemost. 82, 1663–1672 (1999).
Abdollahi, M., Cushman, M. & Rosendaal, F. R. Obesity: risk of venous thrombosis and the interaction with coagulation factor levels and oral contraceptive use. Thromb. Haemost. 89, 493–498 (2003).
Darvall, K. A., Sam, R. C., Silverman, S. H., Bradbury, A. W. & Adam, D. J. Obesity and thrombosis. Eur. J. Vasc. Endovasc. Surg. 33, 223–233 (2007).
Dirven, R. J., Vos, H. L. & Bertina, R. M. The R306G and R506Q mutations in coagulation Factor V reveals additional cleavage sites for Activated Protein C in the R313–R321 region and at R505. Thromb. Res. 125, 444–450 (2010).
Parker, J. et al. Discrepancy between phenotype and genotype on screening for factor V Leiden after transplantation. Blood 97, 2525–2526 (2001).
Allen, D. H. & Tracy, P. B. Human coagulation factor V is activated to the functional cofactor by elastase and cathepsin G expressed at the monocyte surface. J. Biol Chem. 270, 1408–1415 (1995).
Rogers, W. J. et al. Characterization of signal properties in atherosclerotic plaque components by intravascular MRI. Arterioscler. Thromb. Vasc. Biol. 20, 1824–1830 (2000).
Midwood, K. S., Williams, L. V. & Schwarzbauer, J. E. Tissue repair and the dynamics of the extracellular matrix. Int. J. Biochem. Cell Biol. 36, 1031–1037 (2004).
Alberio, L., Safa, O., Clemetson, K. J., Esmon, C. T. & Dale, G. L. Surface expression and functional characterization of alpha-granule factor V in human platelets: effects of ionophore A23187, thrombin, collagen and convulxin. Blood 95, 1694–1702 (2000).
Etingin, O. R., Silverstein, R. L. & Hajjar, D. P. von Willebrand factor mediates platelet adhesion to virally infected endothelial cells. Proc. Natl. Acad. Sci. U S A 90, 5153–5156 (1993).
Nesheim, M. E., Canfield, W. M., Kisiel, W. & Mann, K. G. Studies of the capacity of factor Xa to protect factor Va from inactivation by activated protein C. J. Biol. Chem. 257, 1443–1447 (1982).
Hevessy, Z., Haramura, G., Boda, Z., Udvardy, M. & Muszbek, L. Promotion of the crosslinking of fibrin and alpha 2-antiplasmin by platelets. Thromb. Haemost. 75, 161–167 (1996).
Muszbek, L., Ariens, R. A. & Ichinose, A. Factor XIII: recommended terms and abbreviations. J. Thromb. Haemost. 5, 181–183 (2007).
Poon, M. C. et al. Hemopoietic origin of factor XIII A subunits in platelets, monocytes and plasma. Evidence from bone marrow transplantation studies. J. Clin. Invest. 84, 787–792 (1989).
Esmon, C. T. & Owen, W. G. Identification of an endothelial cell cofactor for thrombin-catalyzed activation of protein C. Proc. Natl. Acad. Sci. U S A 78, 2249–2252 (1981).
Esmon, N. L., Owen, W. G. & Esmon, C. T. Isolation of a membrane-bound cofactor for thrombin-catalyzed activation of protein C. J. Biol. Chem. 257, 859–864 (1982).
Esmon, N. L., Carroll, R. C. & Esmon, C. T. Thrombomodulin blocks the ability of thrombin to activate platelets. J. Biol. Chem. 258, 12238–12242 (1983).
McCachren, S. S., Diggs, J., Weinberg, J. B., Dittman, W. A. Thrombomodulin Expression by Human Blood Monocytes and by Human Synovial Tissue Lining Macrophages. Blood 78, 3128–3132 (1991).
Birschmann, I. et al. Use of functional highly purified human platelets for the identification of new proteins of the IPP signaling pathway. Thromb. Res. 122, 59–68 (2008).
Tsuchiya, S. et al. Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res. 42, 1530–156 (1982).
Badlou, B. A., Wu, Y. P., Smid, W. M. & Akkerman, J. W. Platelet binding and phagocytosis by macrophages. Transfusion 46, 1432–1443 (2006).
Barbieri, S. S., Eligini, S., Brambilla, M., Tremoli, E. & Colli. S. Reactive oxygen species mediate cyclooxygenase-2 induction during monocyte to macrophage differentiation: critical role of NADPH oxidase. Cardiovasc. Res. 60, 187–197 (2003).
Mulder, A. B. et al. Basal tissue factor expression in endothelial cell cultures is caused by contaminating smooth muscle cells. Reduction by using chymotrypsin instead of collagenase. Thromb. Res. 80, 399–411 (1995).
Molema, G. et al. Analysis of In Vitro Lymphocyte Adhesion and Transendothelial Migration by Fluorescent-Beads-Based Flow Cytometric Cell Counting. Cytometry 32, 37–43 (1998).
Joanna, M. et al. Differential effects of NF-κB and p38 MAPK inhibitors and combinations thereof on TNF-α- and IL-1β-induced proinflammatory status of endothelial cells in vitro. Am. J. Physiol. Cell Physiol. 289, C1229–C1239 (2005).
Asgeirsdóttir, S. A. et al. Targeted transfection increases siRNA uptake and gene silencing of primary endothelial cells in vitro--a quantitative study. J. Control. Release 141, 241–51 (2010).
Author information
Authors and Affiliations
Contributions
All authors contributed to the experiments, analysis of the results and writing of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Dashty, M., Akbarkhanzadeh, V., Zeebregts, C. et al. Characterization of coagulation factor synthesis in nine human primary cell types. Sci Rep 2, 787 (2012). https://doi.org/10.1038/srep00787
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00787
This article is cited by
-
Map of thrombogenesis in viral infections and viral-driven tumours
Discover Oncology (2023)
-
Characterization and visualization of murine coagulation factor VIII-producing cells in vivo
Scientific Reports (2021)
-
Local Production of Activated Factor X in Atherosclerotic Plaque Induced Vascular Smooth Muscle Cell Senescence
Scientific Reports (2017)
-
Lipid droplets hypertrophy: a crucial determining factor in insulin regulation by adipocytes
Scientific Reports (2015)
-
Expression of FABP4, adipsin and adiponectin in Paneth cells is modulated by gut Lactobacillus
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
