
Abstract
The full exploration of gene-environment interactions requires model organisms with well-characterized ecological interactions in their natural environment, manipulability in the laboratory and genomic tools. The waterflea Daphnia magna is an established ecological and toxicological model species, central to the food webs of freshwater lentic habitats and sentinel for water quality. Its tractability and cyclic parthenogenetic life-cycle are ideal to investigate links between genes and the environment. Capitalizing on this unique model system, the STRESSFLEA consortium generated a comprehensive RNA-Seq data set by exposing two inbred genotypes of D. magna and a recombinant cross of these genotypes to a range of environmental perturbations. Gene models were constructed from the transcriptome data and mapped onto the draft genome of D. magna using EvidentialGene. The transcriptome data generated here, together with the available draft genome sequence of D. magna and a high-density genetic map will be a key asset for future investigations in environmental genomics.
Design Type(s) | parallel group design • replicate design • strain comparison design • stimulus or stress design • transcription profiling by high throughput sequencing design |
Measurement Type(s) | transcription profiling assay |
Technology Type(s) | Whole Transcriptome Sequencing |
Factor Type(s) | selectively maintained organism • biological replicate role • exposure of material to environment |
Sample Characteristic(s) | Daphnia magna • multicellular organism |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

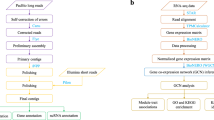

Background & Summary
Illuminating the link between genes and environment is an exciting yet challenging goal. The full exploration of this link requires model organisms with well-characterized ecological interactions in nature, tractability in the laboratory and available genomic tools. The waterflea Daphnia magna Straus satisfies these requirements1,2. D. magna occurs in lakes and ponds in Europe, Africa, Asia and America3,4. It has a prominent ecological role in pelagic freshwater food webs, where it is the primary forage for many vertebrate and invertebrate predators5–7, an efficient grazer of algae8, including cyanobacteria9, a strong competitor for other zooplankters10 and in a constant evolutionary race with parasites11. Experimental tractability is high in Daphnia because of the short generation time, comparable to the genetic model species Drosophila. The small body size enables experimental approaches on large populations, and the cyclic parthenogenetic life cycle enables the parallel analysis of functional and fitness changes in the same genotype in multiple environmental conditions. Moreover, species of the genus Daphnia are renowned models in ecotoxicology and are widely used as indicators of water quality and environmental health12–16. They are also key models in evolutionary biology and the study of adaptive responses to environmental change17–21.
Capitalizing on this unique model system, the STRESSFLEA consortium, a research network funded by the ESF EUROCORES Programme EuroEEFG, generated a comprehensive RNA-Seq data set obtained from two natural genotypes, subsequently inbred in the laboratory, and a recombinant line of D. magna, obtained from the crossing of the two inbred genotypes, exposed to a suite of biotic and abiotic environmental perturbations. The two inbred genotypes were collected from two ecologically different habitats in the species distributional range. One of the inbred strains has been used to obtain the first draft genome of D. magna v2.4 (GenBank LRGB00000000).
Genome-wide transcription profiling was obtained from the three genotypes following environmental perturbations. The EvidentialGene method based on combined RNA-assembly and genome-based modelling of euGenes eukaryote genome informatics (http://eugenes.org/EvidentialGene/)22 was used to generate a public gene set for D. magna with as complete and accurate gene and transcript repertoire as possible. EvidentialGene uses evidence from public gene expression and protein datasets to annotate new genes. Briefly, for each gene, different models are tested and ranked based on quality scores and on deterministic evidence. Selecting the best representative model for a locus from among a large set of models is accomplished over two criteria: (1) gene evidence must pass a minimum threshold score, and (2) the combined score is maximal for all models overlapping the same coding sequence locations. Other criteria and tests are included and used for classification, such as orthology scores, CDS/UTR quality, and expression and intron evidence. The algorithm used for evidence scoring attempts to match expert choices, using base-level and gene model quality metrics. Determining a final gene set is an iterative process that involves evaluation and expert examination of problematic cases, modification of score weights, and reselection.
The data generated here combined with the D. magna draft reference genome and a genetic map available for this species23 will open a new era for environmental genomics. These genome and gene data sets are publicly available in the interactive Daphnia genome database at wFleaBase.org24. This database includes a genome map viewer with an option to display expression data (for example from this study) and genome annotation data from Daphnia pulex and related species, as well as search functions for queries at sequence, gene function, expression, orthology and annotation levels. The RNA-Seq data generated in this study will enable us to disentangle the relative contribution of genetic adaptation and phenotypic plasticity to adaptation in presence of both natural and anthropogenic stressors. Such investigations are possible because of the rich ecological data available for Daphnia, which is arguably the best studied model system in terms of phenotypic and genetic responses to ecological stressors1,2. In combination with the key assets of this model system for experimental work, the transcriptomic data deposited here will enable unprecedented advances in environmental, population and functional genomics.
Methods
Strains
Two inbred genotypes derived from natural strains, and a recombinant line derived from a cross of these two strains, were used to generate the transcriptome of D. magna. The two natural strains were collected from a system of ephemeral rock pools from the northern distributional range of the species (Xinb3, South west Finland 59.833183, 23.260387) and a fish-rearing pond in Southern Germany (Iinb1, Germany, 48.206375, 11.709727), respectively. The Xinb3 genotype was the result of three generations of selfing, and the Iinb1 strain was selfed for one generation, leading to a predicted 87.5 and 50% reduction in their original level of heterozygosity, respectively. The recombinant line is an F2 laboratory strain part of a mapping panel supporting research on the genetic basis of adaptive traits in D. magna23,25. The strains will hereafter be referred to as X- Xinb3, I- Iinb1, and XI-recombinant line.
Environmental perturbations and experimental design
Genome-wide transcription profiles were obtained from the three strains following environmental perturbation by a suite of environmental challenges. Exposures to environmental perturbations on the two inbred strains were conducted at the University of Leuven, Belgium. The sequencing for this experiment was performed at the Finnish Institute of Molecular Medicine (FIMM, Technology Centre, Sequencing unit) at the University of Helsinki. Exposures of the recombinant line were completed at the University of Notre Dame, IN, USA. The sequencing data from this experiment were obtained at the JP Sulzberger Columbia Genome Center (https://systemsbiology.columbia.edu/genome-center). All exposures to environmental perturbations were conducted using the protocol described below. All three genotypes were maintained in the laboratory for several generations after selfing (X and I) or crossing (XI) to reduce interference from maternal effect prior to the exposures to environmental perturbations.
Inbred genotypes (X and I)
For the exposure to environmental perturbations, the genotypes were grown in climate chambers with a fixed long day photoperiod (16 h light/8 h dark) at 20 °C. The first generation was cultured at a density of 10 individuals/l, and increased to 50 individuals/l in the second generation to enable the harvesting of enough offspring for the environmental perturbation exposure. Animals were harvested and exposed in ADaM medium (Aachener Daphnien Medium:26). The medium was renewed every second day in the harvesting phase and the daphnids were fed daily with 150,000 cells Scenedesmus obliquus/ml. The diet changed to a 2:1S. obliquus:Cryptomonas sp. mix from the second generation onwards to provide the animals with optimal food quality. When multiple genotypes were used in the same experimental set up, they were synchronized for at least two generations prior to the actual exposures. The second clutch of the second generation was used for exposures to environmental perturbations. Five-day old juveniles at a density of 100 juveniles/l were exposed for 4 h to the different environmental challenges (Fig. 1). Prior to separating the juveniles for the actual exposures, they were grown in groups of 1,000 in 10 l aquaria for four days. The aquaria were fed daily 150,000 cells per ml in a 2:1 S. obliquus: Cryptomonas sp. Half of the medium was replaced every second day. The animals were not supplied with food during the perturbation exposures to reduce contamination from algae in the sequencing phase. Seven environmental perturbations were imposed on the inbred strains. These consisted of six biotic and one abiotic stressor. The biotic stressors were: kairomone signalling of vertebrate and invertebrate predation, exposure to Pasteuria ramosa parasite spores, crowding, and grazing on toxic and non-toxic cyanobacteria; the abiotic stressor was the pesticide Carbaryl (1-napthyl methylcarbamate, Sigma-Aldrich, Germany) (Fig. 1). To mimic fish predation, Daphnia were exposed to kairomones-enriched medium obtained from growing 19 sticklebacks in 100 l of water. This medium was obtained from aquaria in which fish was reared. Medium in the fish aquaria was refreshed daily, and kairomone-loaded medium was prepared by filtering the medium over a 0.2 μm filter. This kairomone-loaded medium was added to the Daphnia cultures to constitute 10% of the total volume. Similarly, invertebrate predation was mimicked by exposing Daphnia to kairomones-enriched medium obtained from growing an adult tadpole shrimp Triops in 2 l of water. This medium was obtained by filtering the kairomone-loaded medium on a 0.2 μm filter. Similarly to the fish kairomone experiment, the filtered medium was added to the Daphnia cultures to constitute 10% of the total volume. Experimental animals in the parasite treatment were exposed to a solution containing 40,000 spores/ml of P. ramosa, a parasite known to have strong fitness consequences in Daphnia11. Crowding stress was imposed by increasing the number of experimental animals per volume of medium: 100 individuals in 250 ml of medium as compared to 100 individuals in 1 l. Perturbation from cyanobacteria was obtained by feeding Daphnia with a toxic strain of Microcystis aeruginosa (Cyanobacteria, strain MT50) and a non-toxic strain of Microcystis aeruginosa (strain CCAP 1450/1)9. The experimental animals were exposed to the pesticide Carbaryl in a concentration of 8 μgl−1, known to cause appreciable sublethal stress and increased mortality27. The exposures of the inbred strains were completed over two days. For each day a control (no stress) was run in parallel to the environmental perturbations. We performed five biological replicates for each treatment, including controls. Each consisted of ca. 80 sub-adult animals.

Two natural genotypes of D. magna collected from Finland (Xinb3-X) and Germany (Iinb1-I) and a recombinant line (XI) obtained from the cross of the two natural genotypes were used in experimental perturbations. The three genotypes were synchronized for two generations. The second clutch of the second generation was exposed to environmental perturbations. The environmental perturbations for the natural genotypes were as follows: FI: Vertebrate predation mimicked by fish kairomones released by 19 sticklebacks in 100 l water; TR: Invertebrate predation mimicked by kairomones released by 1 adult Triops in 2 l water; PA: exposure to parasite spores by the common parasite Pasteuria ramosa—40,000 sporesml−1; CR: crowding exposure conditions are of 100 individuals/250 ml; BX Toxic Cyanobacteria—strain MT50; BN Non-toxic Cyanobacteria—strain CCAP 1450/1; CA: exposure to the pesticide Carbaryl—8 μgl−1; CO: control. The environmental perturbations for the recombinant line were as follows: CD: Cadmium-6 μgl−1; PB: Lead-278 μgl−1; pH 5.5; UV light; NaCl- 5 gl−1; CO Control.
Recombinant genotype (XI)
The recombinant line was maintained as described above for the parental genotypes with the exception that recombinant Daphnia were maintained in 1 l containers throughout the rearing phases and for the experimental phase, third generation individuals at eight days old were exposed to five abiotic perturbations linked to anthropogenic disturbance. These exposures included: cadmium (Cd), lead (Pb), low pH (5.5), UV light, and sodium chloride (NaCl) (Fig. 1). The experimental treatments included a single control of individuals placed in fresh media without algae for a 24 h period. All treatments and the control included three biological replicates. The metal exposures were maintained for 24 h at concentrations of 6 μgl−1 and 278 μgl−1 for Cd and Pb, respectively. Daphnia were also exposed to pH 5.5 and media supplemented with 5 g/l NaCl for 24 h. UV light treatments were conducted in 250 ml beakers containing 50 ml of media. Beakers were placed 10.5 cm below 30 W, 36-inch Reptisun 5.0 UVB fluorescent light bulbs for 4 h (Zoo Med Laboratories Inc., San Luis Opispo, CA, USA)20. Exposure to UV light was restricted to 4 h to avoid high mortality observed at 24 h. All recombinant line exposures were conducted at 18 °C and RNA collection was timed to occur at the same time period to minimize circadian variation in expression patterns among treatments.
RNA isolation
Inbred genotypes (X and I)
Five biological replicates for each genotype were perturbed with the environmental conditions explained above and RNA-Seq generated from three of the five biological replicates. Having a larger set of exposed biological replicates per genotypes allowed us to choose the three replicates with the highest RNA quality. Total RNA was extracted from pools of ca. 80 juveniles from each genotype and replica by homogenization in the presence of Trizol reagent followed by acidic phenol extraction as described in (ref. 28) and ethanol precipitation. Quality of the isolated RNA was confirmed with Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and only samples showing no RNA degradation were used in subsequent steps. Sequencing was performed on 49 samples: 3 replicates x 2 genotypes x 8 conditions (2 controls were run for genotype X, making the total number of run samples 49, as the environmental exposures were spread over two days).
Recombinant genotype (XI)
Total RNA was extracted from pools of ca. 50 individuals from each replicate (18 samples: 3 replicates x 6 conditions including control) by homogenization in Trizol reagent and isolating RNA using a Qiagen RNeasy column (Qiagen, Valencia, CA, USA) with on column DNase treatment. RNA quality was assessed as above.
Construction of RNA-seq libraries
The experimental procedure from library construction to sequencing and downstream analysis was identical for the three genotypes and was as follows.
Library construction was performed on three biological replicates. 1–3 μg of total-RNA was used for isolating poly-A RNA (Dynabeads mRNA purification kit, Ambion, Life Technologies, AS, Oslo, Norway). The poly-A RNA was reverse transcribed to ds-cDNA (SuperScript Double-Stranded cDNA Synthesis Kit, Life Technologies, Carlsbad, CA, USA). Random hexamers (New England BioLabs, Ipswich, MA, USA) were used for priming the first strand synthesis reaction and SPRI beads (Agencourt AMPure XP, Beckman Coulter, Brea, CA, USA) for purification of cDNA. Illumina compatible Nextera Technology (Illumina, Inc., San Diego, CA, USA) was used for preparation of RNA-seq libraries. 60 ng of ds-cDNA was fragmented and tagged using in vitro cut-and-paste transposition. The fragmented cDNA was purified with SPRI beads. In order to add the Illumina specific bridge-PCR compatible sites and enrich the library, limited-cycle PCR (5 cycles) was done according to instructions of Nextera system with minor modifications. For bar coded libraries, 50X Nextera Adaptor 2 was replaced with a bar coded Illumina-compatible adaptor from the Nextera Bar Codes kit in PCR setup. SPRI beads were used for purification of the PCR-products and the library QC was evaluated by Agilent Bioanalyzer. Libraries were size selected (350–700 bp) in 2% agarose gel, purified with QIAQuick Gel Extraction kit (Qiagen, Valencia, CA, USA) and the library QC was evaluated by Agilent Bioanalyzer.
RNA-seq library sequencing
C-Bot (TruSeq PE Cluster Kit v3, Illumina, San Diego, CA, USA) was used for cluster generation and Illumina HiSeq2000 platform (TruSeq SBS Kit v3 reagent kit) for paired-end sequencing with 101 bp read length. Sequence data for the inbred genotypes were generated by the FIMM sequencing unit at the University of Helsinki, Finland whereas data from the recombinant genotype were generated by the JP Sulzberger Columbia Genome Center (New York, NY, USA).
RNA-Seq quality check
Read sequences were subjected to adapter trimming and quality filtering using Trimmomatic ver.0.33 (ref. 29). RNA-Seq reads were checked for foreign RNA contamination. Human and mouse contaminant sequences were screened and removed by mapping D. magna reads onto ncbigno2014-human.rna and ncbigno2014-mouse.rna using bowtie2 ver.2.1.0 (ref. 30). Finally, 80% of the reads for the inbred genotypes and 99% of the reads for the recombinant genotype were retained (Q>20). Contaminant screening is essential for transcriptome and genome projects; in this study contaminants of 100% RNA identity to mouse, human, and various bacteria genes were found in all source sets, even though not in all replicates. Care should also be taken to avoid false positive contaminant flags, as putative horizontal gene transfer (HGT); one such case was identified in the current dataset. The cleaned reads were mapped onto the reference transcriptome of D. magna obtained from de novo assembly of RNA-Seq data. These data consisted mostly of the Xinb3 inbred genotype data, but also included a subset of data from the Iinb1 genotype and RNA-Seq available in public databases for D. magna at the time of the analysis (mostly31). This reference transcriptome includes only primary transcripts. The mapping of reads from the three genotypes was conducted using Bowtie2 ver.2.1.0 (ref. 30) allowing a maximum edit distance of 3 per read. 74% of the reads mapped on the reference transcriptome and 82% of those mapped to a unique location. The remaining reads mapped to multiple locations suggesting that those are alternative transcripts or incomplete genes that cannot be accurately mapped. These reads will be the object of further investigations.
As an additional assessment of sequence quality, we counted base positions in which more than two allelic variants were present, hence departing from the expectation of a maximum of two alleles at a given position for a diploid organism. For this analysis reads from all treatments for each genotype were pooled and mapped against a reference sequence set of single copy genes from the D. magna consensus transcriptome. The mapping process was performed using bowtie2 ver. 2.1.0 30 reporting up to a maximum of 20 valid alignments per read (-k 20); from this pool, alignments with least edit distance were selected as best hits for a specific read. Allelic variants as compared to the reference consensus sequence were identified using samtools mpileup command (samtools ver. 0.1.19, 45), and a custom parser written in perl. The minimum base quality score required for a variation to be considered was q=20 where q is the threshold measured. Variant calls with frequencies below 1% representing typical Illumina sequencing errors32 were excluded. The variant positions with 2, 3 or 4 allelic variants were counted.
Transcriptome and gene set construction
Transcriptome assembly of RNA
We used EvidentialGene methods from the euGenes.org22 project to assemble RNA-seq, as well as annotate and validate transcripts per strain. After assembling transcripts per strain, we constructed a complete gene set across strains incorporating chromosome assembly data available for D. magna (draft genome assembly 2.4, GenBank LRGB00000000). Paired end RNA-Seq reads, totalling 7.2 billion reads from the current project and 2 billion reads from published data at the time of the analysis31, were assembled de-novo with several RNA assemblers, using multiple options for kmer fragmenting, insert sizes, read coverage, digital normalization, and quality and abundance filtering. De-novo RNA assemblers used include Velvet/Oases33,34 [v1.2.03/o0.2.06], SOAPDenovo-Trans35 [v2011.12.22] using multi-kmer shredding options from 23 to 95 bp, and Trinity36 [v2012.03.17] (with fixed kmer option). Accessory methods used for RNA data processing include GMAP/GSNAP and Bowtie for read and transcript mapping to genome assembly, diginorm of khmer package, and sequence artefact filtering. Additional transcripts were assembled with genome-mapping assistance, using PASA37 [v2.2011], Cufflinks38 [v1.0.3 and v0.8], and EvidentialGene. EvidentialGene tr2aacds software pipeline (http://eugenes.org/EvidentialGene/trassembly.html) was used to process the resulting assemblies obtained from coding sequences. The assemblies were then translated to proteins, scored for gene evidence including CDS/UTR quality and homology, and reduced to a biologically informative transcriptome of primary and alternate transcripts. We submitted to NCBI only the primary transcripts; alternate transcripts are available at wFleaBase.org.
Gene set construction
Gene models were also predicted on the draft D. magna genome assembly with genome-modelling methods, using AUGUSTUS39, and were incorporated in this public gene set version evg7f9b. Accessory gene set annotation, validation and processing methods included NCBI BLAST suite, exonerate (protein alignment), lastz (sequence alignment), GMAP (gene mapper), CD-Hit (sequence clustering), MUMmer (sequence alignment), MCL (markov clustering), Muscle (sequence alignment), RepeatMasker (repeat and transposon finding), rnaexpress, samtools (rna), SNAP (gene modeller), Splign (alignment), and several database extracts of arthropod and eukaryote genes, proteins and other sequences. A set of primary and alternate transcripts per locus was determined with CDS-overlap discrimination and weighted sum of the several gene evidence scores per transcript model. In hybrid gene set constructions, such as the one presented here, errors occur from both genome map modelling and mRNA assembly, and discrepancies between methods need to be resolved from available gene evidence. The algorithm used for this gene set construction was Evidential Gene and includes three stages:
-
1)
Stage 1. Transcript assemblies of mRNA-seq are performed with several de-novo assemblers and parameters, followed by EvidentialGene tr2aacds redundancy removal for each assembly set.
-
2)
Stage 2. Locus/alternate gene classification is performed from assembly sets obtained in stage 1 to produce non-redundant gene assemblies for each strain using several attributes: transcript alignment classification (tr2aacds), genome-map location and consensus map loci, consensus protein homology and quality, and cross-strain transcript consensus (MCL clustering of transcript alignments40).
-
3)
Stage 3. A candidate locus/alternate gene set for the species is produced from the non-redundant strain sets, using several gene consensus measures across strains, expert curation and computational reclassification. Cases of alternate/paralog discrimination and mis-mapping are investigated in this step using consensus of gene structure among strains, protein orthology analyses, and consensus location on D. magna and the sister species D. pulex chromosome assemblies.
Stage1 produced separate RNA assemblies for the three genotypes, amounting to 16.5 M transcripts for X, 9.5 M for I, and 3.7 M for XI, plus a 4th genome-assisted de-novo assembly of 1 M transcripts from weak expressed loci (X genotype). Stage 2 produced 1.0 M non-redundant mRNA transcripts ranging from 35,000 to 270,000 transcripts per set across 7 gene sets obtained from strain and genome-based inferences. The gene set obtained in this second stage is derived from 30 million mRNA assemblies obtained in stage 1. Stage 3 involved cross-strain consensus locus determination, including paralog/alternate discrimination, iterative reclassification and refinements, reducing the total set to 29,128 loci and primary transcripts, with 84,882 alternative transcripts found among 17,473 of those loci.
Gene homology evidence for the gene construction pipeline includes 300,000 proteins from 10 species: the waterfleas Daphnia magna and Daphnia pulex (version 2010, wFleaBase.org), the tiger shrimp Penaeus monodon (2013 EvidentialGene), the flour beetle Tribolium castaneum (2014 NCBI), the beetle Pogonus chalceus (2013 EvidentialGene), the honeybee Apis mellifera (2014 NCBI), the wasp Nasonia vitripennis (2010 EvidentialGene), the fruitfly Drosophila melanogaster (rel5.30 2012), the fish Maylandia zebra (NCBI 2014) and humans (UniProt 2011). Orthology and paralogy criteria were assigned using all versus all reciprocal blastp of these species, followed by OrthoMCL41 alignment clustering of genes (Dmag analysis version arp7bor5 in wFleaBase.org). Gene names were assigned to our models on the basis of homology scores to UniProt proteins. The consensus gene family names were obtained from OrthoMCL orthology analyses, in accordance with UniProt protein naming guidelines42.
The basic approach employed by EvidentialGene is similar to other eukaryote genome annotation methods, including NCBI Eukaryote genome annotation pipeline43 (http://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/), ENSEMBL genome annotation pipeline (http://www.ensembl.org/info/genome/genebuild/genome_annotation.html), TIGR and Broad genome annotation software44, and MAKER45. It differs from these other approaches for its deterministic evidence scoring, detailed per gene annotations, and single-best model/locus approach. A notable divergence from these other methods is the use of hybrid mRNA-assembly and genome modelling which increases the accuracy and completeness of the gene sets generated.
Assessing the gene set completeness
Orthology completeness, presence and full length of orthology genes were assessed with OrthoMCL in several steps of the gene set construction and in particular during stage 3 (Table 1). For an independent quantitative assessment of orthology completeness we used BUSCO (Benchmarking Universal Single-Copy Orthologs, v1.1, http://busco.ezlab.org/46), a recognized benchmark approach for single copy orthologs providing an assessment of orthologs conserved among species. Deviations from completeness are commonly interpreted as technical or, less frequently, biological deviations from the expected gene complement. We compared the gene models of D. magna (dmagset7finloc9b.mRNA gene set) with the BUSCO arthropod profiles. In addition, we compared our gene model with the one of four other arthropod species including Daphnia pulex, Apis mellifera, Tribolium castaneum and Drosophila melanogaster. Our analysis includes also multiple genes sets from the same species. Different genes sets are identified with year and source: 1) Dma_14EV described here using EvidentialGene methods, 2) Dma_11G obtained from genome-modelled D. magna genes from 2011 (this gene set will be described in a separate paper presenting the first draft genome of D. magna), 3) Dpu_10EG and 4) Dpu_07G available for D. pulex; 5) Ame_14EV obtained from Apis mellifera RNA-seq publicly available using EvidentialGene methods; 6) Ame_12G apis45: OGS v3.2 genome genes; 7) Tca_14EV obtained from Tribolium castaneum RNA-seq publicly available using EvidentialGene methods; 8) other Ame and Tca publicly available gene sets; 9) Fly13 and Fly04 generated in 2013 and 2004 for Drosophila melanogaster. An in depth analysis of the different gene sets and discussion of reliability of validation methods will be presented elsewhere.
Data Records
Daphnia magna transcriptome and related data are published under the International Nucleotide Sequence Database Collaboration BioProject PRJNA284518 (http://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA284518). The D. magna consensus transcriptome for each of the three genotypes studied here and the raw data for each library obtained from different environmental perturbations are deposited in GenBank (Data Citation 1, metadata in Supplementary Table 2). RNA-seq read and transcript assemblies of RNA-Seq data can be found at this entry. Transcript assemblies generated separately for the two inbred strains are also available at GenBank (Daphnia magna Xinb3, Data Citation 2; Daphnia magna Inb1, Data Citation 3). The X assembly contains 42,990 loci with 253,834 transcripts.
The I assembly contains 36,935 loci with 271,331 transcripts. The X and I annotated assemblies contain coding-sequence validated for primary and alternate transcripts from stage 2 in the EvidentialGene pipeline above (Table 1, Dapma6tx and Dapma6ti clone sets), with loci determined by shared exons. Links to public gene set IDs are included with each transcript assembly. The complete hybrid mRNA-assembly and genome-modelled D. magna gene set, and draft genome assembly, in standard sequence and GFF annotation data formats, is publicly available at http://wfleabase.org/genome/Daphnia_magna/openaccess/genes/. The D. magna gene proteins are also available at UniProt http://www.uniprot.org/uniprot/?query=taxonomy:35525.
Technical Validation
Metrics of RNA—seq data
A total of 7.2 billion reads were generated, with an average of 107.5 million reads per sample (s.d. 22 million read pairs). The number of reads was 3.5billion (1.75 billion read pairs) for the X, 2.8 billion (1.4 billion read pairs) for I, and 0.8 billion (0.4 billion read pairs) for XI. Of the total number of reads, 77% for X, 81% for I and 77% for XI had quality scores above 30 (analysed with FastQC software47, Table 2). In Table 3 (available online only) we show a detailed analysis of the RNA-Seq data per sample including raw data read pairs before and after trimming quality was applied, as well as insert size. Approximately 70% of the reads retained their full length of 101 bases after trimming (Table 3) (available online only). Insert size for each paired-read library was estimated by mapping a random subsample of 1,000,000 reads per sample to the mitochondrial genome sequence on the reference draft genome (reference genome ver.2.4). The size of the insert for each concordantly mapped read pair was estimated and the average drawn over all such read pairs. The insert size for each sample is shown in Table 3 (available online only).
When using primary transcripts only, the number of reads mapping onto the transcriptome ranged between 61 and 78% (Fig. 2, Table 4 (available online only)). This percentage reached 98% of all reads when primary and alternate transcripts were used (Table 5). If the same read mapped multiple times onto the same transcript, it was counted only once for that transcript. Multiple mapped reads can be alternate transcripts of the same gene or the result of incomplete mapping likely caused by partial sequence of a transcript. The reads mapping to multiple locations will be object of future studies and hence are not discussed further. The read counts per gene ID are shown in Supplementary Table 1.

Percentage of read pairs per samples mapping to unique (black bars) or to multiple locations (grey bars) in the reference transcriptome of D. magna.
The total number of transcripts retained in this study after trimming and quality checks mapped onto 29,128 genes identified with the EvidentialGene model described above. The distribution of read pairs per gene is summarized in Supplementary Table 1. Between 26,508 and 28,187 transcripts were retrieved across the three genotypes (Table 6). The coverage in bp was highest for the X genotype with 5,282.66 and lowest for the XI genotype with 1,952.93 bp (Table 6). The difference in transcript-read map rates indicated in Tables 4 and 5 results from two main factors: (a) alternate transcripts account for 15% of the difference (all versus primary in Table 5) and (b) roughly a 10% difference in mapping of primary transcripts can be observed when different methods are adopted. For example GSNAP trims read ends to facilitate alignment to reference similarly to transcript assembly methods that trim and shred reads, whereas other methods like Bowtie do not trim ends.
Allelic variants
After removing base positions with frequency lower than 1% which can be explained as sequencing errors32, we identified allelic variants with 2 to 4 alleles as compared to the reference set of single copy genes. The large majority of variants had one or two alleles as expected for a diploid organism (Table 7), confirming the high quality of our sequences. A small fraction of variants had 3 and 4 alleles. When a cut-off value of 5% on allelic variant calls was applied these variants were further reduced in number. From visual inspection of the alignment we assessed that these variants interested a very small fraction of the transcriptome.
Reproducibility of biological replicates
A Principal Component Analysis on trimmed transcripts was used to assess the quality of the RNA-Seq data in terms of reproducibility across the biological replicates. The PCA plot inclusive of all data identified the sample I_BN_r3 as an outlier (Fig. 3a). This sample was removed from downstream analysis as it obscured any signal from both the genotype and the treatment. The PCA plots excluding this outlier showed a clear aggregation of replicates per genotype (Fig. 3b). PCA plots produced separately per natural genotype showed a roughly random distribution of the read counts along the two principal components (Fig. 3c,d) with a tendency of the first replica (r1) to cluster apart from the other two replicates. This may be the effect of slightly earlier developmental stage in r1 as compared to the other two replicas. In the PCA plot of the recombinant line (Fig. 3e), three treatments cluster separately from the others contributing more than 20% to the overall variance along both axes. These are the treatments with exposures of 24 h.

(a) PCA plots including all data, three genotypes (X, I, XI) and their biological replicates; genotypes X and I are in green, whereas genotype XI is in orange. The outlier treatment is the non-toxic cyanobacteria treatment on the I genotype (I_BN_r3 in panel a); (b) PCA plot including the three genotypes and their biological replicates except for the outlier HS_BN_Ir3; (c) PCA plot for the genotype X and its biological replicates; (d) PCA plot for the genotype I and its biological replicates; (e) PCA plot for the XI genotype and its replicates. Treatments short names are as in Fig. 1; they are depicted with different colours within each panel. The replicates are identified by different symbols.
Gene models validation
We generated a public gene catalogue for D. magna version evg7f9b1, for release to the scientific community. This hybrid gene set produced from both mRNA and genome gene models is available at wFleaBase.org with components available in International Nucleotide Sequence Database (INSDC).
Of the total 29,128 gene loci identified in D. magna, 26,825 (92%) genes were assembled from mRNA, and 2,296 (8%) were genome-modelled. 22,059 (76%) of the total recovered genes were complete proteins, and 7,068 (24%) partial proteins. All of these loci are supported by mRNA-Seq and/or protein homology evidence; 65% (18,962) of these genes map completely onto the D. magna draft genome assembly 2.4, and 99% (28,127) contains RNA-Seq reads unique to a specific locus. 76% of the total gene loci identified in D. magna show homology to other species (blastp e<=1e-5 to proteins or conserved domains) and 18% (5,170) show homology only to other Daphnia species. Finally, 40% of the recovered genes were orthologs to other species using orthology criteria of OrthoMCL, and 16% were paralogs of orthologs. 44% (12,826) of the total set of identified gene loci do not cluster with other species genes. This proportion can be considered unique or evolved in D. magna, although many genes derive homology from other species. The high number of Daphnia evolved genes is not unexpected considering the large number of eco-responsive genes identified in the related congener D. pulex48 and the fact that Daphnia species are among the first crustaceans with a draft genome sequence obtained from exposures to ecological stressors. We used the draft genome assembly of D. magna v 2.4 as part of the gene construction and validation process. Of this finished gene set, 65% (18,962) map properly onto the assembly with a coverage >=80%; 35% (10,189) of the genes mapped with low quality scores; 12% (3,389) remained un-mapped, 12% (3,386) partially-mapped, and 12% (3,414) showed split-mapping. These mapped genes include hundreds of trans-spliced and anti-sense loci where mRNA/protein and introns have reversed orientation. Finally, 14% of the genes that could be mapped were single-exon loci. Some of the conflicts among the physical map in the genome assembly v2.4, partially mapped genes and other complexity are artifactual results of draft genome missassemblies. Other of these complexities are located on well assembled portions, including the anti-sense transcription, and appear as true biological complexities. An instance of putative horizontal gene transfer, bacteria to Daphnia, was uncovered during contaminant screening. This has been reported in Daphnia pulex49 as a kairomone-stress responsive horizontal gene transfer (HGT) gene, and appears to exist in the draft genome of D. magna, D. pulex, and in D. galeata (personal observation DG). An automated contaminant screening flagged this as a contaminant, but further examination of evidence indicates probable Daphnia genomic source, with a potential ecological relevance of this gene to Daphnia species.
Assessing the transcriptome annotation completeness
Evidence of high quality and completeness of the D. magna genes was provided by both OrthoMCL and the BUSCO analyses (Table 8 and Fig. 4). According to the OrthoMCL assessment, the current D. magna genes are as or more complete than related arthropods gene sets, with few orthologs missing, a higher number of complete genes, and a lower number of fragment outliers detected (Table 8). In the BUSCO analysis D. magna gene set showed the lowest proportion of missing and fragmented single copy orthologs as compared to the other four arthropod but for two other sets: Ame14evg and Tca14evg (Fig. 4). Notably, the species showing the most complete gene sets in our analysis were the ones in which the EvidentialGene methods was applied. A complete analysis of this method’s performance versus other methods is beyond the scope of the present paper and will be discussed elsewhere.

Stacked bar plots showing proportions of gene sets in quality categories for D. magna and 4 other arthropod species.. Two gene sets are represented per species, as described in methods, to show effects of construction methods on quality. The categories of genes are: i) complete single copy BUSCO: genes which match a single gene in the BUSCO reference group; ii) fragmented BUSCOs: genes only partially recovered for which the gene length exceeds the alignment length cut-off; iii) missing BUSCO: not recovered genes. Abbreviation for species names are as follows: Dma=Daphnia magna; Dpu=Daphnia pulex; Ame=Apis mellifera; Tca=Tribolium castaneum; Fly=Drosophila melanogaster. The gene sets sources used for the 4 arthropod species are as follows: 1) Dma_14EV dapmagevg14:, Evigene mRNA+genome, 2014.08; 2) Dma_11G dapmag11: Evigene genome genes, 2011; 3) Dpu_10EG dapplx10evg: Evigene genome genes, 2010; 4) Dpu_07G dapplxjgiv11: genome genes, 2007, doi: 10.1126/science.1197761; 5) Ame_14EV apisevg14: Evigene mRNA assembly 2014.06; 6) Ame_12G apis45: OGS v3.2 genome genes, 2012, 10.1186/1471-2164-15-86; 7) Tca_14EV tribcas4evg2: Evigene mRNA assembly, 2014.12; 8) Tca_12G tribcas4aug: AUGUSTUS genome genes, tcas4.0, 2014; 9) Fly_13 drosmel548n: Flybase release 5.48, 2013; 10) Fly_04 drosmelr4: Flybase release 4.0, 2004.
The STRESSFLEA consortium was a collaborative network of 10 Universities, including 7 European and 2 North American Universities. The effort of this consortium allowed us to produce a comprehensive transcriptome data set and a frozen gene catalogue for the premier model system D. magna. This effort paves the way to powerful discoveries in environmental and functional genomics elevating D. magna to the rank of genomics empowered ecological model species.
Additional Information
Tables 3 and 4 are only available in the online version of this paper.
Supplementary Information accompanies this paper at http://www.nature.com/sdata
Competing financial interests: The authors declare no competing financial interests.
How to cite this article: Orsini, L. et al. Daphnia magna transcriptome by RNA-Seq across 12 environmental stressors. Sci. Data 3:160030 doi: 10.1038/sdata.2016.30 (2016).
References
References
Altshuler, I. et al. An integrated multi-disciplinary approach for studying multiple stressors in freshwater ecosystems: Daphnia as a model organism. Integr. Comp. Biol. 51, 623–633 (2011).
Miner, B. E., De Meester, L., Pfrender, M. E., Lampert, W. & Hairston, N. G. Linking genes to communities and ecosystems: Daphnia as an ecogenomic model. Proceedings of the Royal Society B-Biological Sciences 279, 1873–1882 (2012).
Adamowicz, S. J., Petrusek, A., Colbourne, J. K., Hebert, P. D. & Witt, J. D. The scale of divergence: a phylogenetic appraisal of intercontinental allopatric speciation in a passively dispersed freshwater zooplankton genus. Mol. Phylogenet. Evol. 50, 423–436 (2009).
Ebert, D . Ecology, epidemiology, and evolution of parasitism in Daphnia. Bethesda (MD) National Library of Medicine (US), National Center for Biotechnology, (2005).
Drenner, R. W., Strickler, J. R. & O’Brien, W. J. Capture probability: role of zooplankter escape in selective feeding of planktivorous fish. J. Fish. Res. Board. Can. 35, 1370–1373 (1978).
Louette, G. & De Meester, L. Predation and priority effects in experimental zooplankton communities. Oikos 116, 419–426 (2007).
Rabus, M., Waterkeyn, A., Van Pottelbergh, N., Brendonck, L. & Laforsch, C. Interclonal variation, effectiveness and long-term implications of Triops-induced morphological defences in Daphnia magna. Journal of Plankton Research 34, 152–160 (2012).
Brett, M. T. et al. Species-dependent effects of zooplankton on planktonic ecosystem processes in Castle Lake, California. Ecology 75, 2243–2254 (1994).
Lemaire, V. et al. Genotype x genotype interactions between the toxic cyanobacterium Microcystis and its grazer, the waterflea Daphnia. Evol. Appl. 5, 168–182 (2012).
Loureiro, C., Pereira, J. L., Pedrosa, M. A., Goncalves, F. & Castro, B. B. Competitive outcome of Daphnia-Simocephalus experimental microcosms: salinity versus priority effects. PLoS ONE 8, e70572 (2013).
Decaestecker, E. et al. Host-parasite Red Queen dynamics archived in pond sediment. Nature 450, 870–874 (2007).
Khan, F. R. et al. Accumulation Dynamics and Acute Toxicity of Silver Nanoparticles to Daphnia magna and Lumbriculus variegatus: Implications for Metal Modeling Approaches. Environmental Science & Technology 9, 4389–4397 (2015).
Li, L. Z. et al. A metabolomic study on the responses of Daphnia magna exposed to silver nitrate and coated silver nanoparticles. Ecotoxicology and Environmental Safety 119, 66–73 (2015).
Lee, S. et al. Ecotoxicological assessment of cimetidine and determination of its potential for endocrine disruption using three test organisms: Daphnia magna, Moina macrocopa, and Danio rerio. Chemosphere 135, 208–216 (2015).
Simpson, A. M., Jeyasingh, P. D. & Belden, J. B. Variation in toxicity of a current-use insecticide among resurrected Daphnia pulicaria genotypes. Ecotoxicology 24, 488–496 (2015).
Zhang, L., Liu, J., Liu, H., Wan, G. & Zhang, S. The occurrence and ecological risk assessment of phthalate esters (PAEs) in urban aquatic environments of China. Ecotoxicology 24, 967–984 (2015).
Frisch, D. et al. A millennial-scale chronicle of evolutionary responses to cultural eutrophication in Daphnia. Ecol. Lett. 17, 360–368 (2014).
Latta, L. C., Weider, L. J., Colbourne, J. K. & Pfrender, M. E. The evolution of salinity tolerance in Daphnia: a functional genomics approach. Ecol Lett 15, 794–802 (2012).
Orsini, L., Spanier, K. I. & De Meester, L. Genomic signature of natural and anthropogenic stress in wild populations of the waterflea Daphnia magna: validation in space, time and experimental evolution. Molecular Ecology 21, 2160–2175 (2012).
Scoville, A. G. & Pfrender, M. E. Phenotypic plasticity facilitates recurrent rapid adaptation to introduced predators. Proc. Natl Acad Sci USA 107, 4260–4263 (2010).
Yampolsky, L. Y. et al. Functional genomics of acclimation and adaptation in response to thermal stress in Daphnia. BMC Genomics 15, 859 (2014).
Gilbert, D. G. euGenes, a eukaryote organism genome information service. Nucleic Acids Research 30, 145–148 (2002).
Routtu, J. et al. An SNP-based second-generation genetic map of Daphnia magna and its application to QTL analysis of phenotypic traits. BMC Genomics 15, 1033 (2014).
Colbourne, J. K., Singan, V. R. A. & Gilbert, D. wFleaBase: the Daphnia genome database. BMC Bioinformatics 6, 45 (2005).
Routtu, J., Jansen, B., Colson, I., De Meester, L. & Ebert, D. The first-generation Daphnia magna linkage map. BMC Genomics 11, 508 (2010).
Klüttgen, B., U., D., Engels, M. & T., R. H. Combined effects of 3,4-dichloroaniune and food concentration on life-table data of two related cladocerans, Daphnia Magna and Ceriodaphnia Quadrangula. Chemosphere 32, 2015–2028 (1994).
Coors, A. & De Meester, L. Fitness and virulence of a bacterial endoparasite in an environmentally stressed crustacean host. Parasitology 138, 122–131 (2011).
Ahola, V. et al. The Glanville fritillary genome retains an ancient karyotype and reveals selective chromosomal fusions in Lepidoptera. Nature Communications 5, 4737 (2014).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Labbe, P., McTaggart, S. J. & Little, T. J. An ancient immunity gene duplication in Daphnia magna: RNA expression and sequence analysis of two nitric oxide synthase genes. Developmental and Comparative Immunology 33, 1000–1010 (2009).
Lou, D. I. et al. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proceedings of the National Academy of Sciences of the United States of America 110, 19872–19877 (2013).
Schulz, M. H., Zerbino, D. R., Vingron, M. & Birney, E. Oases: robust de novoRNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092 (2012).
Zerbino, D. R. & Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Research 18, 821–829 (2008).
Xie, Y. et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 1660–1666 (2014).
Grabherr, M. G. et al. Full- length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29, 644–652 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31, 5654–5666 (2003).
Trapnell, C. & Salzberg, S. L. How to map billions of short reads onto genomes. Nature Biotechnology 27, 455–457 (2009).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62 (2006).
Enright, J., Dongen, S. V. & Ouzounis, C. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Research 30, 1575–1584 (2002).
Li, L. Jr., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Research 13, 2178–2189 (2003).
Conesa, A. & Gotz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int J Plant Genomics 2008, 619832 (2008).
Thibaud-Nissen F., Souvorov A., Murphy T., DiCuccio M. & Kitts P. Eukaryotic genome annotation pipeline in The NCBI Handbook, (ed National Center for Biotechnology Information (US) NCBI, http://www.ncbi.nlm.nih.gov/books/NBK169439/, 2013).
Haas, B. J., Zeng, Q., Pearson, M. D., Cuomo, C. A. & Wortman, J. R. Approaches to Fungal Genome Annotation. Mycology 2, 118–141 (2011).
Holt, C. & Yandell, M. D. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Andrews, S. FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Colbourne, J. K. et al. The ecoresponsive genome of Daphnia pulex. Science 331, 555–561 (2011).
Miyakawa, H. et al. Gene up-regulation in response to predator kairomones in the water flea, Daphnia pulex. BMC Developmental Biology 10, 45 (2010).
Data Citations
NCBI Sequence Read Archive SRP059260 (2015)
Gilbert, D. G. GenBank GDIP00000000 (2015)
Gilbert, D. G. GenBank GDIQ00000000 (2015)
Acknowledgements
This research was financially supported by the ESF EUROCORES Programme EuroEEFG, Grant 09-EEFG-FP-040. The KU Leuven team was supported by FWO project STRESSFLEA-B (G061411N) and by the KU Leuven Research Fund (coordination grant and grant PF/2010/07). Kevin Pauwels helped with the exposure experiments; Veerle Lemaire, Aline Waterkeyn, Isabel Vanoverberghe and Nellie Konijnendijck provided material for exposures. The University of Notre Dame team was supported by NIH grant R24-GM078274 to MEP. The University of Helsinki Team was supported by Academy of Finland grants 250444, 284601 and 135291. Sayanty Roy, Kerry Regan, Jihyun Won and Jackie Lopez conducted the exposure experiments and RNA isolation for the recombinant genotype. Seanna McTaggart provided additional RNA-seq transcriptome clone data for the public D. magna gene set. Donald Gilbert has been supported by the National Science Foundation (grant No. 0640462 to DGG), including genomics computational resources via TeraGrid amd XSEDE. Jana Asselman is the recipient of a FWO scholarship.
Author information
Authors and Affiliations
Contributions
L.O. and L.D.M. coordinated the STRESSFLEA consortium and manuscript writing with input from D.G., M.P., M.F., M.J.R., D.E., J.A., J.K.C., and M.J.
L.O. coordinated the research activities, contributed to the exposures on the two natural clones, and coordinated data analysis.
D.G. assembled transcriptomes, constructed annotated gene sets, contributed to expression analyses, provided genome database with these data, coordinated and performed public database submissions
R.P. performed data processing, validation, analyses of transcriptome data, and public database submissions
M.J. coordinated and performed the exposures of the two natural clones
J.B. and O.S.S. were responsible of the downstream analysis of the transcriptome data
K.S. contributed to the downstream analysis
J.K.C. contributed to the paper writing and experimental design
D.B.R. contributed to the data analysis including mapping and downstream analysis
J.A. and D.G. performed the BUSCO analysis
D.E. collected and bred the D. magna clones and produced purified samples of two parasite strains.
J.K. prepared samples for RNA -seq and performed QC
C.L. contributed to experimental design provided invertebrate predators
M.E.P. coordinated the exposures on the recombinant clone and contributed to data analysis.
M.J.F. generated the RNA-Seq data for the natural strains
All authors contributed to manuscript editing and discussion on experimental design.
Corresponding author
ISA-Tab metadata
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0 Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Orsini, L., Gilbert, D., Podicheti, R. et al. Daphnia magna transcriptome by RNA-Seq across 12 environmental stressors. Sci Data 3, 160030 (2016). https://doi.org/10.1038/sdata.2016.30
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2016.30
This article is cited by
-
RNA-seq analysis reveals changes in mRNA expression during development in Daphnia mitsukuri
BMC Genomics (2024)
-
Beyond Daphnia: a plea for a more inclusive and unifying approach to freshwater zooplankton ecology
Hydrobiologia (2023)
-
Nutrient-imbalanced conditions shift the interplay between zooplankton and gut microbiota
BMC Genomics (2021)
-
DNMT3.1 controls trade-offs between growth, reproduction, and life span under starved conditions in Daphnia magna
Scientific Reports (2021)
-
A comprehensive epigenomic analysis of phenotypically distinguishable, genetically identical female and male Daphnia pulex
BMC Genomics (2020)
