
Abstract
Polyadenylation plays an important role in gene regulation, thus affecting a wide variety of biological processes. In the rice blast fungus Magnaporthe oryzae the cleavage factor I protein Rpb35 is required for pre-mRNA polyadenylation and fungal virulence. Here we present the bioinformatic approach and output data related to a global survey of polyadenylation site usage in M. oryzae wild-type and rbp35 strains under a variety of nutrient conditions, some of which simulate the conditions experienced by the fungus during part of its infection cycle.
Design Type(s) | transcription profiling by high throughput sequencing design • stimulus or stress design • replicate design • genetic modification design |
Measurement Type(s) | total RNA |
Technology Type(s) | polyA-selected RNA sequencing assay |
Factor Type(s) | Organism Strain • culture medium • biological replicate |
Sample Characteristic(s) | Magnaporthe oryzae |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
Magnaporthe oryzae is an economically relevant fungal pathogen of cereals, and represents an important threat to food security, particularly for cereal-dependent populations1. It is responsible for the devastating blast disease in rice and other grasses such as wheat, finger millet and maize1. The RNA-binding protein Rbp35 has been characterized in this fungus2,3. No clear orthologues of this protein are present in budding yeast. The rbp35 mutant shows reduced virulence on leaves and roots of rice. Interestingly, this protein interacts with CFI25, the homologue of human CFIm254,5, indicating that Rbp35 is a component of the fungal polyadenylation machinery. In fact, Rbp35 is required for the alternative polyadenylation (APA) of pathogenicity-associated genes3.
The pre-mRNA 3′ end processing occurs in two steps. First, the pre-mRNA is cleaved and, subsequently, a polyadenosine (poly(A)) tail is added6. Unfortunately, standard high throughput RNA sequencing does not allow the identification of polyadenylation sites (PASs) since only a small percentage of sequencing reads contain poly(A) tails. To circumvent this problem, several techniques have been developed to enrich the transcript ends of poly(A)+RNAs (reviewed in Marconi et al.7).
Here, we present the datasets derived from a genome-wide PASs profiling within the M. oryzae wild type isolate Guy11 and the rbp35 mutant using the 3′T-fill method8. The datasets include three biological replicates in four nutritional conditions including nitrogen and carbon starvation. These data will help to identify the coordinated RNA networks that regulate M. oryzae growth in starved cells as well as gene networks regulated by Rbp35. These data also provide useful information for enhancing genome annotations and for cross-species comparisons of PASs and PAS usage within the fungal kingdom. The biological analyses of these data have been published9.
Methods
Fungal media, growth conditions and infection assays
M.oryzae strain Guy11 (WT) and 2D4 (rbp35) were grown on CM (complete medium), MM (minimal medium) and DCM (defined complex medium)10. Starvation conditions were carried out on MM depleted of nitrogen or carbon sources.
RNA extraction, library preparation and RNA sequencing
Fungal material was harvested from 60 hour-old fungal mycelia grown in liquid CM (24 h) and then transferred for an additional 12 h in a fresh CM, MM, MM-N or MM-C. Three biological replicates were independently harvested and extracted for each library preparation. Total fungal RNA was isolated by a standard trizol method11 and treated with RNase-free DNaseI using Turbo DNA-free kit (Ambion). Library preparation was carried out as described by the EMBL (Heidelberg) sequencing service, and similarly, the sequencing libraries and the 3’T-fill reaction were prepared as previously described8. The final libraries were loaded into the cluster station (cBot, Illumina) and the priming buffer was exchanged for the T-fill solution: 101 l water, 20 l Phusion buffer HF (5 ×) (NEB), 3 l dTTPs (10 mM), 0.8 l genomic DNA Sequencing primer V2 (100 M) (Illumina), 3 l non-hot start Phusion polymerase (2 U/l, NEB) and 2 l Taq polymerase E (5 U/l; Genaxxon). The addition of the latter Taq polymerase was crucial for a complete T-fill. After clustering, the samples were sequenced on a HiSeq 2000 (Illumina).
RNA sequencing and PAS detection protocol
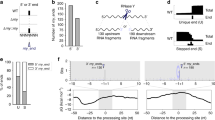
Fig. 1 provides a schematic of the workflow leading to PAS calling. Quality evaluation of sequence reads was performed with FastQC12, with the results shown in Fig. 2. De-multiplexed samples were trimmed to remove poly(A) tails and Illumina adapter sequences using the fastq-mcf utility from the ea-utils package (https://expressionanalysis.github.io/ea-utils/), keeping only reads with at least 17 nucleotides after trimming. Read-mapping was performed using STAR13 with default parameters. The alignments obtained were later filtered removing low-quality mapping (MAPQ < 30, see https://samtools.github.io/hts-specs/SAMv1.pdf), and mappings with high A/T content ( >80%) and potential internal primings (more than seven consecutive As in the first 5’ nucleotides or more than twelve As in the the first sixteen nucleotides, which is the length of the oligodT primers used during the 3′T-fill protocol); these alignments were discarded because they are suspected to be potential internal primings with genomic regions rich in As. In fact, a closer analysis showed that they displayed an unusual nucleotide profile (Fig. 3a and b), confirming that they were not a product of mRNA 3’UTR sequencing. Globally, approximately 2.5% of the alignments were discarded according to the previous reasoning. Alignments that were not discarded during the previous process were further analyzed, and found to mainly align to annotated 3′UTRs regions of the genome (Fig. 3c), supporting the idea that these were in fact genuine sequencing from the 3′end of mRNA transcripts. On the other hand, the discarded alignments usually derived from non-annotated genomic regions, possibly related with transposable elements or other highly variable nucleotide sequences.

After total RNA extraction, Illumina RNA libraries were created following the specifics of the 3′T-Fill protocol8. RNA sequencing was then performed on the Illumina HiSeq2000 platform. FastQ reads were processed by removing sequencing adapters and finally aligned onto the reference genome. Alignments were further processed, filtering out low quality mappings, mappings with high A/T content and potential internal priming. Reads were thereafter assigned to overlapping genes up to 400 bp from the annotated gene end. Finally, identified poly(A) sites were clustered together in order to select high-confidence poly(A) sites.

FastQC report illustrating the average quality scores across all bases of the paired-end datasets for the sample wild-type/complete medium/replicate 1 (SRR6289246). (a,c) Phred quality scores for each nucleotide position are represented as a box and whisker plot. The central red line is the median value. The yellow box represents the inter-quartile range (25–75%). The upper and lower whiskers represent the 10 and 90% points. The blue line represents the mean quality. (b,d) Average quality per read along the read.

(a) Nucleotide profile of final high-confidence PASs. (b) The nucleotide profile around an alignment resulting from products of internal priming. (c) Comparison between mapped location of discarded reads (low quality, high A/T content and potential internal primings) and final kept reads. (d) Distance distribution of aligned reads from the closest annotated stop-codon. Reads rarely mapped greater than 400 bp from the closest annotated stop-codon, indicating that this was the typical 3’UTR length of M. oryzae mRNAs.
Taking all wild-type data sets, 14,593 PASs were reliably assigned to the genomic features listed in the Ensembl Fungi gene annotation for M. oryzae version 27 (http://fungi.ensembl.org/), which contains 13,218 annotated features. Each read was assigned to its overlapping feature (including protein-coding genes and the available ncRNAs), ambiguous cases were assigned to the closest 3’ terminal end. To account for inaccurate (usually short) 3’UTR annotations, which are common even in well-annotated genomes14, we extended the gene annotation by 400 bp. The selection of this supplemental UTR range was justified by the fact that on average reads were at most 400 bp far from the closest annotated stop codon (Fig. 3d). Reads that could not be assigned to any known feature are not included in this dataset. A PAS was considered to be called with high-confidence if it was detected in at least two of the three replicates according to the following rule: its expression is considered distinct from basal noise if its standard score calculated against the whole gene expression (summing up all PASs expression from that gene) has a confidence level >99% (z-score > 2.58 or z-score < 2.58). The standard score was calculated as: z-score = (value-mean)/std, where “value” is the number of supporting reads for the PAS, “mean” is the mean value of supporting reads for all the PASs in the gene, and “std” is the standard deviation of supporting reads for all the PASs in the gene. PASs with less than five supporting reads in every replicate were discarded. High-confidence PASs that fell within 33 bp of one another (roughly the span of the PAS regulatory region, including the A-rich and the U-rich regions upstream of the cleavage site) were resolved to a single PAS - that being, the one that was most highly expressed in that 33 bp region.
Code availability
A Galaxy workflow describing the procedure followed to reproduce the PAS identification is available at (Magnaporthe oryzae polyadenylation sites for wild-type and Δrbp35 mutant, Data Citation 1), together with additional scripts required to run the workflow together with a summary of the pipeline.
Data Records
All sequencing data have been uploaded to the National Center for Biotechnology Information (NCBI) (Alternative polyadenylation controls pathogenicity-associated mechanisms in the rice blast fungus, Data Citation 2), with an overview of the submission provided in Table 1. These data contain paired-end read sequencing for the 24 samples considered: wild-type and rbp35 mutant strains in four different growth conditions (complete medium (CM), minimal medium (MM), nitrogen starvation (MM-N) and carbon starvation (MM-C)), in three replicates. Each archive is stored in FastQ file format.
Illumina deep sequencing read alignments are provided as BAM files (samtools.github.io/hts-specs/SAMv1.pdf), which are sorted and come with the relative index files (Magnaporthe oryzae polyadenylation sites for wild-type and Δrbp35 mutant, Data Citation 1); Bed Graph files were created in order to ease the visualization of PAS-usage levels and are available in the same deposit, together with Generic Feature Format (GFF) files that represent the high-confidence PASs called by our algorithm. All these files are produced from the final filtered alignment, and can be loaded and viewed with a genome browser like IGV15, using version 21 of Ensembl Fungi DNA data and genome annotation (http://fungi.ensembl.org/). The GFF files were used to create the FAIR Linked Data16 that was published in Dydra (https://dydra.com) under a Creative Commons license to enable mechanized query of the results (see Usage Notes below). Data files are stored on Zenodo (Magnaporthe oryzae polyadenylation sites for wild-type and Δrbp35 mutant, Data Citation 1).
Technical Validation
Illumina deep sequencing produced 24 samples providing between between 4,751,592 and 11,517,077 reads per sample (Table 1). FastQC analysis indicated that these reads were generally of high quality (Fig. 2). The correlation between biological replicates, based on calculated gene expression (i.e. number of reads observed for each gene), ranged from 92% to 97%. Correlation matrix analysis revealed a strong similarity between the biological replicates (Fig. 4).

Heat map representing the sequencing correlations between the samples. Correlations were calculated using the Pearson correlation coefficient from the log-transformed raw reads counting per gene.
Approximately 73% of reads could be aligned to the M. oryzae genome, and about 36% were finally assigned to a high-confidence PAS (as described in methods). Paired-end reads were 44 bp long with a mean mapping quality of 36. Only 1% and 4% of reads contained illumina adapters or poly(A) tail residues, respectively.
Internal priming was detected and filtered out from the sequencing data using the algorithm described in the methods section, where about 2.5% of the total reads were found to result from internal priming.
As described in methods, reads were assigned to genomic features after an additional 400 bp was included beyond the annotated 3’UTR to compensate for incorrect genome annotations. We justify this by noting that mapped reads are commonly found to accumulate as far as 400 bp from protein-coding genes' stop codons. The typical upstream A-rich region which contains the polyadenylation signal, was found within 3 bp from the cleavage site. Consequently, high-confidence PAS detection included the clustering of PASs closer than 33 bp.
Usage Notes
The publicly accessible triplestore (https://dydra.com/markw/polyadenylation-sites-in-magnaporthe-oryza/) provides a sample SPARQL query representing the query we anticipate to be most informative (i.e. for a given locus, what PASs are used under which conditions). A Web interface has been constructed to enable visualizing the same query results via a Web browser (http://linkeddata.systems/Magnaporthe/polyA_Sites/). Consistent with the FAIR Principles, machine-readable metadata about this SPARQL endpoint is available in VOiD format by resolving the URL of the endpoint, and machine-readable metadata about the data within the endpoint is available by resolving the named graph URI of the dataset. A track hub was also registered, compatible with genome browsers such as Ensembl17, under the name ID “Magnaporthe oryzae poly(A) sites” (Magnaporthe oryzae poly(A) sites, Data Citation 3).
Additional information
How to cite this article: Marconi, M. et al. Genome-wide polyadenylation site mapping datasets in the rice blast fungus Magnaporthe oryzae. Sci. Data. 5:180271 doi: 10.1038/sdata.2018.271 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Illana, A., Rodriguez-Romero, J. & Sesma, A. Major Plant Pathogens of the Magnaporthaceae Family 45–88 (Springer Berlin Heidelberg, 2013).
RodrÃguez-Romero, J., Franceschetti, M., Bueno, E. & Sesma, A. Multilayer regulatory mechanisms control cleavage factor I proteins in filamentous fungi. Nucleic Acids Research 43 (1), 179–195 (2015).
Franceschetti, M. et al. Fungal virulence and development is regulated by alternative pre-mRNA 3’end processing in magnaporthe oryzae. PLoS Pathog. 7 (12), e1002441 (2011).
Yang, Q., Coseno, M., Gilmartin, G. M. & Doublie, S. Crystal structure of CFIm68 RRM/CFIm25 complex https://doi.org/10.2210/pdb3Q2S/pdb (2011).
Martin, G., Gruber, A. R., Keller, W. & Zavolan, M. Genome-wide analysis of pre-mRNA 3’ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 1 (6): 753–763 (2012).
Proudfoot, N. J. Ending the message: poly(a) signals then and now. Genes Dev. 25 (17): 1770–1782 (2011).
Marconi, M., Rodriguez-Romero, J., Sesma, A. & Wilkinson, M. D. Bioinformatics tools for Next-Generation RNA sequencing analysis. In Ane Sesma and T von der Haar, editors Fungal RNA biology 15, 371–391 (2014).
Wilkening, S. et al. An efficient method for genome-wide polyadenylation site mapping and RNA quantification. Nucleic Acids Res. 41 (5): e65 (2013).
Rodríguez-Romero, J. et al. Virulence- and signaling-associated genes display a preference for long 3’UTRs during rice infection and metabolic stress in the rice blast fungus. New Phytol. https://doi.org/10.1111/nph.15405 (2018).
Foster, A. J., Jenkinson, J. M. & Talbot, N. J. Trehalose synthesis and metabolism are required at different stages of plant infection by magnaporthe grisea. EMBO J. 22 (2): 225–235 (2003).
Galhano, R. et al. Tpc1 is an important Zn(II)2Cys6 transcriptional regulator required for polarized growth and virulence in the rice blast fungus. PLoS Pathogens 13 (7): e1006516 (2017).
Tully, B. A quality control tool for high throughput sequence data http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29 (1): 15–21 (2013).
Muro, E. M. et al. Identification of gene 3’ ends by automated EST cluster analysis. Proc. Natl. Acad. Sci. U.S.A. 105 (51): 20286–20290 (2008).
Nicol, J. W., Helt, G. A, Blanchard, S. G., Raja, A. & Loraine, A. E. The integrated genome browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 25 (20): 2730–2731 (2009).
Wilkinson, M. D. et al. The FAIR guiding principles for scientific data management and stewardship. Scientific Data 3, 160018 (2016).
Kersey, P. J. et al. Ensembl Genomes 2018: an integrated omics infrastructure for non-vertebrate species. Nucleic Acids Research 46 (D1): D802–D808 (2018).
Data Citations
Marconi, M., Rodriguez-Romero, J., Sesma, A., & Wilkinson, M. D. Zenodo https://doi.org/10.5281/zenodo.1168454 (2018)
Sequence Read Archive SRP124953 (2018)
Marconi, M., Rodriguez-Romero, J., Sesma, A., & Wilkinson, M. D. The Track Hub Registry http://trackhubregistry.org/search/view_trackhub/AWGUnQiPPEEPJhVh5mdH (2018)
Acknowledgements
A.S. and M.D.W. are funded by institutional support from the Universidad Politécnica de Madrid, the Spanish Research council (MICINN, grant ref. BIO2014-53211-R and TIN2014-55993-RM), and the Community of Madrid (grant ref. S2013/ABI-2734 and PEJD-2016/BIO-2857). A.S. is also supported by the EC (REA grant agreement n. 304039) J.R.R. is funded by the MICINN grant ref. BIO2014-54233-JIN. M.M. is funded by the Isaac Peral Programme of the Universidad Politécnica de Madrid through a staff award to M.D.W.
Author information
Authors and Affiliations
Contributions
A.S., M.D.W., M.M. and J.R.R. designed and executed the experiments, and analyzed the results. M.L.R.G. and M.D.W. created the Web interface to the poly(A) database. M.D.W. created the FAIR Data versions of the submitted data. All authors contributed to reading and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Marconi, M., Sesma, A., Rodríguez-Romero, J. et al. Genome-wide polyadenylation site mapping datasets in the rice blast fungus Magnaporthe oryzae. Sci Data 5, 180271 (2018). https://doi.org/10.1038/sdata.2018.271
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.271
This article is cited by
-
Template-switching artifacts resemble alternative polyadenylation
BMC Genomics (2019)
-
A database for using machine learning and data mining techniques for coronary artery disease diagnosis
Scientific Data (2019)
