
Abstract
It is of ongoing interest to develop new approaches for efficient and selective modification of cysteine residues on biomolecules. Here we present a comprehensive study on a newly developed isoxazolinium-mediated cysteine modification of peptides and proteins. Using a stoichiometric amount of isoxazolinium reagents generated in situ from a catalytic amount of silver salts, cysteine-containing peptides can be efficiently modified to afford products in nearly complete conversions. With the optimized conditions, free cysteine containing proteins HSA and BSA, as well as a site-directed mutated therapeutic protein (BCArg) can be efficiently and selectively labelled using small amounts of the isoxazolinium reagents. We find that the phenylacyl thioether linkage bearing an alkyne moiety can be rapidly cleaved under irradiation of UV-A light, giving the formation of a thioaldehyde moiety, which can be converted back to cysteine by reduction.
Similar content being viewed by others
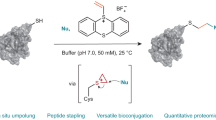
Introduction
Selective modification of peptides and proteins has been recognized as an important tool for biological studies and drug development1,2,3,4,5,6. Among the 20 natural amino acids, lysine and cysteine are prominent targets for chemical modification due to their high nucleophilicity. However, the prevalence of lysine residues on protein surface results in a difficulty to control the level and regioselectivity of the modification, and only a few examples of site-selective lysine labeling have been reported1,7,8,9,10. In comparison, the low abundance (1.7%) and possible incorporation by site-directed mutagenesis allow cysteine to serve as an ideal residue for labeling11,12. Conventional approaches for cysteine modification relied on α-halocarbonyls (via SN2 reaction) and maleimides (via Michael addition). However, the relatively low chemoselectivity of α-halocarbonyls and potential hydrolysis of the maleimide-based conjugates prompted the development of a number of metal-free and transition metal-mediated cysteine modifications in the past decade by Davis, Bernardes, Pentelute, our group, and others13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30. Despite those advances, it is of ongoing interest to develop new cysteine modification methods with high efficiency, excellent selectivity, and using easily accessible reagents under mild reaction conditions.
Development of bioconjugates with cleavable linkers has recently been recognized as an emerging area in chemical biology due to their versatile applications in drug development, proteomics, and in vivo imaging31. Among the reported cysteine-selective modifications, only a few of them are cleavable, such as electron-deficient alkynes reported by our group15, 5-methylene pyrrolones reported by Zhou24, and 4-substituted cyclopentenones reported by Yin25. However, all of those methods are of thiol-induced cleavage and the bioconjugates may undergo exchange reactions with thiols in plasma, limiting their applications in in vivo studies32,33. To overcome this limitation, an ideal approach is to develop a cysteine modification method utilizing a photocleavable and biologically compatible linkage31.
In 1978, Clark and Lowe reported that the phenylacyl thioether linkage, formed by the reaction between cysteine and phenylacyl bromide, underwent photolysis irradiated by UV light (λmax = 342 nm) to form thioaldehyde and subsequently gave a chemically mutated serine residue through hydrolysis and chemical reduction34,35. This is the first example of photocleavable cysteine modification. However, presumably due to the low selectivity of phenylacyl bromide reagents in cysteine modification, a long reaction time (3 h) for the UV-mediated photolysis and the lack of reliable technique to characterize the mutated residue, this method was rarely mentioned afterward. Dichlorotetrazine was reported as an efficient reagent for modification of peptides with two cysteine residues, giving stapled peptides with S,S-tetrazine linkages by Smith et al.21. The S,S-tetrazine linkage was unstapled under irradiation of UV-B light (λmax = 312 nm). However, the efficiency of cleavage under UV-A light (λmax = 365 nm) was much lower, which might lead to the formation of side products21. Recently, Bernardes et al. described that the isobutylene-caged thiols could be efficiently cleaved under mild UV irradiation in the presence of thiol sources and a photoinitiator36. Besides, a UV-mediated photodeprotection of genetic encoded ortho-nitrobenzyl (ONB)-caged cysteine on proteins has been reported by Chin et al.37. Apart from these examples, photocleavable cysteine modification still remains largely unexplored.
Using transition metal-based reagents for cysteine modifications has become attractive recently due to their high efficiency28,29,30. However, employing a stoichiometric amount of organometallic reagents may lead to a relatively high content of transition metal-containing species as side products. In our previous works on modification of the N-terminal α-amino groups of peptides and proteins, we found that ketenes as intermediates generated in situ in manganese-catalyzed oxidative alkyne transformation were the key reagents for the modification38,39. Inspired by this work, it is envisioned that the reactive intermediates generated in transition metal-catalyzed organic transformations can be utilized for the development of new cysteine modification reagents. Along this direction, we have reported that the electrophilic isoxazolinium ions, generated in silver-catalyzed transformations of propargylamine N-oxides, could be employed for efficient cysteine modification of a peptide and a protected cysteine model compound. The modification only required a catalytic amount of silver ions (25 mol%) 40.
In this paper, we report a comprehensive study on an efficient and highly chemoselective cysteine modification with a series of isoxazoliniums generated in situ via silver-catalyzed transformations of propargylamine N-oxides. The modification has been extended to free cysteine-containing peptides and proteins. The enzymatic activities as well as anticancer properties of a modified therapeutic protein (BCArg) have also been studied. In addition, by introduction of an alkyne moiety, the modified bioconjugates bearing phenylacyl thioether linkages can be efficiently cleaved under irradiation of UV-A light (λmax = 365 nm).
Results
Optimization studies
To prepare the isoxazolinium reagents, propargylamines were modularly synthesized via gold-catalyzed three-component coupling reactions of aldehydes, amines, and alkynes developed by Li and us (Supplementary Fig. 1)41,42,43,44. Then, by treatment with a stoichiometric amount of m-CPBA and a catalytic amount of AgNO3 sequentially, the propargylamines were stepwise converted to propargylamine N-oxides and isoxazoliniums (Supplementary Fig. 2)40. The in situ generated isoxazoliniums could be stabilized by hydrogen-bonding interactions in protic solvents45, and the reagents could be stored at −20 °C for repeated usage.
To begin our study, cysteine-containing peptide STSSSCNLSK 1a and isoxazolinium reagent 2a were employed as model substrates for condition screening. By treatment of peptide STSSSCNLSK 1a (0.1 mM) with isoxazolinium reagent 2a (1 equivalent) with 1 mol% of AgNO3 in PBS 7.4 buffer/CH3CN (19:1) at 25 °C for 2 h, following the mechanism depicted in Fig. 1e, modified peptide 3a was afforded in 79% conversion (Table 1, entry 1). Increasing the loading of AgNO3 from 1 to 2.5 and 5 mol% led to an improvement of the conversion to 86 and 96% (Entries 2–3). The total ion chromatogram of the modified mixtures (using conditions depicted in entry 3) by LC-MS analysis indicated the modification was efficient and clean (Fig. 2a). MS/MS analysis of the modified peptide 3a revealed that only the cysteine residue was modified, while other residues remained intact (Fig. 2b). However, further increasing the loading of AgNO3 to 10 mol% resulted in a significant drop of the conversion to 68%, which was attributed to the low stability of isoxazolinium ions with a high content of silver salts. Moreover, within 2 h, addition of excessive equivalents of the isoxazolinium reagent 2a (2–5 equivalents) gave modified peptide 3a in lower conversions (entries 5–8), though nearly complete conversions could be achieved after 24 h.

Reagents and strategy for cysteine modification. a Classical reagents used for cysteine modification. b Reagents used for thiol-induced cleavable cysteine modification. c Reagents used for photocleavable cysteine modification. d General strategy for cysteine modification using isoxazoliniums. e Reaction mechanism of cysteine modification with isoxazolinium reagents

Modification of peptide STSSSCNLSK 1a with isoxazolinium reagent 2a. a The total ion chromatogram by LC-MS analysis of the modified mixtures, afforded by treatment of peptide STSSSCNLSK 1a (0.1 mM) with isoxazolinium reagent 2a (1 equivalent) containing 5 mol% of AgNO3 in 50 mM PBS 7.4 buffer/CH3CN (19:1) at 25 °C for 2 h. b MS/MS spectrum of cysteine-modified peptide 3a. c Time course experiments of the formation of cysteine-modified peptide 3a at different pH values. d Time course experiments of the formation of cysteine-modified peptide 3a at different temperatures
We next attempted to study the effects of pH values and temperature on the modification using time course experiments. Screening reactions in PBS buffer with different pH values indicated that the reaction could be conducted with good to excellent conversions (88–99%) from slightly acidic to basic media (pH 5.3 to 9.0), and basic conditions would further increase the conversion (entries 9–12 and Fig. 2c). This observation could be explained by the pKa value of the thiol group (~8.5) of cysteine. Basic conditions would promote the deprotonation of the thiol group, leading to stronger nucleophilicity, which facilitated the modification. Time course experiments at different temperatures were also performed, suggesting that the reaction proceeded even faster at 37 °C, giving modified peptide 3a in >80% conversion after 15 min, while low conversion (<50%) was afforded after 4 h when the reaction was performed at 4 °C (entries 13–14 and Fig. 2d). Control experiments indicated that isoxazolinium ions were the key reagents for the modification (entries 15–16).
We also studied the compatibility of the modification under different aqueous solutions. With the optimized conditions (using 1 equivalent of isoxazolinium reagent 2a with 5 mol% of AgNO3), the modification proceeded smoothly under PBS, Tris-HCl, imidazole-HCl, citric acid–Na2HPO4, or NaCl medium, giving conversions in 88–97% (Supplementary Methods). These findings implied that this cysteine modification approach could proceed efficiently in various buffers at physiological pH (~7.4) requiring only a stoichiometric amount of isoxazolinium reagent and a catalytic amount (5 mol%) of silver salts.
We then sought to investigate the regioselectivity of the modification. Treatment of cysteine-containing peptides STSSSCNLSK 1a, AYEMWCFSQR 1b, and KSTFC 1c with a stoichiometric amount of isoxazolinium reagent 2a gave modified peptide 3a, 4a, and 5a in 96, 99, and 99% conversion, respectively (Table 2, entries 1–3). MS/MS analysis revealed that only the cysteine residues on the peptides were modified, while other residues remained intact. Control experiments using peptides 1d–i without free cysteine residue resulted in no modification (entries 4–9), suggesting that this modification was highly chemoselective toward the thiol moiety of the cysteine residue in the presence of other nucleophilic residues, such as N-terminus, lysine, histidine, tryptophan, and methionine, etc.
Scope of the isoxazolinium reagent
As propargylamines could be easily accessed by modular synthesis, we moved on to study the structure–reactivity relationship of the isoxazolinium reagents (Supplementary Fig. 3). Isoxazoliniums 2a–d bearing amine moieties with different ring sizes were first screened for the modifications. The results indicated that amine moiety with six-membered ring gave the highest conversion in 96% (Table 3, entry 1). The conversion was lower (87%) when the amine moiety with five-membered ring was utilized (entry 2). Moreover, conducting the modifications with amine moieties bearing larger ring sizes caused significantly drop of the conversions and the formation of β-thio-substituted ketone product 3aa via Michael addition (entries 3–4)12. Keeping the optimal six-membered ring size on the amine moiety, isoxazoliniums with different combinations of R1 were screened. Isoxazoliniums 2e–f with R1 bearing alkyl groups were well compatible with the modifications giving the formation of 3a in 98–99% conversions (entries 5–6). However, when isoxazolinium reagent 2g (R1 = aryl) was employed, 3a was afforded in 61% conversion, while α-thio-substituted enone product 3ab was given in 27% conversion via amine elimination (entry 7). When isoxazolinium reagent 2h (R1 = H) was used, only amine elimination product 3ac was obtained in 98% conversion (entry 8). These findings indicated that different substituents on R1 would lead to the formation of switchable product profiles. Since R2 was incorporated on the resulting bioconjugates while the amine moiety and R1 were cleaved, we further screened the scope of R2 with various substituents. Isoxazolinium reagents 2i–n with electron-donating groups (R2 = OCH3, OCH2CH3, CH3) and electron-withdrawing groups (R2 = F, Br, COCH3) were well tolerated with the modifications giving modified peptides 3b–g in 97–99% conversions (entries 9–14). By changing the benzene moiety to a naphthalene moiety, the formation of 3h was achieved in 99% (entry 15). Isoxazolinium reagents 2p–q bearing alkyne moieties, with potential applicability for sequential modifications using click reactions, were also conducted for the modifications, resulting in the formation of 3i–j in 97 and 98% conversions, respectively (entries 17–18).
To further demonstrate the utility of this cysteine modification, we attempted to expand the scope of this modification by using functional isoxazolinium reagents 2t–v (Fig. 3d). Employment of coumarin-derived isoxazolinium 2t (1.5 equivalents) and fluorescein-derived isoxazolinium 2u (2.5 equivalents) gave the corresponding modified peptides 3k in 84% conversion and 3l in 86% conversion. In addition, PEGylated peptide 3m could be afforded by using a stoichiometric amount of PEG-derived isoxazolinium 2v.

Scope of the isoxazolinium reagents and modified peptides. a Scope of the isoxazolinium reagents 3a–j. b Scope of the modified peptides 3a–j and 3ab–ac. c scope of the stapled peptides 3n-o (conversion). d Scope of the modified peptides 3k–m (conversion) with functional tags
Peptide stapling
Peptide stapling with covalent linkages via macrocyclization reactions has been demonstrated to be an important strategy for constraining the peptide conformations, leading to a potential improvement of their proteolytic stability and cell permeability. However, the approaches for stapling peptides using native amino acids as handles still remained limited46. With this efficient and selective cysteine modification using isoxazolinium reagents in hand, we synthesized bis-isoxazolinium reagent 2u for macrocyclization of peptides YCKEACAL 1j and YCKEAGGACL 1k with two cysteine residues, respectively. Stapled products 3n (i, i + 4) and 3o (i, i + 7) were afforded in moderate-to-good conversion, indicating that the bis-isoxazoliniums were useful for the construction of covalently stapled peptides via macrocyclization (Fig. 3c).
Stability studies
Stability of the modified bioconjugates was evaluated by treatment of the modified peptide 3a with excessive thiol-containing reagents, reducing reagents and oxidizing reagents. Investigations were conducted by treatment of modified peptide 3a with 500 equivalents of L-cysteine, DL-homocysteine, glutathione (GSH), and dithiothreitol (DTT), respectively. After 3 h, LC-MS analysis of the resulting mixtures revealed that the modified peptide 3a still remained intact. Treatment of the modified peptide 3a with 500 equivalents of common reducing reagents, TCEP as well as sodium ascorbate also led to no interference with the modified bioconjugates. These findings implied that the phenylacyl thioether linkage formed after the modification was stable toward environments with thiol-containing reagents and common reducing reagents. H2O2 as an oxidizing reagent was also examined. Treatment of 500 equivalents of H2O2 with modified peptide 3a oxidized the thioether moiety to the corresponding sulfoxide moiety as confirmed by LC-MS/MS analysis. Under the same conditions, using 500 equivalents of Oxone (potassium peroxymonosulfate) as oxidant, the thioether moiety on the modified peptide 3a was further oxidized to the corresponding sulfone moiety. These findings implied that the stability of the thioether linkage toward oxidants was consistent to that on methionine residue, which was reported to be oxidized by addition of oxidizing reagents47.
To provide more insights on this newly developed cysteine modification, we compared this method with the conventional approaches using α-halocarbonyls and maleimides, and a previously reported method using electron-deficient alkynes (Supplementary Fig. 4)15. Treated with 1 equivalent of 2-bromoacetophenone 6, AYEMWCFSQR 1b was converted to modified peptide 4a in 99% conversion which was comparable with modification using isoxazolinium reagent 2a (99% conversion as depicted in Table 3, entry 2). However, if 5 equivalents of 6 was employed, di-modified peptide 4aa with a second modification on methionine residue was afforded in 10% conversion after 2 h. After 3 days, di-modified peptide 4aa conversion was increased to 53%. In contrast, by treatment of 1b with 5 equivalents of isoxazolinium reagent 2a for 3 days, apart from cysteine, no other residues were modified, suggesting that the isoxazolinium reagent was highly chemoselective for cysteine modification. A mixture of 1 equivalent of N-benzylmaleimide 7 with AYEMWCFSQR 1b gave modified peptide 4b in 99% conversion in 2 h. After 3 days, it was found that 52% hydrolyzed derivative 4ba was afforded. Under the same conditions, modified product 4a afforded using isoxazolinium reagent 2a still remained intact. Treatment of AYEMWCFSQR 1b with 1 equivalent of electron-deficient alkyne 1-phenyl-2-propyn-1-one 8 gave modified peptide 4c in 99% conversion in 2 h. By addition of excessive thiol-containing reagent L-cysteine (50 equivalents), cleavage product 1b was afforded in 30%, which was consistent with our previous observation that the vinyl sulfide linkage could be cleaved by addition of excess thiol-containing reagents15. In contrast, modified product 4a afforded through the modification using isoxazolinium reagent 2a still remained intact, supporting the aforementioned results that the phenylacyl thioether linkage formed was stable toward excess thiols.
Application to protein modification
After a comprehensive study on the efficiency, chemoselectivity, scope, and stability of this isoxazolinium-mediated cysteine modification, we further explored its applicability for protein bioconjugation. Bovine serum albumin (BSA) and human serum albumin (HSA) with a single free cysteine residue were utilized for bioconjugation. Treatment of HSA or BSA (0.1 mM) with isoxazolinium reagent 2a (1 equivalent) with 5 mol% of AgNO3 in PBS 7.4 buffer/CH3CN (19:1) at 25 °C for 2 h afforded modified protein HSA-1 in 84% or BSA-1 in 94% conversion by LC-MS analysis (Fig. 3c–e). Upon trypsin digestion, the modification was found on Cys34 residue on peptide fragment GLVLIAFSQYLQQCPFDEHVK of HSA-1 or ALVLIAFSQYLQQCPFDEHVK of BSA-1, while other residues still remained intact. For non-cysteine-containing proteins, insulin, RNaseA, and lysozyme, under the same reaction conditions, no modification was found. These results indicated that the isoxazolinium-mediated modification could be conducted with high efficiency and chemoselectivity in protein modification.
Arginase is a family of enzymes that converts L-arginine to L-ornithine. By arginine depletion, arginase has been investigated to possess anticancer effects toward a broad spectrum of cancer types48. The first-generation non-site-specific lysine PEGylated human arginase I with a prolonged circulating half-life is undergoing phase II clinical trials49. Bacillus Caldovelox arginase (BCArg) is a type of arginase with high production yields, and it can be simply purified50. We attempted to proceed a site-specific modification of BCArg with our newly developed cysteine modification using isoxazolinium reagents. A free cysteine residue was mutated on Ser161 via site-directed mutagenesis (Supplementary Fig. 5). Treatment of the mutated BCArg (0.1 mM) with with isoxazolinium reagent 2a (2.5 equivalents) with 5 mol% of AgNO3 in Tris-HCl 7.4 buffer/CH3CN (19:1) at 25 °C for 2 h afforded modified protein BCArg-1 in 83% conversion (Fig. 4c). Upon trypsin digestion, the modification was found on Cys161 residue on peptide fragment LGVIWYDAHG-DVNTAETSPSGNIHGMPLAASLGFGHPALTQIGGYCPK of BCArg-1. Circular dichroism (CD) measurement of BCArg and BCArg-1 revealed that the secondary structures retained after the modification (Supplementary Fig. 6, Supplementary Table 1). In addition, as determined by ICP-MS analysis, only 1.8 mol% of the silver content was found from the BCArg-1 sample after a filtration, suggesting it would be promising to examine the enzyme activities and the antitumor efficiencies of the modified proteins (Supplementary Tables 2–4). Using isoxazolinium reagent 2p, an alkyne handle was incorporated on BCArg to give the formation of alkyne-functionalized BCArg-2 in 87% conversion. After washing out the excessive reagents, the enzymatic activities of the resulting modified proteins BCArg-1 and BCArg-2 were found to be comparable with BCArg (Table 4). The anticancer properties of the native and modified BCArg were examined using a breast cancer cell line MDA-MB-231 and a lung cancer cell line NCI-H23 (Supplementary Fig. 7, 8). The IC50 values measured indicated that the antitumor efficacies of the modified proteins BCArg-1 and BCArg-2 were similar to that of BCArg (Supplementary Table 5). These findings revealed that the therapeutic proteins still retained their anticancer properties after the modification.

Modification of proteins with isoxazolinium reagents. a General scheme of modification of proteins with (or without) free cysteine residues. b Fluorescent labeling of proteins. c Mass deconvolution spectrum of HSA-1 (84% conversion). d Mass deconvolution spectrum of BSA-1 (94% conversion). e Mass deconvolution spectrum of BCArg-1 (83% conversion)
Labeling of proteins with fluorescent tags provides an opportunity for convenient protein staining in SDS-PAGE gel using fluorescent analysis, and also has potential for in vivo tracking the uptake and physiological parameters5. As the isoxazolinium-mediated cysteine modification could be successfully applied on modification of proteins, we next demonstrated the utility of this reaction for fluorescent labeling of proteins. Using isoxazolinium reagent 2p, alkyne handles were first incorporated on the free cysteine residues of HSA, BSA, and BCArg with good conversions. After washing out the excessive reagents, the alkyne-functionalized proteins (HSA-2, BSA-2, and BCArg-2) were rapidly labeled with red fluorescent rhodamine dyes with good conversions by azide-alkyne Huisgen cycloadditions (Fig. 4b). SDS-PAGE analysis revealed that, rhodamine-labeled proteins (HSA-3, BSA-3, and BCArg-3) were observed using fluorescent analysis, while no fluorescent signal was observed for native proteins (HSA, BSA, and BCArg) or alkyne-functionalized proteins (HSA-2, BSA-2, and BCArg-2). Employing Coomassie blue staining on the same gel, deep blue color signals of native, alkyne-functionalized as well as rhodamine-labeled proteins were observed, indicating that the fluorescent tags have been labeled on the proteins using the isoxazolinium-mediated cysteine modification and a sequential azide-alkyne click reaction (Supplementary Fig. 9, 10).
Photolysis studies
Finally, the photocleavable properties of the modified peptides and proteins were investigated (Fig. 5; Supplementary Table 6). Under irradiation of UV-A light (λmax = 365 nm) for 15 min, cysteine-modified peptide 3a was converted to thioaldehyde product 10 in 41% conversion, while the phenylacyl moiety was cleaved via a Norrish type II photolysis reaction (Supplementary Fig. 11). By introduction of an alkyne handle, modified peptide 3i could be cleaved more efficiently, leading to the formation of 10 in >99% conversion in 15 min. We monitored the photolysis reaction using time course experiments (Supplementary Fig. 12, 13). The results revealed that >90% of peptide 3i was cleaved in 10 min, while only 24% of peptide 3a was converted in the same period. Control reaction in the dark gave no photolysis of 3i. Conducting the reaction in a higher concentration of peptide 3i gave little influence on the efficiency of the photolysis reaction. Interestingly, by treatment of the cleavage peptide 10 in solution with NaBH4 (50 mM) for 30 min, the peptide 10 could be reduced to give native peptide 1a in 97% conversion. We also investigated the photolysis using fluorescent labeled proteins HSA-3 and BSA-3. Under irradiation for 30 min, >60% of the linkages on the proteins were cleaved.

Modification photolysis and sequential reduction of the modified peptide 3i. Reduction regenerates the original cysteine residue
Discussion
In summary, we have presented a comprehensive study on a newly developed cysteine modification using isoxazoliniums generated in situ via silver catalysis. The modification could proceed efficiently with high chemoselectivity toward cysteine residue on peptides and proteins. In most cases, only a stoichiometric amount of isoxazolinium reagents with a catalytic amount of silver salts were needed to give high-to-excellent conversions. Besides, easily accessible isoxazolinium reagents with versatile functional groups were compatible with this modification. Fluorescent tags could be efficiently labeled on the cysteine-containing peptides and proteins by directly employing fluorescent tag-derived isoxazolinium reagents or by sequential modification using the azide-alkyne click reaction. The resulting phenylacyl thioether linkage was stable toward various thiol-containing reagents and reductants.
Investigation of the effect of the modification on a therapeutic protein (BCArg) revealed that the incorporated tag had little influence on the enzymatic activity and anticancer property of the protein, which suggested that the isoxazolinium reagents could be potentially employed as promising reagents for labeling bioactive proteins in the future.
We have also found that incorporation of an alkyne moiety on the phenylacyl thioether linkage would induce a rapid photolysis under irradiation of UV-A light (λmax = 365 nm), and the resulting thioaldehyde moiety could be reduced to be thiol moiety rapidly. To the best of our knowledge, this is the first time to use LC-MS/MS analysis to monitor the formation of thioaldehyde by photolysis of phenylacyl thioether linkage in a peptide sample. Ongoing interest is to employ this photocleavable cysteine modification for applications on proteomic analysis and drug development.
Methods
Synthesis and characterization
The synthetic procedures and characterization for compounds are depicted in Supplementary Methods, and references are listed in Supplementary Table 7. Chromatography and mass spectrometry data are presented in Supplementary Fig. 14–163. NMR spectra are presented in Supplementary Fig. 164–176.
Preparation of isoxazolinium reagents 2a-u
For preparation of the isoxazolinium reagents 2a–t, a mixture of propargylamines I-a-t (0.05 mmol, 1 equiv.) and meta-chloroperoxybenzoic acid (m-CPBA, 0.05 mmol, 1 equiv.) was conducted in a solution of CH3CN (0.50 mL) and H2O (0.40 mL) at 25 °C for 15 min, giving the formation of propargylamine N-oxides II-a–t in situ. After that, 0.10 mL of AgNO3 solution (25 mM in H2O) was added to the mixture and the reaction was conducted at 25 °C for 2 h to afford the isoxazolinium reagents 2a–t (50 mM in CH3CN/H2O (1:1)). The reagents were further diluted to 5 mM in CH3CN/H2O (1:1) and stored at −20 °C for repeated usage. For preparation of isoxazolinium reagents 2u, the mixture of propargylamine I-u, m-CPBA, and AgNO3 was treated using the aforementioned conditions except for two equivalents of m-CPBA was used.
Modification of peptides using isoxazolinium reagents 2a–t
A mixture of 10 µL of peptides 1a–i (1 mM in H2O), 2 µL of isoxazolinium regents 2a–q (5 mM in CH3CN/H2O containing 5 mol% AgNO3), 4 µL of CH3CN, 4 µL of H2O, and 80 µL of 50 mM pH 7.4 PBS buffer was treated in a 1.5 -mL Eppendorf tube at 25 °C for 2 h. The modified product was characterized by LC-MS and LC-MS/MS analysis. For modification of cysteine-containing peptide 1a with coumarin-derived isoxazolinium regent 2r, the modification was conducted using the aforementioned conditions except that 3 µL of isoxazolinium regents 2r (5 mM in CH3CN/H2O containing 5 mol% AgNO3), 3.5 µL of CH3CN, and 3.5 µL of H2O were added. For modification of cysteine-containing peptide 1a with fluorescein-derived isoxazolinium regent 2s, the modification was conducted using the aforementioned conditions, except that 5 µL of isoxazolinium regents 2s (5 mM in CH3CN/H2O containing 5 mol% AgNO3), 2.5 µL of CH3CN and 2.5 µL of H2O were added.
Macrocyclization of peptides using isoxazolinium reagents 2u
A mixture of 10 µL of peptides 1j-k (1 mM in H2O), 2 µL of isoxazolinium regents 2u (5 mM in CH3CN/H2O containing 5 mol% AgNO3), 4 µL of TCEP (12.5 mM in H2O), 4 µL of CH3CN, and 80 µL of 50 mM pH 7.4 PBS buffer was treated in a 1.5 -mL Eppendorf tube at 25 °C for 2 h. The modified product was characterized by LC-MS analysis.
Time course studies on the modification of peptide STSSSCNLSK 1a
A mixture of 20 µL of peptides 1a (1 mM in H2O), 4 µL of isoxazolinium regent 2a (5 mM in CH3CN/H2O containing 5 mol% AgNO3), 8 µL of CH3CN, 8 µL of H2O, and 160 µL of 50 mM PBS buffer with different pH values was treated in a 1.5 -mL Eppendorf tube at different temperatures for 0–4 h. At each time point, 10 µL of the resulting mixture was collected and mixed with 10 µL of L-cysteine (50 mM in H2O) to quenched the modification. The resulting mixture was characterized by LC-MS and LC-MS/MS analysis to determine the conversion.
Modification of proteins using isoxazolinium reagents 2a and 2p
A mixture of 10 µL of proteins (HSA, BSA, insulin, RNaseA, or lysozyme) (1 mM in 50 mM pH 7.4 PBS buffer), 2 µL of isoxazolinium reagents 2a or 2p (5 mM in CH3CN/H2O containing 5 mol% AgNO3), 4 µL of CH3CN, 4 µL of H2O, and 80 µL of 50 mM pH 7.4 PBS buffer was treated in a 1.5 -mL Eppendorf tube at 25 °C for 2 h. The modified product was characterized by LC-MS analysis. For modification of the therapeutic protein BCArg, a mixture of 100 µL of BCArg (1 mM in 50 mM pH 7.4 Tris-HCl buffer), 50 µL of isoxazolinium reagents 2a or 2p (5 mM in CH3CN/H2O containing 5 mol% AgNO3), 25 µL of CH3CN, 25 µL of H2O, and 800 µL of 50 mM pH 7.4 Tris-HCl buffer was treated in a 1.5 -mL Eppendorf tube at 25 °C for 2 h. The modified product was characterized by LC-MS analysis. Before sequential modification and analysis of biological properties, the modified proteins in solution were added into the filter of a Millipore Amicon® Ultra-4 or −15 10 -K centrifugal device. After that, the filter was filled with 15% CH3CN/50 mM pH 7.4 PBS buffer (or Tris-HCl buffer in consistent to the buffer used in the modification). The Amicon® Ultra device was centrifuged under 4000 RCF for 20 min by a BOECO CENTRIFUGE C-28A bench-top centrifuge. The purification process was repeated for three times. The 50 mM pH 7.4 buffer was used instead of 15% acetonitrile/50 mM pH 7.4 buffer at the last time. Modified proteins (0.1 mM) in 50 mM pH 7.4 buffer were collected.
Sequential modification of proteins via Huisgen azide-alkyne cycloaddition
A mixture of 50 µL of alkyne-functionalized proteins HSA-2 or BSA-2 (0.1 mM in 50 mM pH 7.4 PBS buffer), 5 µL of rhodamine azide 9 (5 mM in DMSO), 5 µL of TBTA (5 mM in DMSO), 5 µL of TCEP (5 mM in H2O), 5 µL of CuSO4 solution (5 mM in H2O) and 30 µL of pH 7.4 PBS buffer was treated in a 1.5 -mL Eppendorf tube at 25 °C for 1 h. The modified product was characterized by LC-MS analysis. For modification of alkyne-functionalized proteins BCArg-2, 50 mM pH 7.4 Tris-HCl buffer was used instead of PBS buffer.
Procedure for photocleavage of modified peptide and proteins
Photolysis experiments were performed using a MAXIMA™ ML-3500S/FB Ultra-High Intensity UV-A Lamp (365 nm, 230 V, 50 Hz, 0.75 AMP). For photocleavage of modified peptides, a mixture of 50 µL of modified peptides 3a or 3i (0.1 mM in 50 mM pH 7.4 PBS buffer/CH3CN (19:1)), and 950 µL of 50 mM pH 7.4 PBS buffer in a well of the Thermo Scientific™ Nunc™ Cell-Culture Treated 24-well plate was irradiated by a UV-A Lamp (λmax = 365 nm) on an ice bath for 0–40 min. The resulting mixture was characterized by LC-MS and LC-MS/MS analysis. For time course experiments, at each time point, 20 µL of the mixture was collected for LC-MS analysis. For photocleavage of modified proteins, a mixture of 250 µL of modified proteins HSA-3 or BSA-3 (0.1 mM in 50 mM pH 7.4 PBS buffer/CH3CN (19:1)) and 750 µL of 50 mM pH 7.4 PBS buffer in a well of the Thermo Scientific™ Nunc™ Cell-Culture Treated 24-well plate was irradiated by a UV-A Lamp (λmax = 365 nm) on an ice bath for 30 min. The resulting mixture was characterized by LC-MS analysis.
Data availability
All principal data with detailed experimental procedure and characterization of this work are included in this article, and its Supplementary Information or are available from the corresponding author upon reasonable request.
References
Stephanopoulos, N. & Francis, M. B. Choosing an effective protein bioconjugation strategy. Nat. Chem. Biol. 7, 876–884 (2011).
Spicer, C. D. & Davis, B. G. Selective chemical protein modification. Nat. Commun. 5, 4740 (2014).
Boutureira, O. & Bernardes, G. J. L. Advances in chemical protein modification. Chem. Rev. 115, 2174–2195 (2015).
Koniev, O. & Wagner, A. Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 44, 5495–5551 (2015).
Krall, N., da Cruz, F. P., Boutureira, O. & Bernardes, G. J. L. Site-selective protein-modification chemistry for basic biology and drug development. Nat. Chem. 8, 103–113 (2016).
Chudasama, V., Maruani, A. & Caddick, S. Recent advances in the construction of antibody–drug conjugates. Nat. Chem. 8, 114–119 (2016).
Choi, S., Connelly, S., Reixach, N., Wilson, I. A. & Kelly, J. W. Chemoselective small molecules that covalently modify one lysine in a non-enzyme protein in plasma. Nat. Chem. Biol. 6, 133–139 (2010).
Asano, S., Patterson, J. T., Gaj, T. & Barbas, C. F. Site-selective labeling of a lysine residue in human serum albumin. Angew. Chem., Int. Ed. 53, 11783–11786 (2014).
Nanna, A. R. et al. Harnessing a catalytic lysine residue for the one-step preparation of homogeneous antibody-drug conjugates. Nat. Commun. 8, 1112 (2017).
Matos, M. J. et al. Chemo- and regioselective lysine modification on native proteins. J. Am. Chem. Soc. 140, 4004–4017 (2018).
Chalker, J. M., Bernardes, G. J. L., Lin, Y. A. & Davis, B. G. Chemical modification of proteins at cysteine: opportunities in chemistry and biology. Chem. Asian J. 4, 630–640 (2009).
Gunnoo, S. B. & Madder, A. Chemical protein modification through cysteine. ChemBioChem 17, 529–553 (2016).
Bernardes, G. J. L., Chalker, J. M., Errey, J. C. & Davis, B. G. Facile conversion of cysteine and alkyl cysteines to dehydroalanine on protein surfaces: versatile and switchable access to functionalized proteins. J. Am. Chem. Soc. 130, 5052–5053 (2008).
Chalker, J. M., Lin, Y. A., Boutureira, O. & Davis, B. G. Enabling olefin metathesis on proteins: chemical methods for installation of S-allyl cysteine. Chem. Commun. 7, 3714–3716 (2009).
Shiu, H.-Y. et al. Electron-deficient alkynes as cleavable reagents for the modification of cysteine-containing peptides in aqueous medium. Chem. Eur. J. 15, 3839–3850 (2009).
Smith, M. E. B. et al. Protein modification, bioconjugation, and disulfide bridging using bromomaleimides. J. Am. Chem. Soc. 132, 1960–1965 (2010).
Li, G.-L. et al. Multifunctional bioconjugation by Morita–Baylis–Hillman reaction in aqueous medium. Chem. Commun. 48, 3527–3529 (2012).
Toda, N., Asano, S. & Barbas, C. F. Rapid, stable, chemoselective labeling of thiols with Julia-Kocieński-like reagents: a serum-stable alternative to maleimide-based protein conjugation. Angew. Chem. Int. Ed. 52, 12592–12596 (2013).
Spokoyny, A. M. et al. A perfluoroaryl-cysteine SNAr chemistry approach to unprotected peptide stapling. J. Am. Chem. Soc. 135, 5946–5949 (2013).
Abbas, A., Xing, B. & Loh, T. P. Allenamides as orthogonal handles for selective modification of cysteine in peptides and proteins. Angew. Chem. Int. Ed. 53, 7491–7494 (2014).
Zhang, C. et al. π-Clamp-mediated cysteine conjugation. Nat. Chem. 8, 120–128 (2016).
Brown, S. P. & Smith, A. B. Peptide/protein stapling and unstapling: introduction of s-tetrazine, photochemical release, and regeneration of the peptide/protein. J. Am. Chem. Soc. 137, 4034–4037 (2015).
Bernardim, B. et al. Stoichiometric and irreversible cysteine-selective protein modification using carbonylacrylic reagents. Nat. Commun. 7, 13128 (2016).
Zhang, Y. et al. Thiol specific and tracelessly removable bioconjugation via Michael addition to 5-methylene pyrrolones. J. Am. Chem. Soc. 139, 6146–6151 (2017).
Yu, J., Yang, X., Sun, Y. & Yin, Z. Highly reactive and tracelessly cleavable cysteine-specific modification of proteins via 4-substituted cyclopentenone. Angew. Chem. Int. Ed. 57, 11598–11602 (2018).
Chan, A. O.-Y. et al. Gold-mediated selective cysteine modification of peptides using allenes. Chem. Commun. 49, 1428–1430 (2013).
Kung, K. K.-Y. et al. Cyclometalated gold(III) complexes for chemoselective cysteine modification via ligand controlled C–S bond forming reductive elimination. Chem. Commun. 50, 11899–11902 (2014).
Vinogradova, E. V., Zhang, C., Spokoyny, A. M., Pentelute, B. L. & Buchwald, S. L. Organometallic palladium reagents for cysteine bioconjugation. Nature 526, 687–691 (2015).
Rojas, A. J. et al. Divergent unprotected peptide macrocyclisation by palladium-mediated cysteine arylation. Chem. Sci. 8, 4257–4263 (2017).
Messina, M. S. et al. Organometallic gold(III) reagents for cysteine arylation. J. Am. Chem. Soc. 140, 7065–7069 (2018).
Leriche, G., Chisholm, L. & Wagner, A. Cleavable linkers in chemical biology. Bioorg. Med. Chem. 20, 571–582 (2012).
Saito, G., Swanson, J. A. & Lee, K.-D. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv. Drug Deliv. Rev. 55, 199–215 (2003).
Cal, P. M. S. D., Bernardes, G. J. L. & Gois, P. M. P. Cysteine-selective reactions for antibody conjugation. Angew. Chem. Int. Ed. 53, 10585–10587 (2014).
Clark, P. L. & Lowe, G. Conversion of the active-site cysteine residue of papain into a dehydro-serine, a serine and a glycine residue. Eur. J. Biochem. 84, 293–299 (1978).
Wright, T. H., Vallée, M. R. J. & Davis, B. G. From chemical mutagenesis to post-expression mutagenesis: a 50 year odyssey. Angew. Chem. Int. Ed. 55, 5896–5903 (2016).
Sun, S., Oliveira, B. L., Jiménez-Osés, G. & Bernardes, G. J. L. Radical-mediated thiol-ene strategy: photoactivation of thiol-containing drugs in cancer cells. Angew. Chem. Int. Ed. 57, 15832–15835 (2018).
Nguyen, D. P. et al. Genetic encoding of photocaged cysteine allows photoactivation of TEV protease in live mammalian cells. J. Am. Chem. Soc. 136, 2240–2243 (2014).
Chan, W.-K., Ho, C.-M., Wong, M.-K. & Che, C.-M. Oxidative amide synthesis and N-terminal α-amino group ligation of peptides in aqueous medium. J. Am. Chem. Soc. 128, 14796–14797 (2006).
Chan, A. O.-Y. et al. Modification of N-terminal α-amino groups of peptides and proteins using ketenes. J. Am. Chem. Soc. 134, 2589–2598 (2012).
Cui, J.-F., Kung, K. K.-Y., Ko, H.-M., Hui, T.-W. & Wong, M.-K. Silver-catalyzed transformation of propargylic amine N-oxides to enones and acyloxy ketones via isoxazolinium intermediates. Adv. Synth. Catal. 356, 2965–2973 (2014).
Wei, C. & Li, C.-J. A highly efficient three-component coupling of aldehyde, alkyne, and amines via C−H activation catalyzed by gold in water. J. Am. Chem. Soc. 125, 9584–9585 (2003).
Lo, V. K.-Y., Liu, Y.-G., Wong, M.-K. & Che, C.-M. Gold(III) salen complex-catalyzed synthesis of propargylamines via a three-component coupling reaction. Org. Lett. 8, 1529–1532 (2006).
Lo, V. K.-Y., Kung, K. K.-Y., Wong, M.-K. & Che, C.-M. Gold(III) (C^N) complex-catalyzed synthesis of propargylamines via a three-component coupling reaction of aldehydes, amines and alkynes. J. Organomet. Chem. 694, 583–591 (2009).
Kung, K. K.-Y. et al. Cyclometallated gold(III) complexes as effective catalysts for synthesis of propargylic amines, chiral allenes and isoxazoles. Adv. Synth. Catal. 355, 2055–2070 (2013).
Mucsi, Z., Szabó, A., Hermecz, I., Kucsman, Á. & Csizmadia, I. G. Modeling rate-controlling solvent effects. The pericyclic meisenheimer rearrangement of n-propargylmorpholine N-oxide. J. Am. Chem. Soc. 127, 7615–7631 (2005).
Lau, Y. H., de Andrade, P., Wu, Y. & Spring, D. R. Peptide stapling techniques based on different macrocyclisation chemistries. Chem. Soc. Rev. 44, 91–102 (2015).
Kung, K. K.-Y., Wong, K.-F., Leung, K.-C. & Wong, M.-K. N-terminal α-amino group modification of peptides by an oxime formation–exchange reaction sequence. Chem. Commun. 49, 6888–6890 (2013).
García, D., Uribe, E., Salgado, M., Martínez, M. P. & Carvajal, N. Mutagenic and kinetic support for an allosteric site in arginase from the extreme thermophile Bacillus caldovelox, which allows activation by arginine. Biochimie 108, 8–12 (2015).
Cheng, P. N.-M. et al. Pegylated recombinant human arginase (rhArg-peg5,000mw) inhibits the in vitro and in vivo proliferation of human hepatocellular carcinoma through arginine depletion. Cancer Res. 67, 309–317 (2007).
Yau, T. et al. Preliminary efficacy, safety, pharmacokinetics, pharmacodynamics and quality of life study of pegylated recombinant human arginase 1 in patients with advanced hepatocellular carcinoma. Invest. New Drugs 33, 496–504 (2015).
Acknowledgements
We are grateful for the financial support from the Hong Kong Research Grants Council (PolyU 153031/14P), State Key Laboratory of Chemical Biology and Drug Discovery, Department of Applied Biology and Chemical Technology, and the University Research Facility in Life Sciences (ULS) of the Hong Kong Polytechnic University. We thank Dr. B.C.-B. Ko and Mr. W.-C. Chan for providing access to the ultra-high intensity UV-A lamp for photocleavage experiments, Dr. Alston H.-W. Lee for ICP-MS analysis, and Dr. Q. Zhao for advice on SDS-PAGE analysis.
Author information
Authors and Affiliations
Contributions
M.-K.W. and Y.-C.L. conceptualized and supervised the study. J.-R.D., B.Y., J.-F.C. and K.-W.L. performed the organic synthesis in this work. J.-R.D., W.-M.Y. and K. K.-Y.K. performed the bioconjugation experiments and mass spectroscopy analysis of this work. S.-F.C., A.S.-L.L. and M.-C.C. purified and measured the biological activities of the anticancer proteins. Z.Z. performed the SDS-PAGE analysis. J.-R.D. performed the photolysis experiments. M.-K.W., Y.-C.L., J.-R. D. and W.-M.Y. prepared this paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, JR., Chung, SF., Leung, A.SL. et al. Chemoselective and photocleavable cysteine modification of peptides and proteins using isoxazoliniums. Commun Chem 2, 93 (2019). https://doi.org/10.1038/s42004-019-0193-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-019-0193-5
This article is cited by
-
Recent advances in chemical protein synthesis: method developments and biological applications
Science China Chemistry (2024)
-
Site-selective photocatalytic functionalization of peptides and proteins at selenocysteine
Nature Communications (2022)
-
N-Terminal selective modification of peptides and proteins using 2-ethynylbenzaldehydes
Communications Chemistry (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
