
Abstract
Introduction:
We report a case of spinal cord infarct as a result of suspected fibrocartilaginous embolism (FCE).
Case presentation:
A 23-year-old man presented with sudden onset cervical and upper back pain followed by progressive weakness in his extremities after throwing a baseball. History, neurologic examination and spinal cord imaging were consistent with spinal cord infarct. We believe the cause was from FCE.
Discussion:
Though rare, physicians should be familiar with this diagnosis and the proposed mechanisms. There is no specific treatment for FCE-related spinal cord infarct and long-term prognosis is largely dependent on the degree of spinal cord injury.
Similar content being viewed by others
Introduction
There are many known causes of acute spinal cord infarct including prolonged hypotension, aortic dissection, spinal cord vascular malformation, cardiac embolism, and as a complication of surgery. Fibrocartilaginous embolism (FCE) is a rare and under-recognized cause of spinal cord infarct that was first described in 1961 by Naiman et al.1 We report a 23-year-old man who presented with suspected FCE-related spinal cord infarct. Consent was obtained from the patient for case report publication.
Case presentation
A 23-year-old Amish man with a family history of factor V Leiden mutation was playing baseball in the morning when he suddenly developed right shoulder pain and felt a ‘pop’. He continued to play, but stopped 2 h later when he developed cervical and upper back pain that progressed over 15 min. He then climbed into his horse-drawn buggy for the 10 min ride home. Getting out of the buggy, he felt weak and wobbly. Symptoms worsened over the next hour as he laid at home with an ice pack on his back. He was seen by a chiropractor for spinal manipulation without relief. By the afternoon, he felt tingling in his hands and feet, and was unable to urinate. At a local hospital, a Foley catheter was placed for urinary retention, however, he refused admission and left. The following morning, his extremities were much weaker and he presented to our hospital.
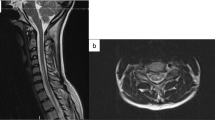
On presentation, he was afebrile and other vital signs were normal. Neurologic exam revealed mild distal weakness in both upper extremities (hand grip 4/5 bilaterally on his muscle strength grading scale) and mild proximal weakness in both lower extremities (hip flexion 4/5 bilaterally). Sensory exam showed decreased pain and temperature sensation below the T2 level but preserved vibratory sensation. Muscle stretch reflexes were normal. His gait was unsteady. American Spinal Injury Association/ International Standards For Neurological Classification Of Spinal Cord Injury (ASIA/ISCoS) exam chart was grade D (Motor Incomplete). Cervical spine magnetic resonance imaging (MRI) showed abnormal increased intramedullary signal in the anterior spinal cord on T2-weighted images and short tau inversion recovery (STIR) sequences extending from the C3/4 level caudally through the C7/T1 level. There was no contrast enhancement, and no evidence for a vascular fistula or malformation (Figure 1). Brain MRI with and without contrast was normal. Lumbar puncture and cerebrospinal fluids (CSF) analysis showed 4 nucleated cells per μl, 1000 red blood cells per μl, total protein 43 mg dl−1 and glucose of 65 mg dl−1. Myelin basic protein was elevated at 7.8 μ l−1, although no oligocolonal bands or aquaporin 4 antibodies were detected. Tests for Varicella zoster virus, West Nile virus, Herpes simplex virus, JC virus and Lyme were negative.

(a) Axial cervical T2 MRI image with increased signal at C5 level mainly in the anterior cord. (b) Sagittal cervical T2 MRI image extending from the C3/4 level caudally through the C7/T1 level mainly in the anterior cord.
The patient was initially treated for possible transverse myelitis with intravenous methylprednisolone, however, because anterior spinal cord infarction was suspected, methylprednisolone was stopped and low-dose aspirin was started. Cholesterol testing was normal. He was heterozygous for factor V Leiden mutation and homozygous for MTHFR mutation, while testing for prothrombin 20210 mutation was negative. Methylmalonic acid and homocysteine levels were normal. Further workup included CRP, HgbA1c, B12, fibrinogen, syphilis IgG, anti-cardiolipin antibodies, ANA, anti-double-stranded DNA, anti-ribosomal P, anti-Jo, anti-chromatin, anti-scleroderma 70, anti-SSA, anti-SSB, and anti-Sm/RNP antibodies, protein C, protein S, anti-thrombin—all of which were negative or within normal limits. Computed tomography chest angiogram showed no evidence for aortic dissection. The patient’s motor and sensory exam remained stable throughout the hospital course (4 days), but hyperreflexia with ankle clonus developed. Upon discharge, he was urinary catheter-dependent. ASIA/ISCoS exam chart remained grade D (incomplete motor). On a 2-month follow-up exam as an outpatient, his strength continued to improve. His Foley catheter was removed. He returned to his job as a logger. He still had a T2 sensory level to temperature. His ASIA/ISCoS exam remained grade D on his 5-month follow-up exam, with normal strength in his lower extremities and minimal weakness in his hand grip 4/5, with no sensory level, and improved gait to normal.
Discussion
FCE is a rare and under-recognized cause of spinal cord infarct.2 Though well described in animals, particularly dogs,3 there have only been 67 cases of FCE-related spinal cord infarct reported in humans (41 histopathologically confirmed and 26 clinically suspected).4 In these cases, patients ranged from 14–78 years old with a female predominance. Most developed symptoms after a minor traumatic event, that is, falls, playing tennis, dancing, basketball, and so on.4,5 In 76% of cases, sudden back pain was the initial symptom followed by weakness that progressed over minutes to hours to paralysis.4 Our patient presented with sudden right shoulder pain and a feeling of a ‘pop’ while playing baseball followed by cervical and upper back pain over 15 min. He then started to feel weak within an hour. This temporal progression is important to differentiate FCE-related spinal cord infarct from other causes of weakness. Weakness from inflammatory disease, for example, usually progresses over days not hours. And while increased CSF protein can be found in patients with spinal cord infarct, they usually do not have pleocytosis or increased IgG index, the presence of which would also be more suggestive of an inflammatory process. The neurologic exam is also crucial in making the diagnosis of spinal cord infarct. Posterior spinal cord infarct secondary to FCE has been reported,6 but most patients, like ours, have anterior spinal cord syndrome with preserved posterior column function (proprioception and vibration). Differential diagnosis also includes a traumatic spinal cord contusion that may develop over minutes to an hour. Our patient, however, did not have a history of trauma. The clinical feeling of a ‘pop’ with sudden back pain and progression to a weakness over an hour are more consistent with FCE-related spinal cord injury.
Typical MRI findings of spinal cord infarct include diffusion restriction, spinal cord edema, increased T2 signal changes in a vascular distribution pattern and absence of contrast enhancement. Our patient’s cervical spine MRI showed abnormal increased intramedullary signal on T2-weighted images and STIR sequences extending from the C3/4 level caudally through the C7/T1 level mainly in the anterior cord, with no enhancement consistent with spinal cord infarct. The cervical, thoracic and lumbar vertebral bodies are in anatomic alignment. There are small Schmorl’s nodes noted along the endplates within the lower thoracic and lumbar region. No focal bone marrow signal abnormality or ligamentous injury is noted. There is a loss of cervical lordosis. The sensory level in our patient was at T2 level, although the MRI changes were higher. This could be explained by testing the distribution of C4, which has a close dermatome proximity to T2, or a more severe injury in the spinothalamic tracts at T2 level. Patient had a T2 level to temperature on a 2-month outpatient follow-up, which resolved on a 5-month follow-up exam.
Several mechanisms have been proposed to explain FCE-spinal cord-related infarcts. The most common is migration of nucleus pulposus material into the vasculature resulting in embolization into spinal cord vessels. The anterior vertebral body and spinal cord are supplied by a single anterior spinal cord artery. The posterior vertebral body is supplied by radicular artery branches and the posterior spinal cord by paired posterior spinal cord arteries. The vasculature of the intervertebral disc is well developed in infancy, but starts to regress around 2 months of age and typically disappears entirely by age 11–16 (though rarely it can persist into the early 20 s).7 Neovascularization can occur, however, around age 50 and earlier in patients with degenerative disease.8 With degeneration, the nucleus pulposus may also extend into the vertebral body (Schmorl’s nodes).
Emboli can gain access to any of these routes—persistent intervertebral disc vasculature, neovascularization or Schmorl’s nodes.4 Specifically, minor trauma is thought to result in increased intervertebral pressure, which then causes migration of fibrocartilaginous embolic material into this vasculature via the arterial or venous route. In the arterial route, the emboli migrate retrograde to the radicular artery, then anterograde to the spinal cord leading to infarction. In the venous route, emboli travel through the caval system, then retrograde to Batson plexus and then to the spinal cord.9
AbdelRazek et al.4 proposed diagnostic criteria for the diagnosis of FCE-related spinal cord infarct that includes a history consistent with the diagnosis, examination and imaging consistent with spinal cord infarct, and exclusion of other causes. Though our patient was heterozygous for factor V Leiden mutation and homozygous for MTHFR mutation, his history of feeling a ‘pop’ while playing baseball followed by pain and progression of weakness over hours, neurological examination and imaging findings are all consistent with the diagnosis of FCE-related spinal cord infarct.
Unfortunately, there is no specific surgical or medical treatment for FCE-related spinal cord infarct. In general, patients are treated according to spinal cord injury guidelines including maintenance of mean arterial pressure between 85–90 mm Hg, prevention of respiratory complications and intensive physical therapy.10 In the literature, FCE-related spinal cord infarct has been associated with a high mortality rate, with surviving cases only reported after 1996.5 Death from respiratory complications (pulmonary embolism, pneumonia and aspiration) has been reported in 40% of cases.4 For survivors, long-term prognosis is largely dependent on the degree of spinal cord injury.
Conclusion
We described a case of a young man with suspected FCE-related spinal cord infarct. Our patient’s presentation was typical in term of history, symptoms, physical examination and imaging findings. Though rare, physicians should be familiar with this diagnosis and the proposed mechanisms. There is no specific treatment for FCE-related spinal cord infarct and long-term prognosis is largely dependent on the degree of spinal cord injury.
References
Naiman JL, Donohue WL, Prichard, JS . Fatal nucleus pulposus embolism of spinal cord after trauma. Neurology 1961; 11: 83–87.
Mateen FJ, Monrad PA, Hunderfund AN, Robertson CE, Sorenson EJ . Clinically suspected fibrocartilaginous embolism: clinical characteristics, treatments, and outcomes. Eur J Neurol 2011; 18: 218–225.
De Risio L, Platt SR . Fibrocartilaginous embolic myelopathy in small animals. Vet Clin North Am Small Anim Pract 2010; 40: 859–869.
AbdelRazek MA, Mowla A, Farooq S, Silvestri N, Sawyer R, Wolfe G . Fibrocartilaginous embolism: a comprehensive review of an under-studied cause of spinal cord infarction and proposed diagnostic criteria. J Spinal Cord Med 2016; 39: 146–154.
Cuello JP, Ortega-Gutierrez S, Linares G, Agarwal S, Cunningham A, Mohr JP et al. Acute cervical myelopathy due to presumed fibrocartilaginous embolism: a case report and systematic review of the literature. J Spinal Discord Tech 2014; 27: E276–E281.
Bansal S, Brown W, Dayal A, Carpenter JL . Posterior spinal cord infarction due to fibrocartilaginous embolization in a 16-year-old athlete. Pediatrics 2014; 134: e289–e292.
Boos N, Weissbach S, Rohrbach H, Weiler C, Spratt KF, Nerlich AG . Classification of age-related changes in lumbar intervertebral discs: 2002 Volvo Award in basic science. Spine 2002; 27: 2631–2644.
Roberts S, Evans H, Trivedi J, Menage J . Histology and pathology of the human intervertebral disc. J Bone Joint Surg Am 2006; 88 (suppl 2): 10–14.
Vuia O, Alexianu. M . Arteriovenous shunt in the spinal cord circulation. Acta Neurol Scand 1969; 45: 216–223.
Ryken TC, Hurlbert RJ, Hadley MN, Aarabi B, Dhall SS, Gelb DE et al. The acute cardiopulmonary management of patients with cervical spinal cord injuries. Neurosurgery 2013; 72: 84–92.
Author information
Authors and Affiliations
Contributions
AMA has contributed to writing the manuscript. DW has contributed to writing the case presentation. MADG has contributed to the conceptualization of the manuscript, its writing and revisions for intellectual content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Alkhachroum, A., Weiss, D., Lerner, A. et al. Spinal cord infarct caused from suspected fibrocartilaginous embolism. Spinal Cord Ser Cases 3, 17027 (2017). https://doi.org/10.1038/scsandc.2017.27
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/scsandc.2017.27
This article is cited by
-
Fibrocartilagenous embolism case series: is it a zebra?
Spinal Cord Series and Cases (2021)
