
Abstract
Experimental evidences are presented showing unusually large and highly anisotropic vibrations in the “simple cubic” (SC) unit cell adopted by calcium over a broad pressure ranging from 30–90 GPa and at temperature as low as 40 K. X-ray diffraction patterns show a preferential broadening of the (110) Bragg reflection indicating that the atomic displacements are not isotropic but restricted to the [110] plane. The unusual observation can be rationalized invoking a simple chemical perspective. As the result of pressure-induced s → d transition, Ca atoms situated in the octahedral environment of the simple cubic structure are subjected to Jahn-Teller distortions. First-principles molecular dynamics calculations confirm this suggestion and show that the distortion is of dynamical nature as the cubic unit cell undergoes large amplitude tetragonal fluctuations. The present results show that, even under extreme compression, the atomic configuration is highly fluxional as it constantly changes.
Similar content being viewed by others


Introduction
High-density polymorphs of calcium show structural and electronic properties dissimilar to those of other elemental solids. Under compression at room temperature, Ca transforms successively, from the face-centered cubic structure (FCC, phase I) to body-centered cubic structure (BCC, phase II) at 19 GPa1,2 and, with further compression, to a simple cubic structure (SC, phase III) at 32 GPa3. Three more phases, beyond that of the SC phase stability field, were recently characterized from 100 to 300 GPa4. Unlike alkaline Group II Sr and Ba5,6, an incommensurate phase in Ca was only found very recently at a pressure in excess of 200 GPa4,7. Moreover, the SC structure, a rare occurrence in metallic elements, was found to be remarkably stable over a large pressure range from 32 to 100 GPa6. Density functional calculations employing generalized gradient approximation (GGA) have been performed in order to understand the stability of the SC phase8,9,10,11,12,13. It was concluded that the SC structure is dynamically unstable8 and, instead, a tetragonal I41/amd structure9,10,11,12 was found to be substantially lower in energy. More elaborate meta-dynamics9 and molecular dynamics (MD)11 calculations suggested the SC phase could be thermally stabilized, although the technical validity of the calculations has been raised10. Nevertheless, experiments by Gu et al.14 showed that at room temperature, even after sample annealing, the powder x-ray diffraction patterns could be indexed to an SC structure, irrevocably. In contrast, low temperature x-ray diffraction experiments have led to the conclusion of the existence of an orthorhombic Cmmm structure near 40 GPa at 4K2,15. Density functional GGA calculations showed that the latter structure is dynamically unstable and presents a higher enthalpy than that of the I41/amd structure13. Diffusion Quantum Monte Carlo calculations at GGA-optimized geometries, however, suggested that the relative stability of I41/amd and Cmmm phases at 50 GPa may be reversed but at low temperature the SC phase would still have a higher enthalpy13. A recent theoretical study16 indicated that anharmonic vibrations might help to stabilize the SC structure16. However, in spite of extensive theoretical and experiment efforts, no satisfactory explanation on the electronic origin of the very flat and anharmonic potential energy surface and, equally significant, the reason why the enthalpy most stable I41/amd phase has not observed are forthcoming. The discrepancy between theory and experiment is resolved and presented in the present work. Present results on low-temperature high-pressure x-ray diffraction experiments and first principles MD calculations suggest the s → d hybridization of Ca leads to a dynamical lattice distortion of the SC structure rooted in an electronic instability.
To resolve this problem, the pressure-temperature phase diagram of Ca was determined near the bcc → sc transition. Powder x-ray diffraction experiments were performed using He as the pressure transmitting medium. Pure Ca samples (Sigma-Aldrich) were loaded in an inert gas atmosphere. Experiments were carried out at beamline BL10XU at SPring-8 using 0.4137Å radiation, focused at the sample by a compound refractive lens17. Quasi-hydrostatic pressures were obtained from the calibrated spectral shifts of Al2O3:Cr3+ luminescence and first-order Raman line of the diamond (anvil), measured and corrected at each temperature. Pressure and temperature uncertainties were 0.1 GPa and 1 K, respectively. Several pressure-temperature paths were followed to explore systematically the phase diagram with pressures ranging from 30–50 GPa and while varying the temperature from ambient to 14 K.
Results
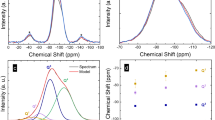
The FCC → BCC transition was observed between 19.3 and 20.4 GPa at room temperature, in agreement with the reported value1. Then the Ca sample was cooled to 14K and further compressed up to 53 GPa. The onset of a structural transition of the BCC phase occurs at 39 GPa, which is much higher than the 32 GPa observed under ambient temperature2. In a narrow pressure range above 40 GPa and at 14 K, a series of structural transformations indicated by changes in x-ray diffraction patterns were observed suggesting the occurrence of possibly more than one intermediate phases (Fig. 1). At pressures above 42 GPa, the x-ray diffraction patterns became simpler (Fig. 1) and indexed to a SC unit cell. Hence at low temperature, Ca does not transform directly from the BCC to SC structure, but goes through a sequence of intermediate structural configurations. A distinct feature of the x-ray diffraction patterns is the full width at half maximum (FWHM) of the (110) Bragg reflection which is significantly larger than both the (100) and (200) reflections (Fig. 1). The observed line broadening cannot be attributed to non-hydrostatic conditions, as the width of diffraction lines corresponding to similar d-spacings, measured at 14 K and 300 K and around 40 GPa, differ marginally, indicating that He is still a good quasi-hydrostatic pressure transmitting medium. Also there is no evidence that the unusually large width of the (110) line is due to a splitting of the Bragg peak resulting from a symmetry change. A sequence of x-ray diffraction patterns was also recorded by warming the sample from 14 to 100 K, at around 42 GPa. No strong temperature dependence on the FWHM of the (100), (110) and (200) reflections was observed. In fact, the (110) line width reduced slightly from 0.67° to 0.64° (FWHM) with an increase of temperature from 14 to 100 K at 42 GPa, as measured to ± 0.01° in our experiments. In comparison, the FWHM for the (100) and (200) reflections remained to be 0.10° and 0.18°, respectively. Normally, the width of a Bragg reflection increases with the diffracted angle18. However, the exceptionally large line width and the fact that the (110) Bragg peak diffracts at a lower angle than the (200) reflection, rule out this explanation. The anomalous preferential broadening in the (hh0) Bragg reflections corroborates the fact that the Ca atoms in the [110] lattice plane are indeed disordered. Other possible effects like deviatoric stresses are not ruled out but considered unlikely given the peculiar line broadening has been observed repeatedly for pressures below 43 GPa and above 39 GPa at very low temperature (14 K) and not in other phases of Ca, namely phases II and III, which exist under close temperature and pressure conditions. Another usual observation is the line width of the (110) Bragg peak apparently decreases slightly with increasing temperature (Fig. 1). This anomaly will be discussed below (vide infra).

(a) Selected x-ray diffraction patterns of Ca recorded at specified (P, T) conditions. (b) The measured line width for the (100), (110) and (210) Bragg reflections of the SC phase. (c) Variation of the unit cell with temperature at 42 GPa.
The simple cubic unit cell parameters as a function of temperature along the 42 GPa isobar are calculated using the measured (100), (110) and (200) reflections. The results presented in Fig. 1 show a negative thermal expansion from 40 to 100 K. A negative thermal expansion is indicative of large anharmonicity in lattice vibrations19. Moreover, at temperatures above 80 K and 42 GPa, the estimated coefficient of linear thermal expansion of Ca of 2.1×10−4/K is an order of magnitude larger than that of the FCC phase at ambient pressure (2.23×10−5/K)20.
The peculiar behaviour of Ca at low temperature and high pressure gives rise to a different and confined field in the phase diagram, nested between that of phases II and III. This new stability field in the phase diagram is identified as that of phase III'. A schematic diagram for Ca is presented in Fig. 2. Within the phase III' field, at the lowest temperature reached in our experiment (14 K), a sequence of changes in the x-ray diffraction patterns was observed. None of the x-ray diffraction patterns could be successfully fitted to the predicted I41/amd structure, a solution which has been definitely ruled out. The Cmmm unit cell identified earlier2,15 as the best description of the Ca at 44 GPa and 4 K (close to the phase III-III' boundary) could not be confirmed as an unique and non-ambiguous structural solution to our x-ray diffraction results. It is plausible that small residual stress could help to stabilize the intermediate structures and the discrepancy from previous work could be due to the quasi-hydrostatic conditions.

The schematic phase diagram of high pressure Ca.
Phase boundaries are the results of the many temperature and pressure paths explored in this study.
Discussion
Why the structural distortion only occur in the [110] plane of the SC structure? This peculiar phenomenon can be explained from a simple crystal field picture. When compressed, alkali metals often transform between simple close packed structures5. At higher density, the nearest-neighbour atoms form confining potential localizing empty orbitals of higher angular momentum and then mixed with the valence state21. The s → d transition in Cs, first recognized by Sternheimer22, provided a good example of hybridization. In alkali and alkaline earth metals with low-lying d-orbitals the full realization of the s → d transition is usually preceded by an intermediate pressure regime whereby unusually complex commensurate or incommensurate structures are observed23. A common feature of these high-density structures is their tendency to adopt planar packing of atoms5,21,23,24. The participation of d-orbitals results in the localization of electron density in the interstitial region. This point of view, although empirical, has helped to rationalize the structural features, transformation sequences and charge distribution in Rb and Cs21.
A previous calculation indicated that the d-character of the valence band increases substantially accompanying the BCC → SC structural transformation in Ca11. Therefore, it is reasonable to assume that the promotion of the 4s electron into the 3d orbital of Ca is the cause for the transformation. Since each Ca atom in the SC unit cell is surrounded by an octahedral arrangement of nearest neighbours, under the crystal field25 of the surrounding atoms, the d-orbital manifold splits into triply degenerate t2g and doubly degenerate eg sets of orbitals. As there are at most two valence 4s electrons, the promotion of an electron from 4s to 3d (4s2 → 4s3d) gives rise to a 2D state. Double excitation (4s2 → 3d2) will result in an 3F state. Both electronic states are orbitally degenerated and, according to the Jahn-Teller (JT) theorem26, a lower-energy structure can result from the distortion of the local octahedral environment to a lower symmetry. The lattice distortion, however, can be either static (permanent) or dynamic (fluctuating)26. Consider the case of a tetragonal distortion for which the Ca atoms in the axial positions move further away from the center of the octahedron; in that case, the schematic energy level diagram (Fig. 3) shows that the energy degeneracy of the t2g set of orbitals is lifted and the resulting degenerate 3dxz and 3dyz orbitals then have the lowest energy. Thus, as these orbitals were filled, the electron-electron interactions are dominated by the equatorial Ca atoms located in the plane. To test this hypothesis, the electron density of a two-dimensional (2D) square net of Ca was calculated. Indeed, it was found that the electrons do occupy the Ca 3dxz and 3dyz orbitals and the net charge density is localized above and below the plane of the atoms (Fig. 3). As an extension to the tri-dimensional case11, the electron density maximum in the center of the cube can be obtained from the stacking of these 2D networks in a proper way. It is worth noting that the structure27 and electron topology of tetragonal Cs IV28 can be rationalized as the result of an s → d transition followed by a static JT distortion,

A schematic representation of possible Jahn-Teller distortion of Ca in the SC structure under the effect of the crystal field (for explanation, see text).
The JT theorem26 does not provide guidance per se as whether the lattice distortion is either static or dynamic. To this end, constant-pressure/constant temperature (NPT) ab initio MD simulations using the pseudopotential planewave method29 were carried out at several temperatures for the pressure of interest, namely, 42 GPa. A common observation arose from the calculations: the time-averaged structure remains cubic however significantly large lattice distortions are observed in the time domain. A representative trajectory obtained at 140 K is shown in Fig. 4: in this case, the 2×2×2 simulation cell fluctuates around the SC structure with an average cell length of 5.2 Å. Several lattice distortion events indicated by arrows in Fig. 4 were captured in during the course of the simulation. Note that for these events, the cell axes and angles did not distorted randomly. In all cases, when one of the cell angles deviated from 90° to 110°, a concomitant elongation of two cell axes to 5.4 Å was recorded. It is conceivable that on time average, all cell axes and angles are sampled equally and the resulting cell maintains indeed the cubic symmetry. We note that, as a significant result, the lattice distorts with the approximate cell parameters of a = b = 5.4Å, c = 5.1Å and α = β = 90°, γ = 110° which, incidentally, are close to the cell parameters of the predicted I41/amd structure8. The potential energy surface of the JT distortion is known to resemble a Mexican hat25, thus, inter-conversion dynamics as observed in the MD trajectories, sampled the potential energy surface without accessing the higher energy SC structure. The consequence of the dynamical fluctuations in dense Ca is that the cubic unit cell will present significant disorder in the [110] plane. As the atomic configuration is constantly changing, it is considered as highly fluxional. In agreement with previous ab initio studies9,10,11,12 the simple cubic structure is unstable at 0 K. Finite temperature lattice dynamics calculations, however, show the anharmonicity is reduced with increasing temperature and the simple cubic structure becomes dynamically stable at temperature higher than 200 K (Fig. 5). The observed small narrowing on the width of the (110) Bragg peak is consistent with the theoretical results.

Temporal variation of the simulated cell angle (a) and lattice parameter (b).
The changes in the cell dimensions can be interpreted as dynamic fluctuations of a cubic unit cell in the [110] plane. (c), (d) and (e) illustrates the “fluxional” distortions in the ab plane of the simple cubic cell. In a simple cubic cell, all the three crystal axes are formally equivalent. In the molecular dynamics simulation, one can labelled the axes as x, y and z (4c). The figures show, schematically, that at a given instance, pair of the crystal axes, i.e. either the (x, y) or (x,z) axes are distorted simultaneously as expected for dynamical Jahn-Teller distortion of a cubic system.

Phonon band structure of the simple cubic structure at 0, 150, 175 and 200 K calculated with self-consistent ab initio lattice dynamics method.
In summary, experimental and theoretical results show unambiguously that the SC structure of Ca above 39 GPa is an "average" structure, presenting dynamic lattice distortions. There is no need to hypothesize that the SC structure should be energetically competitive to the predicted I41/amd structure at high temperatures9,10,11,12,13. In fact, the energetically less favourable SC structure should not be sampled in the lattice fluctuations. At very low temperature from 39–42 GPa, due to structural fluctuations and local stress on the crystalline environment, it is conceivable that a plethora of crystal structures of lower symmetry than the SC structure can be stabilized and observed2,15. The results presented although confirm the theoretical prediction16,30 that the motions of Ca in phase III' are rather facile, however, the nature of structural distortion is different. In addition, an intuitive chemical principle was provided to explain on the origin of the large anharmonicity30. This insight should be applicable to the interpretation of the structure and dynamics of elemental solids under high pressure.
Methods
Electronic calculations of the charge densities were performed with the VASP code31,32using the projected augmented potential33 for Ca with semi-core 3s and 3p orbitals treated as valence states. Constant pressure-constant volume molecular dynamics calculations were performed with the Quantum Espresso code (www.pwscf.org)34 using the pseudopotential reported in ref. 24 in which the semi-core electrons 3s and 3p together with those in the 4s orbital were treated as valence in the construction of the pseudopotential. A 2×2×2 supercell model was employed for the calculation. The Brillouin zone was sampled with a 4×4×4 k-point mesh.
Phonon dispersion curves were calculated at 0, 100, 150 and 200 K using the self-consistent ab initio lattice dynamics method35. Anharmonicity in the force constants are considered explicitly in the finite temperature simulation. The phonon calculations employed a 4×4×4 supercell with a 4×4×4 k-point mesh.
References
Jayaraman, A., Klement, Jr, W. & Kennedy, G. C. Phase Diagrams of Calcium and Strontium at High Pressures. Phys. Rev. 132, 1620–1624 (1963).
Olijnyk, H. & Holzapfel, W. B. Phase transitions in alkaline earth metals under pressure. Phys. Lett. 100A, 191–194 (1984).
Nakamoto, Y., Sakata, M., Shimizu, K., Fujihisa, H., Matsuoka, T., Ohishi, Y. & Kikegawa, T. Ca-VI: A high-pressure phase of calcium above 158 GPa. Phys. Rev. B. 81, 140106(R) (2010).
Fujihisa, H., Nakamoto, Y., Shimizu, K., Yabuuchi, Y. & Gotoh, Y. Crystal Structures of Calcium IV and V under High Pressure. Phys. Rev. Lett. 101, 095503 (2008).
Nelmes, R. J., Allan, D. R., McMahon, M. I. & Belmonte, S. A. Self-Hosting Incommensurate Structure of Barium IV. Phys. Rev. Lett. 83, 4081–4084 (1999).
McMahon, M. I. & Nelmes, R. J. Incommensurate crystal structures in the elements at high pressure. Z. Kristall. 219, 742–748 (2004).
Yabunchi, T., Matsuoka, T., Nakamoto, Y. & Shimizu, K. Superconductivity of Ca Exceeding 25 K at Megabar Pressures. J. Phys. Soc. Japan. 75, 083703 (2006).
Gao, G., Xie, Y., Cui, T., Ma, Y., Zhang, L. & Zou, G. Electronic structures, lattice dynamics and electron-phonon coupling of simple cubic Ca under pressure. Solid State, Commun. 146, 181–186 (2008).
Yao, Y., Klug, D. D., Sun, J. & Martoňák, R. Structural Prediction and Phase Transformation Mechanisms in Calcium at High Pressure. Phys. Rev. Lett. 103, 055503 (2009).
Teweldeberhan, A. M. & Bonev, S. A. Comment On “Structural Prediction and Phase Transformation Mechanisms in Calcium at High Pressure”. Phys. Rev. Lett. 104, 209601 (2010).
Yao, Y., Martoňák, R., Patchkovskii, S. & Klug, D. D. Stability of simple cubic calcium at high pressure: A first-principles study. Phys. Rev. B 82, 094107 (2010).
Oganov, A. R., Ma, Y., Xu, Y., Errea, I., Bergara, A. & Lyakhov, A. O. Exotic behavior and crystal structures of calcium under pressure. Proc. Natl. Acad. Sci. U.S.A. 107, 7646–7651 (2010).
Teweldeberhan, A. M., Dubois, J. L. & Bonev, S. A. High-Pressure Phases of Calcium: Density-Functional Theory and Diffusion Quantum Monte Carlo Approach, Phys. Rev. Lett. 105, 235503 (2010).
Gu, F., Krauss, G., Grin, Y. & Steurer, W. Experimental confirmation of the stability and chemical bonding analysis of the high-pressure phases Ca-I, II and III at pressures up to 52 GPa. Phys. Rev. B 79, 134121 (2009).
Mao, W. L. et al. Distortions and stabilization of simple-cubic calcium at high pressure and low temperature. Proc. Natl. Acad. Sci. U.S.A. 107, 9965–9968 (2010).
Errea, I., Rousseau, B. & Bergara, A. Anharmonic Stabilization of the High-Pressure Simple Cubic Phase of Calcium. Phys. Rev. Lett., 106, 165501 (2011).
Ohishi, Y., Hirao, N., Sata, N., Hirose, K. & Takata, M. Highly intense monochromatic X-ray diffraction facility for high-pressure research at SPring-8. High. Press. Res. 28, 163–178 (2008).
Sabine, T. M. A powder diffractometer for a synchrotron source. J. Appl. Crystallogr. 20, 173–178 (1987).
Ma, Y. & Tse, J. S. Ab initio determination of crystal lattice constants and thermal expansion for germanium isotopes. Solid State Commun 143, 161–165 (2007).
Bernstein, B. T. & Smith, J. F. Coefficients of thermal expansion for face-centered cubic and body-centered cubic calcium. Acta Cryst. 12, 419–420 (1959).
Tse, J. S. Crystallography of selected high pressure elemental solids. Z. Kristall. 220, 521–530 (2005).
Sternhiemer, R. M. On the Compressibility of Metallic Cesium. Phys. Rev. 78, 235–243 (1950).
McMahon, M. I. & Nelmes, R. J. High-pressure structures and phase transformations in elemental metals. Chem. Soc. Rev. 35, 943–963 (2006).
Yao, Y., Tse, J. S., Song, Z., Klug, D. D., Sun, J. & Le Page, Y. Structures and superconducting properties of the high-pressure IV and V phases of calcium from first principles. Phys. Rev. B 78, 054506 (2008).
Bersuker, L. Jahn-Teller Effect, Cambridge University Press (2006).
Jahn, H. & Teller, E. Stability of Polyatomic Molecules in Degenerate Electronic States. I, Electronic States. Proc. Roy. Soc. London. Series A 161, 220–235 (1937).
Takemura, K., Minomura, S. & Shimomura, O. X-Ray Diffraction Study of Electronic Transitions in Cesium under High Pressure. Phys. Rev. Lett. 49, 1772–1775 (1982).
von Schnering, H. G. & Nesper, R. How Nature Adapts Chemical Structures to Curved Surfaces. Angew. Chem. Intl. Ed 26, 1059–1080 (1987).
Marx, D. & Hutter, J. Ab Initio Molecular Dynamics Basic Theory and Advanced Methods, Cambridge University Press (2009).
Errea, I., Martinez-Canales, M., Oganov, A. R. & Bergara, A. Fermi surface nesting and phonon instabilities in simple cubic calcium. High Press. Res. 28, 443–448 (2008).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, J. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter. 21, 395502 (2009).
Souvatzis, P., Eriksson, D., Katsnelson, M. I. & Rudin, S. P. The self-consistent ab initio lattice dynamical method. J. Comp. Mat. Sci. 44, 888–894 (2009).
Acknowledgements
We thank Mr. M. Wu for the finite temperature lattice dynamics calculations.
Author information
Authors and Affiliations
Contributions
J.S. Tse and S. Desgreniers planned and executed the experiment, analyzed the data and wrote the paper. T. Matsuoka and Y. Ohishi helped to perform the experiment and with the discussion of the results. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Tse, J., Desgreniers, S., Ohishi, Y. et al. Large amplitude fluxional behaviour of elemental calcium under high pressure. Sci Rep 2, 372 (2012). https://doi.org/10.1038/srep00372
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00372
This article is cited by
-
Melting line of calcium characterized by in situ LH-DAC XRD and first-principles calculations
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
