
Abstract
Microbes induce innate immune responses in hosts. It is critical to know how different microbes control adaptive responses through innate pathways. The impact of gram-positive bacteria on the innate and adaptive responses is unclear. Herein we report that Staphylococcus aureus induces IL-10, Th17-inducing cytokines IL-6 and IL-23, chemokines and regulates dendritic cell markers. S. aureus inhibits T-cell IL-2 responses through modulation of HLA-DR, CD86 and PD-L1. IFN-gamma, Src kinase inhibitors, or TLR2 antibodies prevented the down-modulation of HLA-DR by S. aureus. Our data demonstrate that innate TLR signaling induces multi-dimensional inhibition of adaptive immune responses, which may contribute to the lack of protective immunity to bacteria or microbe tolerance. IL-10 and PD-L1 antagonists may boost immunity to vaccines for S. aureus and other microbes.
Similar content being viewed by others

Introduction
Some pathogens, such as the DNA viruses varicella and smallpox and the RNA virus measles result in life-long immune protection following infection. However, other pathogens infect hosts repeatedly either due to frequent mutations, antigenic shift/drift of the pathogens that permit immune escape (e.g., HIV-1 or influenza), or possibly, due to an inability to generate pathogen-specific immunity by the host. Understanding why some pathogens are able to infect the same host repeatedly1,2 and gaining insight into the mechanisms that permit or prevent establishment of protective immunity during the initial infection is important.
Toll-like receptors, a major subgroup of the Pattern-Recognition Receptors (PRRs), play critical roles in the initial interaction between microbes and the host. Following infection, gram-negative bacteria signal via TLR43, while TLR2 is the key receptor responsible for mediating host interaction with gram-positive bacteria3,4. Although bacteria-PRR interactions can induce harmful inflammatory cytokines, data from studies in knockout mice indicate that toll-like receptors, such as TLR4 and TLR2, actually play a protective role during infection. Although studies of S. aureus signaling are limited, it is known for other TLRs that in addition to signaling through adaptor molecules, such as MyD88, TIRAP(Mal), TRAM and triggering NF-κB activation5,6,7,8, toll-like receptors may be phosphorylated by tyrosine kinases.
Invasive and recurrent infections with Staphylococcus aureus, are increasing in both hospital and community-settings. S. aureus is a gram positive extracellular bacterium that can repeatedly infect people for which there is no effective preventive vaccine. Antibiotic treatment has been complicated by the development of resistant strains such that methicillin-resistant S. aureus (MRSA) infection is now a major public health problem9,10,11,12. S. aureus has been associated with serious diseases such as endocarditis, bacteremia, pneumonia, osteomyelitis, cellulitis and septic shock. S. aureus is the leading cause of death due to infection in the US exceeding the number of deaths attributed to HIV-1, hepatitis and influenza combined13. Although the host innate and adaptive responses involving neutrophils, macrophage elastase and reactive oxygen species can control or limit bacterial infection in certain situations14,15, proper antibiotic treatment is required to prevent fatalities associated with sepsis or pneumonia.
Some factors that contribute to disease pathogenesis following infection with gram positive bacteria are known. Bacterial cell wall components from gram-positive bacteria and gram negative bacteria initiate shock and multiple organ failure, by up-regulating TNF-α production even in the absence of enterotoxins or exotoxins. In addition, during sepsis caused by gram-negative bacteria antigen presenting cell (APC) function may be lost16,17 due to release of soluble mediators. Monocytes are one of the major sources of endogenous cytokines induced during sepsis which in turn may play a critical role in determining APC function. Some studies have reported a direct correlation between monocyte HLA-DR expression and survival in severe gram negative sepsis18 and septic shock19, indicating that monocyte function may be a biomarker for survival. Although some gram positive bacteria produce superantigens that can interfere with immune responses by polyclonal T cell activation and contribute to pathogenesis, many strains of gram-positive bacteria do not produce superantigens. Therefore, the mechanisms responsible for recurrent or chronic gram-positive infection largely remain undefined. We hypothesize that certain gram-positive pathogens, such as S. aureus, may disrupt antigen presentation and abrogate signaling required for co-stimulation and inhibit establishment of immunity during initial and subsequent infections. Since monocytes, the precursors of dendritic cells, provide a crucial link between the innate immune response and the initiation of adaptive immunity, we have examined the effects of S. aureus on innate and adaptive responses mediated by human monocyte/macrophages with a focus on the expression and molecular control of cell surface markers involved in antigen presentation and co-stimulation (or co-inhibition) and have characterized cytokine production by human primary monocytes/macrophages following exposure to S. aureus.
Results
Modulation of MHC Class II and co-stimulatory molecules by S. aureus in human monocytes
Monocytes were isolated from peripheral blood mononuclear cells by counter-current elutriation and then cultured in the presence or absence of killed S. aureus cells (SAC), harvested and then stained for cell surface expression of markers critical to innate and adaptive responses. Fixed bacteria were used to avoid live bacterial contamination in the cultures, a common practice by many research groups. As shown in Fig. 1a and 1b, SAC down-regulated the expression of MHC class II antigen HLA-DR and the major co-stimulatory molecule CD86, but induced the expression of co-stimulatory molecules CD40 and CD80, dendritic cell marker CD83 and chemokine receptor CCR7. The expression of MHC class I molecule HLA-ABC and innate toll-like receptors TLR2 and TLR4 were not changed. The abundance of cell surface HLA-DR and CD86 molecules in SAC treated cultures was dose-dependent and inversely related to the concentration of SAC used (Fig.1c and 1d). The expression levels of HLA-DR and CD86 vary with donors. The down modulation of HLA-DR and CD86 by S. aureus was statistically significant (p values are less than 0.05) as analyzed with paired t test(Fig. 1e).

Effects of S. aureus on the expression of HLA-DR and CD86 in human monocytes.
(a) and (b). Monocytes were cultured in macrophage-SFM (Med) or in the presence of SAC (0.0075% or 0.0075g/100mL). After 40 hours, cells were analyzed for the expression of indicated cell surface molecules. Each marker was assessed for 3 to 10 times with similar results. The number at corner of each histogram is the value of GMFI (geometric mean fluorescence intensity). (c) and (d) Monocytes were cultured in the presence of different doses of SAC and stained for HLA-DR and CD86. Cells were subject to acquisition and analysis on flow cytometer. (e) Statistic analysis of the down modulation of HLA-DR and CD86 (data from 8 donors) by S. aureus. (f). Monocytes were cultured in medium and stimulated with LTA, or LPS for 40 hours.
CD36 is essential for macrophage function since it is needed to clear apoptotic cells in vitro20 and in vivo21. CD36 is also a co-receptor for TLR222. Monocytes/macrophages in untreated cultures express cell surface CD36, however SAC treated cells were negative for surface CD36 expression (Fig. 1b).
Down-modulation of HLA-DR expression by LTA from gram- positive bacteria
Since lipoteichoic acid (LTA) is a ligand of TLR2 and one of the major cell wall components shared by strains of S. aureus, we tested LTA for its ability to down-modulate HLA-DR in human primary monocytes. Monocytes were stimulated with LTA for 40 hours. As shown in Fig. 1f, LTA from S. aureus, down-modulated HLA-DR, indicating that LTA may be an important component contributing to S. aureus mediated down-modulation of HLA-DR, consistent with a previous report of LTA inducing IL-1023.
To examine whether the finding of HLA-DR down-modulation by SAC and LTA on monocytes is specific for Staphylococcus aureus or extends to other bacteria, we then tested the effects on HLA-DR expression of LTA from the gram-positive bacteria, Streptococcus faecalis. LTA of Streptococcus faecalis also reduced HLA-DR expression in monocytes (Fig. 1f). The data show that bacterial components from multiple gram-positive bacteria can control HLA-DR expression. LPS from multiple gram-negative bacteria also down-modulate HLA-DR.
Induction of inflammatory and inhibitory cytokines by S. aureus
It was then determined whether SAC modified the cytokine profile of treated monocytes. The cytokine profile included a potential mediator of septic shock (TNF-α), a Th1 promoting cytokine (IL-12) and an anti-inflammatory and suppressive cytokine (IL-10). Since pretreatment or cytokine priming is required for LPS induced IL-12 production, the production of cytokines by human monocytes induced by SAC with or without IFN-γ pretreatment was also investigated. Monocytes were primed with IFN-γ for 16 hours, followed by stimulation with SAC for 24 hours.
As shown in Fig. 2a and 2b, monocytes activated by SAC produced high levels of TNF-α and IL-10, but not IL-12. Pretreatment with IFN-γ dramatically increased SAC induced IL-12 and TNF-α production. In the contrary, pretreatment with IFN-γ reduced IL-10 secretion following SAC stimulation.

Induction of cytokines and chemokines in human monocytes by S. aureus.
Secretion of cytokines. Monocytes were pre-stimulated with or without IFN-γ for 16 hours, then stimulated with SAC (0.0075%) overnight. Cytokines were measured by specific ELISAs with supernatants from 5 to 8 donors. Results represented as mean (pink line) of each group in box plots. (a) TNF-α and IL-12. (b) IL-10. (c) Transcriptional Induction of cytokines and chemokines by SAC. Monocytes (5 x 106 /well) were stimulated with SAC for 5 hours. Total RNA was isolated and RT-PCR was carried out as described in Materials and Methods. Data were from one donor representing three donors with similar results. (d) Transcriptional induction of cytokines by SAC for indicated times in the absence or presence of IFN-γ.
Transcriptional induction of cytokines and chemokines by SAC
To determine the impact of S. aureus on transcriptional regulation of cytokines and chemokines in human monocytes, we performed RT-PCR for the pro-inflammatory cytokines IL-6 and IL-8, anti-inflammatory cytokine IL-10 and CC chemokines CCL3/MIP-1α and CCL4/MIP-1β, which are ligands of HIV-1 co-receptor CCR5 and secreted by both monocytes and T lymphocytes. As shown in Fig. 2c, messages for IL-10, IL-6 and IL-8 were induced by SAC in monocytes. CCL4 mRNA was induced and the level of CCL3 mRNA was further enhanced by SAC.
SAC induced p40 mRNA, which is shared by IL-12 and IL-23, in the presence or absence of IFN-γ at 2 and 5 hours after stimulation. The pattern of IL-12 p35 transcriptional expression induced by SAC was quite different from that of p40. IL-12 p35 mRNA was induced significantly only after IFN-γ priming plus SAC stimulation, similar to the induction of p35 by LPS that requires IFN-γ24 and consistent with the dependence of optimal IL-12 p70 protein secretion on the presence of IFN-γ. Since IL-23 is important for IL-17 production and Th17 development25, we also examined the induction of IL-23 p19 mRNA by SAC. In contrast to induction of p35, IL-23 p19 mRNA was induced directly by SAC (Fig. 2d) and was not enhanced by IFN-γ, instead a slight reduction of IL-23 p19 was observed in some samples treated with IFN-γ and S. aureus when compared to S. aureus alone treated samples. PCR data from two donors were consistent with each other.
TLR2 dependent downmodulation of MHC Class II molecules
The innate and adaptive responses are key components of host responses, especially to microbes. However, it has yet to be determined whether innate immune receptors control adaptive immune responses. Herein we showed that anti-TLR2, but not anti-TLR4, blocking antibodies restored the expression of HLA-DR, a key molecule of the adaptive system, in S. aureus treated cells (Fig. 3). These data along with LTA data demonstrate that TLR2 is a key receptor for S. aureus in human primary monocytes.

Role of TLR2 in the downmodulation of MHC Class II molecules.
Human primary monocytes were treated for 40 hours with S. aureus with/without one hour pre-incubation with indicated blocking antibodies. First row: isotype control. Cells from donor A were also used for experiments shown in Figure 1f.
Effects of S. aureus or IL-10 on HLA-DR expression were overcome by IFN-γ
IFN-γ is one of the host factors that enhance HLA-DR expression. To test whether IFN-γ could overcome the negative effects of SAC, or IL-10 on the expression of HLA-DR, monocytes were cultured in the presence of IL-10, or SAC with and without IFN-γ for 40 hours and then stained for HLA-DR (Fig. 4a). As expected, HLA-DR expression was reduced in cultures of IL-10, or SAC. However, when IFN-γ was added into SAC exposed cultures, the expression of HLA-DR was enhanced significantly. Thus IFN-γ can antagonize the negative effects of gram-positive or gram negative bacteria on HLA-DR expression. Although IFN-γ can counter-act the effect of IL-10 on the down-modulation of HLA-DR, the effect of IFN-γ in reversing SAC induced HLA-DR down-modulation was most likely through the reduction of IL-10 production.

HLA-DR down-modulation by S. aureus: role of IL-10 and kinases.
Monocytes were cultured in medium in the presence of: (a) indicated reagents (IL-10, 20 ng/mL; IFN-γ, 100 ng/ml and SAC) for 40 hours. Data is representive of results with 3 donors. (b) in the presence of SAC with or without anti-IL-10 neutralizing antibodies or anti-TNF-α neutralizing antibodies for 40 hours. Cells were stained, washed and analyzed on flow cytometer for the expression of CD86, HLA-DR and CD36. Two additional experiments have been done with similar results. (c) Monocytes were cultured in the presence or absence of SAC for 60 hours. After fixation and permeablization cells were stained with indicated antibodies on coverslips and subject to analysis with the confocal microscope equipped with an objective lens of 63x. Overlay: FITC, DAPI, DIC. (d) Monocytes were cultured in medium and pretreated with genistein (G, 74 μM), herbimycin A (H, 1 μg/ml), or tyrphostin (T, 1 μg/ml) for one hour, then stimulated with SAC for 40 hours. Data were pooled data from three donors. (e) Monocytes were pretreated with indicated inhibitors for five hours, then treated with SAC for 40 hours. Cells were stained with indicated antibodies and subject to analysis on flow cytometer. HerbA, herbimycin A; Src KI, Src kinase inhibitor II; GMFI, geometric mean fluorescence intensity. (f) Induction of NF-κB nuclear translocation in human monocytes by S. aureus. (A) Monocytes (25 x 106/well) were stimulated with either LPS, or SAC for 1 hour. Nuclear extracts (NE) were made and assayed according to Materials and Methods. Data were from one donor representing two with similar results. (B) Nuclear extracts were made as (A) and anti-p50 antibody or anti-p65 antibody was added to the DNA binding reactions and assayed according to Materials and Methods.
SAC induced down-modulation of HLA-DR and CD86, but not CD36, is mediated through induction of IL-10
IL-10 is known to decrease expression of HLA-DR and reduce CD86 on dendritic cells26. High levels of IL-10 protein and mRNA have been associated with diminished HLA-DR expression on circulating monocytes during septic shock19,27,28. However, it is not clear whether SAC directly down-modulates HLA-DR and CD86 expression or whether it is mediated through IL-10 induction. We therefore examined the role of IL-10 in modulating the expression of HLA-DR and CD86 following SAC stimulation by testing whether anti-IL-10 neutralizing antibody could block SAC-induced changes in cell surface marker expression. As shown in Fig. 4b, anti-IL-10 neutralizing antibody blocked SAC induced down-modulation of HLA-DR and CD86, whereas anti-TNF-α antibody only weakly affected HLA-DR expression and had no impact on the expression of CD86.
The down-modulation of CD36 by SAC was not significantly affected by either anti-TNF-α neutralizing antibody or by anti-IL-10 neutralizing antibody, suggesting that down-modulation of CD36 by SAC was IL-10 independent.
Down-modulation of HLA-DR was due to sequestration
The expression of HLA-DR is tightly regulated by class II transactivator CIITA, which is controlled by promoters of CIITA29,30. Alternatively, HLA-DR on the cell surface could undergo endocytosis. To determine the possible mechanism of HLA-DR down-modulation by SAC, monocytes treated with SAC were fixed, stained with antibodies and examined by confocal microscopic analysis. As shown in Fig. 4c, in untreated monocytes HLA-DR molecules were located on cell surface. In contrast, in SAC treated macrophages HLA-DR molecules were localized in cytoplasm in granule-like structures, indicating that cell surface HLA-DR molecules were sequestered in or internalized into the cytoplasm after SAC treatment.
Inhibition of SAC induced HLA-DR down-modulation and IL-10 production by tyrosine kinase inhibitors
TLR signaling might involve tyrosine kinases. We, therefore, examined the impact of tyrosine phosphorylation in SAC induced HLA-DR internalization and down-modulation in monocytes. To examine the effect of tyrosine phosphorylation in SAC induced internalization of HLA-DR, monocytes were pretreated with tyrosine kinase inhibitor herbimycin A for one hour, then stimulated with or without SAC for 40 hours. Cells were fixed and permeablized, then stained with HLA-DR specific antibodies and counter-stained with nuclear dye DAPI. As shown in Fig. 4c, HLA-DR molecules were internalized in SAC treated cultures. SAC failed to induce internalization of HLA-DR in cultures pretreated with herbimycin A. Since tyrosine kinases may target either the production of IL-10, or the function and signaling of IL-10, we examined the impact of tyrosine phosphorylation on SAC induced IL-10 production from monocytes. As shown in Fig. 4d, IL-10 production after SAC stimulation was significantly reduced by tyrosine kinase inhibitors herbimycin A and genistein but not by tyrphostin, indicating that tyrosine phosphorylation is essential in SAC induced IL-10 production in human primary monocytes.
Restoration of cell surface HLA-DR expression by inhibitors of Src kinases
Src family members are protein tyrosine kinases. To determine the role of Src kinases in S. aureus induced down-modulation of HLA-DR, monocytes were treated with Src kinase inhibitor II or herbimycin A for 5 hours, then cells were further cultured for 40 hours in the presence or absence of SAC. As shown in Fig. 4e, Src kinase inhibitor II or hermbimycin A prevented SAC induced down-modulation of HLA-DR, while Src kinase inhibitors only slightly reduced HLA-DR expression directly. The results indicate that Src kinases play essential roles in S. aureus induced down-modulation of HLA-DR.
Induction of NF-κB by S. aureus
NF-κB plays an important role in inflammation and immune responses31,32,33. We, therefore, examined the effects of SAC on nuclear translocation of NF-κB in human primary monocytes. As shown in Fig. 4f, SAC was a potent inducer of NF-κB nuclear translocation. While TNF-α can induce NF-κB nuclear translocation in human primary monocytes, induction of TNF-α by SAC takes several hours (data not shown). The rapid induction and translocation of NF-κB (within 1 hour) is consistent with a direct effect of SAC signaling, rather than through induction of TNF-α.
One of the classical NF-κB pathways is mediated by p50/p65 nuclear translocation after IκB-α phosphorylation and degradation. We therefore examined whether SAC induced NF-κB complex contains p50 and p65 by antibody supershift. Supershift assays with anti-p65 and anti-p50 antibodies showed that SAC induced a NF-κB complex containing p65 and p50 subunits (Fig. 4f).
Induction of co-inhibitory molecule PD-L1 by S. aureus
The PD-L1 (CD274) and PD-1 (CD279) pathway delivers negative signals to balance T cell activation and tolerance34. To test whether microbes like S.aureus also impact negative regulatory pathways we measured the expression of PD-L1 and PD-L2 co-inhibitory molecules on monocytes after S. aureus treatment. As shown in Fig. 5a, PD-L1, but not PD-L2 expression, was significantly upregulated by S. aureus in human primary monocytes whereas HLA-DR was down-modulated. The induction of PD-L1 by S. aureus was dose dependent (Fig. 5b). High levels of PD-L1 were found at 24 hours and persisted to 96 hours (data not shown). The induction of PD-L1 by S. aureus was not affected by anti-IL-10 antibodies, indicating an IL-10 independent process of PD-L1 induction by S. aureus (Fig. 5a). IL-10 added to cultures did not upregulate PD-L1 expression, supporting the idea that PD-L1 upregulation by S. aureus in monocytes is IL-10 independent. The induction of CD40 and CD80 expression by S, aureus was enhanced by neutralizing IL-10. In contrast, the modest upregulation of PD-L2 expression by S. aureus was not affected by anti-IL-10 antibodies.

Induction of PD-L1 by SAC.
Monocytes were treated with SAC (0.75%) and stained with indicated markers. (a) Expression of HLA-DR and co-stimulatory or co-inhibitory molecules in the presence or absence of indicated reagents. The data presented are from one donor, are representive of results from three different donors. (b) Dose dependent induction of PD-L1 by SAC.
Inhibition of T cell IL-2 response by S. aureus
To study the functional impact of S. aureus induced phenotypic changes on T cell responses, mixed leukocyte reactions (MLR) were carried out with monocytes pre-treated with SAC. In these experiments, T cell IL-2 responses (Fig. 6a) were inhibited in cultures with monocytes exposed to SAC with some donor variation in absolute level of inhibition.

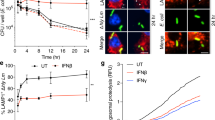
Control of T cell IL-2 response by SAC and restoration by anti-IL-10 and anti-PD-L1.
PBLs and allo-macrophages from multiple pairs of donors were cultured as described in Materials and Methods. (a) T cell IL-2 responses at 24 hours of mixed leukocyte reaction in the presence or absence of SAC as determined by specific IL-2 ELISA. (b) and (c) T cell IL-2 responses at 24 hours of mixed leukocyte reaction in the presence or absence of SAC and indicated neutralizing anti-IL-10 antibodies, or anti-PD-L1, anti-PD-L2 blocking antibodies. (d) Proposed model of microbe tolerance.
Restoring T cell IL-2 responses by anti-PD-L1 and anti-IL-10 antibodies
To study the role of PD-L1 or IL-10 in S. aureus-mediated inhibition of T cell IL-2 responses, either anti-IL-10 neutralizing or anti-PD-L1 blocking antibodies were added with SAC to cultures before MLR and IL-2 production was measured by ELISA. As shown in Fig. 6b and 6c, T cell IL-2 responses were restored by the combination of anti-IL-10 neutralizing antibodies and anti-PD-L1 blocking antibodies, although IL-2 production was enhanced to varying degrees by anti-IL-10 neutralizing antibodies or anti-PD-L1 antibodies alone. Overall, the results indicate that the combination of antagonists of IL-10 and PD-L1 could best restore T cell IL-2 responses.
Discussion
Long lasting protective immunity depends on classic T helper dependent B and T cell mechanisms35. Specific T cell responses to antigen can be regulated by at least three mechanisms: 1) The formation of TCR/antigen/MHC II complexes can be controlled by the amount of MHC II expression on APCs when sufficient TCRs and antigen are present. Factors that affect the expression of MHC II molecules will prevent full conversion of monocytes to efficient antigen presenting dendritic cells, thereby influencing the initiation of T cell responses, 2) Regulation of the expression of co-stimulatory molecules like CD86 or co-regulatory molecules like PD-L1 on APC will dictate whether T cell responses are induced or aborted/anergized and 3) production of cytokines, such as IL-2, IL-4, IL-6, IL-12 and IL-23 are required to support proliferation or differentiation of T cells after TCR-induced activation and, if limited, will abrogate T cell responses.
Here we present data showing that Staphylococcus aureus has the potential to disable host immunity at the time of infection through: 1) inhibition of MHC II/HLA-DR and CD86 expression by inducing IL-10, 2) induction of co-inhibitory molecule PD-L1 and 3) inhibition of T cell IL-2 response. We also show that HLA-DR is down-modulated by high doses of TLR2 ligand LTA from gram-positive bacteria, indicating that anti bacterial immunity might be controlled via inhibition of adaptive responses through activation of innate Toll-like receptor signaling, repression of critical stimulatory molecules and induction of inhibitory molecules.
Lack of cell surface expression of MHC class II in patients with bare lymphocyte syndrome due to genetic defects in class II transactivator CIITA or RFX regulatory factors is associated with frequent and severe infections by bacteria, viruses and fungi29, demonstrating an essential role of MHC class II molecules in protection against infection. Along these lines, several host factors have been shown to down-regulate expression of MHC class II molecules. Herein we showed that microbes, like S. aureus, are able to control key adaptive molecule MHC Class II through innate toll-like receptor 2, demonstrating a link between the innate system and the adaptive response that disables antigen presentation and may increase susceptibility to infection.
S. aureus treatment of monocytes initiates partial antigen presenting cell differentiation as seen in upregulation of CD40, CD80, CD83 together with a slight increase in CCR7. These changes could be a direct effect of TLR signaling along with feedback responses to secreted inflammatory cytokines. In the presence of reduced HLA-DR and CD86, the partial induction of APC markers and chemokine receptors that allow cells to migrate to lymphoid tissues and engage T cells, could contribute to blunting of T cell activation and partial anergy. Alternatively, upregulation of CD40 and CD80 might contribute to an enhanced T cell response after HLA-DR has been restored by anti-IL-10 antibodies.
S. aureus also inhibited T cell IL-2 response to alloantigen. A previous report showed that T cell IL-2 responses induced by superantigen SEE were inhibited within 20 hours by S. aureus and PGN36,37, suggesting that the bacteria might limit the adverse effects of a superantigen (or toxin) produced by S. aureus. The key issue to the current study is why humans have no protective immunity after S. aureus or other bacterial infections. Herein we assessed S. aureus effects on host immune responses by studying the impact of S. aureus on IL-2 production during mixed leukocyte reactions and found that T cell IL-2 responses induced by alloantigens were inhibited by S. aureus through down regulation of HLA-DR and CD86 in an IL-10 dependent manner and up-regulation of PD-L1 in an IL-10 independent fashion.
Since PD-L1 was dramatically induced before 24 hours, while HLA-DR and CD86 were not significantly downmodulated at 24 hours (data not shown), the inhibition of IL-2 by S. aureus could happen at (1) an early phase mediated by induction of PD-L1 and at (2) a late phase mediated by IL-10 dependent downmodulation of HLA-DR and CD86 expression, also accompanied by persistent high level expression of PD-L1. Together, the data suggest that T cell IL-2 responses are affected by S. aureus through modulation of monocyte/macrophage APC function.
When monocytes encounter antigens during infection or after immunization they may become dendritic cells, the most potent professional APCs, expressing high levels of HLA-DR (class II) and other co-stimulatory molecules (Fig. 6d). After interacting with these activated APCs, strong T cell responses promote both humoral and cellular immunity that often lead to immune protection. However, when monocytes/macrophages are targeted by microbes (especially bacteria and some IL-10 producing/inducing viruses), such as Staphylococcus aureus, which cause recurrent infections, high levels of IL-10 as well as Th17 promoting IL-6 and IL-23, but not IL-2 may be produced. High levels of IL-10 might inhibit the innate immune response by decreasing the release of inflammatory cytokines and simultaneously inhibiting antigen presentation by down-modulating MHC class II molecules and co-stimulatory molecules, such as CD86. The strong induction of PD-L1 by S.aureus as shown herein together with the data of HLA-DR and CD86 regulation by IL-10 demonstrate a multi-dimensional control of adaptive response that contributes to immune-suppression and lack of protective adaptive immune responses during and after infection.
PD-L1 was induced in monocytes by S. aureus, but the mechanism has not been determined, except that anti-IL-10 does not block induction. Although induction of PD-L1 in primary monocytes by LPS has not been reported, a study found that PD-L1 induction by LPS required both IL-6 and IL-10, in monocyte-derived immature DCs treated with GM-CSF and IL-438. We observed PD-L1 induction in monocytes by LPS (data not shown), but not by IL-10 directly. In monocytes, PD-L1 induction by S. aureus may result directly from TLR activation or require additional cytokine feedback independent of IL-10. Together these findings suggest that cell differentiation plays a role in antigen presenting cell susceptibility to direct and feedback signals. As a result of negative regulatory pathways, immunity to bacterial pathogens could be aborted for short or long-term due to the inability of microbe-exposed macrophages, to effectively stimulate T cells resulting in failure to establish pathogen specific immunity. Cells with defective APC function could also contribute to tolerance induction. Effective preventative vaccines may need to abrogate the ability of these organisms to induce inhibitory molecules like IL-10 and PD-L1 in order to avoid the negative effects these molecules exert on the development of a protective adaptive immune response.
S. aureus induced not only pro-inflammatory cytokines, but also cytokines implicated in Th17 cell differentiation. Th17 T cells are controlled by IL-6, TGF-β and IL-23 in mice and IL-6, IL-23 and IL-1 in humans39,40,41,42. S. aureus induces IL-17A in mice43. Here we show that S. aureus induced IL-6 and IL-23 directly from human primary monocytes. Therefore, S. aureus might promote the development of naïve T cells into IL-17 secreting Th17 cells in humans as well. However, the exact role of induction of IL-6 and IL-23 and Th17 responses during S. aureus infection has yet to be determined and we do not know if Th17 responses are protective or if they interfere with protection during infection.
In contrast, S. aureus failed to induce IL-12 directly, suggesting an initial bias away from Th1 differentiation. In the presence of IFN-γ, IL-12 is induced by S. aureus in monocytes/macrophages while IL10 is suppressed, leading to expression of high levels of MHC class II and co-stimulatory molecules. This would support intense APC-T cell interaction and Th1 differentiation. Adjuvants that support Th1 cell differentiation in combination with inhibitors of IL-10 and PD-L1 might enhance immunity to S. aureus.
Herein we also show that tyrosine phosphorylation is essential for the induction of IL-10 by S. aureus, which signals through TLR2, as shown by blocking S. aureus induced IL-10 secretion with tyrosine kinase inhibitors. The restoration of HLA-DR expression in S. aureus stimulated cultures by tyrosine kinase inhibitors might primarily act to prevent IL-10 production. Interfering with phosphorylation of STAT3 induced by IL-10, or preventing S. aureus induced HLA-DR internalization or exocytosis might be other possibilities. Src kinase inhibitor restored HLA-DR expression, indicating the essential role of Src family kinases in the down-modulation of HLA-DR by S. aureus.
TLR signaling is complex8. Interestingly, studies in TLR2 and MyD88 knockout mice demonstrated that TLR2 and MyD88 were protective in S. aureus induced death4. Likewise, clinical data from patients with MyD88 or IRAK4 deficiency indicate that TLR2 signaling was important in protecting individuals from severe bacterial infections, including S. aureus44,45. LTA and PGN, the major cell wall components of S. aureus, are known to signal via TLR2. Down-modulation of HLA-DR by TLR2 ligand LTA and blocking HLA-DR down-modulation by anti-TLR2, but not anti-TLR4, antibodies demonstrate that TLR2 is essential in S. aureus mediated inhibiton of HLA-DR in human monocytes. Our data also indicate that anti-TLR2 antibodies have the potential to be used as therapeutics during gram-positive bacteria infection in combination with proper antibiotics.
It is evident that S. aureus and possibly other microbes (Streptococcus faecalis, Salmonella typhosa, Pseudomonas aeruginosa, Klebsiella pneumoniae, Salmonella abortus equi, Mycobacterium tuberculosis, etc) that cause recurrent or persistent human infection may interfere not only with innate responses, but also with the adaptive T cell immune responses through (1) IL-10 dependent down-modulation of HLA-DR and CD86, (2) induction of PD-L1 and (3) inhibition of T cell IL-2 response. Thereby, both humoral and cell-mediated immune responses may be affected, leading to a sepsis-induced immunosuppressive state and microbe-tolerance in which fully protective immunity might not develop. Controlling microbe-induced immune suppressive factors including IL-10 and PD-L1, may have profound clinical value, especially during vaccinations, although the enhancement of inflammatory responses might be a concern for the therapeutic value during infection. In combination with adjuvants to support Th1 differentiation and IFN-γ production, it may provide mechanisms for inducing protective immunity to prevent infections and prevent microbe tolerance.
Methods
Monocytes and cell culture
Human monocytes were isolated by counter-current centrifugal elutriation of single-donor peripheral blood leukocyte preparations, as described previously24, from leuko-packs received from NIH Department of Transfusion Medicine under an approved protocol for experimental use and volunteer consent. These studies were approved by DTP/OBP/CDER under approved projects. Monocytes were cultured in macrophage-serum free medium (Invitrogen, Carlsbad, CA) and seeded into polystyrene tissue culture 24 well-plates (Costar, Cambridge, Mass.) with the following reagents respectively: Staphylococcus aureus cells (SAC; Cowan I strain, heat-inactivated, from Calbiochem Corporation, La Jolla, CA), LTA (lipoteichoic acid from S. aureus and Streptococcus faecalis, Sigma), LPS (lipopolysaccharide from Escherichia coli K235; List Biological Laboratories, Inc., Campbell, CA.), LPS (from Salmonella typhosa, Pseudomonas aeruginosa, Klebsiella pneumoniae, and Salmonella abortus equi, 5 μg/ml, Sigma), IFN-γ(Genentech), anti-human IL-10 neutralizing antibody (10 μg/ml, Invitrogen), anti-human TNF-α neutralizing antibody (10 μg/ml, Endogen, Inc., Cambridge, MA), mouse IgG1 and IgG2a (eBioscience), herbimycin A, genistein, Src kinase inhibitor II (Calbiochem).
Cell surface immunofluorescence staining
Monocytes/macrophages were collected by scraping and centrifuging, then washed with ice-cold PBS and pre-incubated with 1:10 diluted normal rabbit serum (Invitrogen) on ice for 30 minutes. 5−10 X 105 cells were incubated with indicated antibodies on ice for one hour and washed with ice-cold PBS three times then fixed with 1% paraformaldehyde in 1X PBS. The expression of cell surface antigens was assayed by immunofluorescence flow cytometry with a FACScan flow cytometer (Becton-Dickinson, Sunnyvale, CA) and analyzed with FACScan and CellQuest software (Becton Dickinson) or FlowJo software. Monocytes were labeled with direct-labeled antibodies (according to manufacture’s suggestion) including: anti-HLA-DR, anti-HLA-ABC, anti-CD86 (clone IT2.2), anti-PD-L1, anti-PD-L2, anti-CD40, anti-CD80, anti-CD83, anti-CD36, anti-CD14, anti-CCR7 (Pharmingen, San Diego, CA), anti-TLR2, anti-TLR4 (eBiosciences), or isotype controls (Sigma).
Confocal scanning microscope
For assessment of endocytosis, cells on coverslips were fixed and permeablized, then stained with indicated FITC-labeled antibodies. Cells were counter-stained with DAPI (Excitation 358 nm, emission 461 nm, Molecular Probe) solution for 5 minutes. After washing with PBS, cells were subjected to confocal microscope analysis and images collected using a x63 oil objective and Zeiss LSM 510 confocal microscope equipped with a META spectral scanning head.
Cytokine secretion measured by ELISA
Monocytes (2 x 106 cells/ml/well) were cultured in medium alone, or primed with IFN-γ (100 ng/ml) for 16 hours, then stimulated by SAC or LPS for 24 hours. Supernatant from cultures following stimulation were collected and assayed for the indicated cytokines by ELISA. The cytokine-specific ELISAs were: IL-12 (p70), R&D system (Minneapolis, MN), IL-10, TNF-α, Biosource (Camarillo, CA). The detection limits of cytokine-specific ELISA were: IL-12, 5.0 pg/ml; IL-10, 5.0 pg/ml; and TNF-α, 1.0 pg/ml.
Reverse transcriptase-PCR analysis
Total cellular RNA of human monocytes was isolated using Trizol reagent and then treated with DNase I. Equal amounts of total RNA were subject to first-strand cDNA synthesis using RNase minus Superscript II reverse transcriptase (Life Technologies, Rockville, MD). cDNA was amplified by 30 cycles of PCR with human specific primers: IL23p19sense TTCACAGAAGCTCTGCACACT, IL23p19 antisense AACATCATTT GTAGTCTCTTCATCTC, IL10 sense CTGCCTAACATGCTTCGAGA, IL10 antisense GTTGTCCAGCTGATCCTTCAT, IL12/IL23 p40 sense GTCCAGGGCAAGAGCAAGA, antisense GCAGATGACCGTGGCTGA; IL12p35 sense GGCTCAGCATGTGTCCAG, antisense CAGGTTTTGGGAGTGGTGAA; IL-8 sense AACTGAGAGTGATTGAGAGTGGA, antisense CTCTTCAAAAACTTCTCCACAAC; IL-6 sense TGCTTCCAATCTGGATTCAAT GA, antisense CTCTGGCTTGTTCCTCACTACTC; CCL4 sense TGCTCTCCAGCGCTCTCA, antisense AGTAATTCTACCACAAAGTTGCGA; GAPDH sense CTCCTGCACCACCAACT GCT, antisense TGGCAGTGATGGCATGGACT; and actin sense TGCAAGGCCGGCTTCGC GGGCGACGA, antisense AATCCTTCTGACCCATGCCCACCAT. PCR products were separated and stained with SYBR Green 1 (Molecular Probes, Eugene, OR) and analyzed with a FluorImager scanning densitometer and ImageQuant software (Amersham Pharmacia Biotech).
Nuclear extracts and electrophoretic mobility shift assay (EMSA)
Monocytes were stimulated with the indicated reagents for 1 hour. Nuclear extracts from stimulated monocytes were prepared as described previously and 5 μg of nuclear extracts were added directly to DNA-protein binding reactions (10 mM Tris (pH 7.5), 1 mM EDTA, 1 mM DTT, 2 mg poly(dI:dC), 10% glycerol and 0.5 ng32P-labeled NF-κB oligonucleotides and kept at room temperature for 10 min. Reaction products were evaluated on 6% non-denaturing polyacrylamide gels with 0.53 Tris-borate-EDTA buffer at room temperature. Gels were dried and exposed to x-ray film at −70°C.
Cytokine neutralization assays
Indicated neutralizing antibodies were added at the beginning of cultures at concentration of 30 μg/ml. Monocytes in the presence or absence of neutralizing antibodies were stimulated with SAC. After 40 hours cells were stained for indicated markers and subject to flow cytometry analysis.
T cell IL-2 response
PBLs (1 x 106 cells/ml of 10% FBS-RPMI 1640) were added to monocytes (5 x 105/0.5 ml in macrophage-serum free medium with M-CSF, data shown in Fig. 6a or in 10% FBS-RPMI 1640, data shown in Fig. 6b and 6c) pretreated with SAC, with/without anti-IL-10 neutralizing antibodies (3 μg/ml) for 40 hours. Anti-PD-L1, or anti-PD-L2 blocking antibodies (3 μg/ml) were added to cultures one hour at room temperature prior to the addition of PBLs. Supernatants from 24 hour cultures were assayed for IL-2 production by ELISA (BD Biosciences).
References
Lowy, F. D. Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532 (1998).
David, M. Z., Mennella, C., Mansour, M., Boyle-Vavra, S. & Daum, R. S. Predominance of methicillin-resistant Staphylococcus aureus among pathogens causing skin and soft tissue infections in a large urban jail: risk factors and recurrence rates. J Clin Microbiol. 46, 3222–3227 (2008).
Takeuchi, O. et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 11, 443–451 (1999).
Takeuchi, O., Hoshino, K. & Akira, S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 165, 5392–5396 (2000).
Kawai, T., Adachi, O., Ogawa, T., Takeda, K. & Akira, S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 11, 115–122 (1999).
Leendertse, M. et al. TLR2-dependent MyD88 signaling contributes to early host defense in murine Enterococcus faecium peritonitis. J. Immunol. 180, 4865–4874 (2008).
Santos-Sierra, S. et al. Mal connects TLR2 to PI3Kinase activation and phagocyte polarization. EMBO J. 28, 2018–2027 (2009).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 (2010).
Moran, G. J. et al. Methicillin-resistant S. aureus infections among patients in the emergency department. N. Engl. J. Med. 355, 666–674 (2006).
Grundmann, H., Aires-de-Sousa, M., Boyce, J. & Tiemersma, E. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet. 368, 874–885 (2006).
Klevens, R. M. et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 298, 1763–1771 (2007).
Castaldo, E. T. & Yang, E. Y. Severe sepsis attributable to community-associated methicillin-resistant Staphylococcus aureus: an emerging fatal problem. Am. Surg. 73, 684–687 (2007).
Miller, L. S. & Cho, J. S. Immunity against Staphylococcus aureus cutaneous infections. Nat. Rev. Immunol. 10, (2011).
Foster, T. J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 3, 948–958 (2005).
Houghton, A. M., Hartzell, W. O., Robbins, C. S., Gomis-Ruth, F. X. & Shapiro, S. D. Macrophage elastase kills bacteria within murine macrophages. Nature. 460, 637–641 (2009).
Docke, W. D. et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat. Med. 3, 678–681 (1997).
Docke, W. D. & Volk, H. D. Impaired antigen presentation by human monocytes during endotoxin tolerance. Blood. 96, 218–223 (2000).
Lekkou, A., Karakantza, M., Mouzaki, A., Kalfarentzos, F. & Gogos, C. A. Cytokine production and monocyte HLA-DR expression as predictors of outcome for patients with community-acquired severe infections. Clin. Diagn. Lab Immunol. 11, 161–167 (2004).
Monneret, G. et al. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med. 32, 1175–1183 (2006).
Fadok, V. A., Warner, M. L., Bratton, D. L. & Henson, P. M. CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (alpha v beta 3). J. Immunol. 161, 6250–6257 (1998).
Greenberg, M. E. et al. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 203, 2613–2625 (2006).
Nilsen, N. J. et al. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J. Leukoc. Biol. 84, 280–291 (2008).
Wang, J. E. et al. Peptidoglycan and lipoteichoic acid from Staphylococcus aureus induce tumor necrosis factor alpha, interleukin 6 (IL-6) and IL-10 production in both T cells and monocytes in a human whole blood model. Infect. Immun. 68, 3965–3970 (2000).
Hayes, M. P., Wang, J. & Norcross, M. A. Regulation of interleukin-12 expression in human monocytes: selective priming by interferon-gamma of lipopolysaccharide-inducible p35 and p40 genes. Blood. 86, 646–650 (1995).
Boniface, K., Blom, B., Liu, Y. J. & de Waal, M. R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol. Rev. 226, 132–146 (2008).
Corinti, S., Albanesi, C., la, S. A., Pastore, S. & Girolomoni, G. Regulatory activity of autocrine IL-10 on dendritic cell functions. J Immunol. 166, 4312–4318 (2001).
Abe, R. et al. Up-regulation of interleukin-10 mRNA expression in peripheral leukocytes predicts poor outcome and diminished human leukocyte antigen-DR expression on monocytes in septic patients. J Surg. Res. 147, 1–8 (2008).
Fumeaux, T. & Pugin, J. Role of interleukin-10 in the intracellular sequestration of human leukocyte antigen-DR in monocytes during septic shock. Am. J. Respir. Crit Care Med. 166, 1475–1482 (2002).
Reith, W. & Mach, B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu. Rev Immunol. 19, 331–373 (2001).
Wang, J., Roderiquez, G., Jones, T., McPhie, P. & Norcross, M. A. Control of in vitro immune responses by regulatory oligodeoxynucleotides through inhibition of pIII promoter directed expression of MHC class II transactivator in human primary monocytes. J. Immunol. 179, 45–52 (2007).
Hoffmann, A. & Baltimore, D. Circuitry of nuclear factor kappaB signaling. Immunol. Rev. 210, 171–186 (2006).
Guan, E. et al. T cell leukemia-associated human Notch/translocation-associated Notch homologue has I kappa B-like activity and physically interacts with nuclear factor-kappa B proteins in T cells. J. Exp. Med. 183, 2025–2032 (1996).
Wang, J. et al. Human Notch-1 inhibits NF-kappa B activity in the nucleus through a direct interaction involving a novel domain. J. Immunol. 167, 289–295 (2001).
Keir, M. E., Butte, M. J., Freeman, G. J. & Sharpe, A. H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704 (2008).
Withers, D. R. et al. T cell-dependent survival of CD20+ and CD20- plasma cells in human secondary lymphoid tissue. Blood. 109, 4856–4864 (2007).
Chau, T. A. et al. Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nat. Med. 15, 641–648 (2009).
Frodermann, V. et al. A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus. J Infect. Dis. 204, 253–262 (2011).
Wolfle, S. J. et al. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur. J Immunol. 41, 413–424 (2011).
McGeachy, M. J. & Cua, D. J. Th17 cell differentiation: the long and winding road. Immunity. 28, 445–453 (2008).
Annunziato, F. & Romagnani, S. Do studies in humans better depict Th17 cells? Blood. (2009).
Lyakh, L., Trinchieri, G., Provezza, L., Carra, G. & Gerosa, F. Regulation of interleukin-12/interleukin-23 production and the T-helper 17 response in humans. Immunol. Rev. 226, 112–131 (2008).
Ivanov, I. I., Zhou, L. & Littman, D. R. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 19, 409–417 (2007).
Kudva, A. et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. 186, 1666–1674 (2011).
Picard, C. et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore). 89, 403–425 (2010).
von, B. H. et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 321, 691–696 (2008).
Acknowledgements
The authors are grateful to Drs. Kathleen Clouse,-Strebel, Gerald Feldman, Jennifer Swisher, Gunther Boekhoudt, Mark Paciga, Sylvia Bacot and Ms. Linda Tiffany and Jun Gao for elutriation of human monocytes and PBLs and to Dr. Judy Beeler, Dr. Daniela Verthelyi and Dr. Amy Rosenberg for critical readings of the manuscript.
Author information
Authors and Affiliations
Contributions
JW - Study design, performed experiments, analysis of results, preparation of the manuscript; MN - Study design, analysis of results, preparation of the manuscript; GR – performed experiments.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Wang, J., Roderiquez, G. & Norcross, M. Control of Adaptive Immune Responses by Staphylococcus aureus through IL-10, PD-L1 and TLR2. Sci Rep 2, 606 (2012). https://doi.org/10.1038/srep00606
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00606
This article is cited by
-
The induction of natural competence adapts staphylococcal metabolism to infection
Nature Communications (2022)
-
Complement C5a induces PD-L1 expression and acts in synergy with LPS through Erk1/2 and JNK signaling pathways
Scientific Reports (2016)
-
Cytokine profile in severe gram-positive and gram-negative abdominal sepsis
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
