
Key Points
-
Highlights that despite several publications on the topic, there still exists terminological confusion in terms of dentinogenesis imperfecta.
-
Attempts to elucidate this issue and provide clarity within the dental profession.
-
Suggestions of the use of appropriate terminology in terms of HDD in OI
Abstract
Hereditary dentine dysplasias (HDD) such as dentinogenesis imperfecta (DI) and dentine dysplasia (DD) are a group of genetic conditions characterised by an abnormal dentine structure due to disturbances in the formation, composition, or organisation of the dentine matrix. Either the primary or both primary and secondary dentition are affected to varying degrees. These disorders result from mutations in the genes encoding the major protein constituents of dentine, notably collagens and phosphoproteins. The clinical and radiological features of the hereditary dentine dysplasias (HDD) are relevant to clinical dentistry, in particular osteogenesis imperfecta (OI) which is a well-known heterogeneous genetic disorder. OI is currently the focus of considerable academic attention and involvement of the teeth is a frequent and variable manifestation. In this analysis, the literature related to the classification, clinical features, and molecular pathogenesis of heritable structural tooth diseases affecting dentine formation is reviewed. The definition, history of the terminology and the development of the current classification is outlined and discussed in detail with the aim to address semantic confusion that has arisen in the literature on HDD and to provide clarity on the use of appropriate terminology in the context of OI.
Similar content being viewed by others

Introduction
Heritable dentine defects (HDD) such as dentinogenesis imperfecta (DI) and dentine dysplasia (DD) are genetic conditions characterised by an abnormal dentine structure. Several genes are necessary for the fabrication of enzymes that catalyse post-translational modification and assembly of collagen.1 Rare genetic bone dysplasias, such as osteogenesis imperfecta (OI), are caused by defects in these genes and DI is often a feature.2
Severe autosomal recessive (AR) OI type III is relatively common in the indigenous Black African population of South Africa. With the cooperation of medical colleagues, a series of 64 Black African persons with OI III in South Africa were dentally assessed as a component of a PhD investigation. The aim of this study was to document the dental manifestations of these individuals with a view to improving dental care. Arising from this endeavour, an extensive appraisal and review of the relevant terminology was undertaken and the outcome forms the subject of this article.
There is considerable current interest in the dental manifestations of OI, but this is confounded to some extent by confusion in the terminology of DI. In an attempt to elucidate this issue and to provide clarity within the dental community, an analysis of the relevant literature pertaining to inherited dentine defects in OI is presented and discussed in this article.
Discussion
Dentine and dentinogenesis
Dentine is the hard tissue that encloses the pulp and forms the bulk of the tooth. It is mineralised tissue that constitutes the body of the tooth and serves as a support for the overlying enamel and cementum. Dentine is approximately 70% mineral, 20% organic matrix and 10% water.
The development of a tooth results from the reciprocal interaction between oral epithelium and ectomesenchyme. Ectomesenchyme is embryonic mesenchymal tissue together with neural crest cells. The commencement of dentinogenesis is the differentiation of the ectomesenchymal cells at the periphery of the pulp chamber, into odontoblasts.1
These cells secrete the organic components of the dentine extracellular matrix similar to those secreted by osteoblasts and consists of approximately 86% of type I collagen. Type III, type V and type VI collagens form minor components of the collagenous matrix.
Non-collagenous proteins such as dentin sialoprotein (DSP), dentin glycoprotein (DGP) and dentin phosphoprotein (DPP) constitute a further 10% of the organic matrix of dentine. These proteins are encoded by the dentin sialophosphoprotein gene, DSPP. The proteins, DSP, DGP and DPP undergo post-translational modifications which control mineralisation of dentine.2
Non-syndromic genetic abnormalities of dentine are associated with mutations in the DSPP gene.3
Clinical appearance of hereditary dentine dysplasias
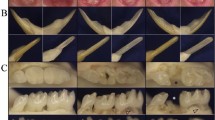
The affected teeth are amber, brown or blue in colour (Figs 1 and 2). Marked attrition and abrasion may result from hypomineralisation of the dentine and the consequent reduction in microhardness.4,5 Periapical abscess formation without pulpal exposure, tooth mobility and early loss of teeth due to periodontal involvement are important complications.

The primary teeth show clinical features of DI and the anterior teeth are amber and translucent at the incisal third. The lower canines have a bluish hue

The primary and secondary teeth are yellow and moderately translucent
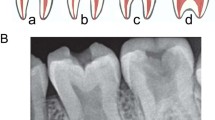
Dentine may be exposed due to shearing of the tooth enamel resulting from an abnormal dentine-enamel junction (DEJ) (Fig. 3). Electron microscopic studies have shown abnormal scalloping at the DEJ and a consequent weaker bond between dentine and enamel. There are also abnormalities in the structure and mineralisation of dentine.6

The lower permanent teeth are amber and there are focal areas with loss of surface enamel (1). The first lower premolar is translucent with a bluish hue (2)
Diagnosis of hereditary dentine dysplasias
The diagnosis of HDD is based on a family history with a pedigree construction and a comprehensive clinical examination. Molecular diagnosis may be useful in the future as several disease causing mutations have been identified. Syndromic HDD is not often associated with OI, in which bone fragility predisposes to frequent fractures. Other rare genetic disorders in which HDD may be a component are listed in Table 1. A knowledge and awareness of these conditions and their possible dental manifestations is necessary by dental clinicians in order to understand the pathogenesis and aid in the dental management of affected persons.
Other disorders that may have clinical features similar to HDD, but differ at a radiological and histological level, include congenital erythropoeitic porphyria, cyclic neutropenia, Histiocytosis X and discolouration due to tetracycline administration.
Classification of hereditary dentine dysplasia
The Shields classification proposed that HDD be divided into five types, specifically, three types of DI and two types of DD.7
The five divisions of the Shields classification are summarised below:
Dentinogenesis imperfecta type I
This dental phenotype is only described in persons affected with OI. Clinically the teeth are amber and translucent with marked attrition in both primary and secondary dentition.
Radiographically, there is pulpal obliteration due to dentine hypertrophy. This process occurs just prior or soon after eruption. A spectrum of change is often seen, even within a single individual, ranging from total pulp obliteration to apparent normal dentine. The teeth often have short constricted roots.
Dentinogenesis imperfecta type II
There are many radiographic and clinical similarities to DI type I. Bulbous crowns with marked cervical constriction are a feature. Sensorineural hearing loss has been reported as an infrequent syndromic manifestation.8 All teeth of both the dentitions are involved and mutations in the gene encoding dentin phosphoprotein and dentin sialoprotein are responsible for this condition. There is complete penetrance of this isolated AD trait and OI is not a component.
Dentinogenesis imperfecta type III
This AD type of isolated DI is known as the 'Brandywine' form and had originally been recognised in a tri-racial isolated population in Maryland and Washington DC. The clinical features are variable and resemble those seen in DI type I and II. Radiographically, the teeth appear hollow due to dentine hypotrophy and the primary teeth often show pulp exposures.9
Dentine dysplasia type I
This dentinal defect is extremely rare and the teeth are clinically unremarkable. Radiographically the roots are conical with an apical constriction or absent in severe cases. The first sign of this condition is tooth mobility which leads to premature exfoliation of teeth. Periapical lesions are common without any associated pathology. The causative gene anomaly has not as yet been identified and the pathological process is not understood.
Dentine dysplasia type II
The primary dentition shows features resembling DI type II yet the permanent dentition is either unaffected or shows mild radiographic anomalies such as 'thistle tube' deformity of the pulp. Pulp stones are often present.
The Shields classification was based on clinical phenotypes and lacked any molecular genetic information concerning the inherited disorders of dentine.
Following the availability of molecular data regarding the cause of each dentine disorder, confusion arose both in the literature and among dental practitioners.
It was suggested at the OI-consensus conference in Norway10 (1999) that abnormal dentine in association with OI should be termed 'DI-like' and that this designation should be used instead of 'DI type I (Shields classification). The term 'DI-like' would refer to affected persons whose teeth were clinically and radiologically aberrant. It would also include the presence of other, less apparent, dentinal aberrations. This latter point is further substantiated by the comment that 'the correspondence and linking between OI and DI is still to be worked out'.11
These terminological shortcomings were subsequently addressed and a revision of the classification was suggested.2 After molecular genetic evaluation of HDD, these authors reasoned that conditions DI type II, DI type III and DD type II are variations in severity of the same pathological process. Shields DI I was not included in the proposed new classification of HDD since these authors also considered the pathogenesis of Shields DI I to be different to that of Shields DI II, DI III and DD II.
This classification, with modifications by the authors is presented in Table 2.
Dentinogenesis imperfecta (DI)
Dentinogenesis imperfecta is an inherited disorder affecting dentine. The resulting defective dentine manifests as discoloured teeth that are prone to attrition and fracture.
Witkop12 proposed a classification for DI which suggested that DI associated with OI is a unique entity. A recent review on the aetiology and nomenclature of DI and a presentation of a case of Shields DI II further highlights the fact that DI associated with OI is a distinct disorder which is clinically, radiologically and histologically similar to DI associated with a mutation in the DSPP gene.13 These authors suggested that the most appropriate classification was that described by Levin14 in 1978 and is published in Oral Pathology textbooks.15,16
These authors13 propose the use of the term 'DI' only in the syndromic context such as when abnormalities in the dentine are recognised clinically and/or radiologically in individuals with OI.
DI associated with AD OI occurs in a significant proportion of individuals in whom mutations in the COL1A1 (17q21.31 – q22.05) or COL1A2 (7q22.1) have been identified. These genes encode the pro-alpha-1 or pro-alpha-2 chains of collagen type I respectively.
DI associated with AR OI is a variable manifestation in individuals with mutations in the following genes, FKBP10, LEPRE1, CRTAP and PPIB.2,13
Radiographical features of hereditary dentine dysplasias
Following the perusal of the literature,2,6,8 the authors propose that a spectrum of radiological manifestations can be identified depending on the severity of the condition, HDD. The following list has been formulated to highlight the radiological features of abnormal dentine:
-
Crown dysmorphology (bulbous crowns to mild occlusal abnormalities)
-
An accentuated constriction at the cemento-enamel junction
-
Variable obliteration of the pulp chambers (narrowed roots to abnormally large root canals)
-
Taurodontism
-
Periapical radiolucency with or without pulp exposure.
Spectrum of the clinical and radiographical features of hereditary dentine dysplasias
The clinical and radiological characteristics of HDD, in particular DI, vary according to the degree of severity of expression of the disorder. These features are summarised in Table 3.
Conclusion
Various authors have made recommendations regarding the terminology of abnormal dentine.2,13 Nevertheless, confusion still exists and the published literature has not as yet provided clarity on the accepted use of the appropriate nomenclature. The authors support the recently proposed classification13 which is well founded since OI is a genetically heterogeneous condition and it is apparent that the genotypes are consistent with the phenotype. The authors also suggest that these disparities should be addressed and that it would be appropriate for internationally agreed guidelines to be formulated.
References
Thesleff I . Developmental Biology and Building a tooth. Quintessence Int 2003; 34: 613–630.
de La Dure-Molla M, Fourier B P et al. Isolated Dentinogenesis imperfecta and dentin dysplasia: revision of the classification. Eur J Hum Genet 2014; 1–7.
Kim J W, Simmer J P . Hereditary dentin defects. J Dent Res 2007; 86: 392–397.
Lygidakis N A, Smith R, Oulis C J . Scanning electron microscopy of teeth in osteogenesis imperfecta type 1. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1996; 81: 567–572.
Teixeira C S, Santos Felippe M C, Tadeu Felippe W et al. The role of dentists in diagnosing osteogenesis imperfecta in patients with Dentinogenesis imperfecta. JADA 2008; 139: 906–914.
Biria M, Fatemeh M A, Mozaffer S et al. Dentinogenesis imperfecta associated with osteogenesis imperfecta. Dent Res J 2012; 9: 489–494.
Shields E D, Bixler D, el-Kafrawy A M . A proposed classification for heritable human dentine defects with a description of a new entity. Arch Oral Biol 1973; 18: 543–553.
Barron M J, McDonnell S T, MacKie I et al. Hereditary dentine disorders: Dentinogenesis imperfecta and dentine dysplasia. Orphanet J Rare Dis 2008; 20: 31.
Witkop C Jr. Manifestations of genetic diseases in the human pulp. Oral Surg 1971; 32: 278–316.
OI-consensus conference. TAKO-Centre. Resource Centre for Oral Health in Rare Medical Conditions. Norway. 1999.
Bloch-Zupan A, Sedano H, Scully C . Dento/Oro/Craniofacial anomalies and genetics, 1st ed. London: Elsevier, 2012.
Witkop C Jr. Hereditary defects of dentin. Dent Clin North Am 1975; 19: 25–45.
Devaraju D, Yashoda Devi B K, Vasudevan V et al. Dentinogenesis imperfecta type I: A case report with literature review on nomenclature system. J Oral Maxillofac Pathol 2014; 18: 131–134.
Levin L S, Salinas C F, Jorgenson R J . Classification of osteogenesis imperfecta by dental characteristics. Lancet 1978; 11: 332–333.
Neville B W, Damm D D, Bauquot J E, Allen C . Oral and Maxillofacial Pathology, 2nd ed. Amsterdam: Elsevier, 2005.
Shafer W G, Hine M K, Levy BM . Text Book of Oral Pathology, fifth ed. Amsterdam: Elsevier, 2006.
Author information
Authors and Affiliations
Corresponding author
Additional information
Refereed Paper
Rights and permissions
About this article

Cite this article
Chetty, M., Roberts, T., Stephen, L. et al. Hereditary dentine dysplasias: terminology in the context of osteogenesis imperfecta. Br Dent J 221, 727–730 (2016). https://doi.org/10.1038/sj.bdj.2016.915
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bdj.2016.915
