
Abstract
Semiconductors are the basis of many vital technologies such as electronics, computing, communications, optoelectronics, and sensing. Modern semiconductor technology can trace its origins to the invention of the point contact transistor in 1947. This demonstration paved the way for the development of discrete and integrated semiconductor devices and circuits that has helped to build a modern society where semiconductors are ubiquitous components of everyday life. A key property that determines the semiconductor electrical and optical properties is the bandgap. Beyond graphene, recently discovered two-dimensional (2D) materials possess semiconducting bandgaps ranging from the terahertz and mid-infrared in bilayer graphene and black phosphorus, visible in transition metal dichalcogenides, to the ultraviolet in hexagonal boron nitride. In particular, these 2D materials were demonstrated to exhibit highly tunable bandgaps, achieved via the control of layers number, heterostructuring, strain engineering, chemical doping, alloying, intercalation, substrate engineering, as well as an external electric field. We provide a review of the basic physical principles of these various techniques on the engineering of quasi-particle and optical bandgaps, their bandgap tunability, potentials and limitations in practical realization in future 2D device technologies.
Similar content being viewed by others

Nearly all modern semiconductor devices employ some type of band-structure-engineered configuration, through the use of heterostructures1, superlattices2, strain3, alloying4, or other effects. Such band structure engineering can lead to profoundly different properties for the constituent materials. For instance, strain engineering in silicon has allowed transistors with up to fourfold higher hole mobilities as compared with unstrained devices5, and strain engineering in lasers enables vastly reduced threshold currents to be achieved, and allows precise control over the emission wavelength6. Multilayer heterostructures have been demonstrated to provide charge carrier and optical confinement to improve transistor performance7 and realize lasers and light-emitting devices8, while miniband formation in superlattice structures can enable groundbreaking new device concepts such as quantum cascade lasers9. Over the past few decades, heterostructures have also led directly to advances in fundamental physics, such as quantum tunneling phenomena10 and the fractional quantum Hall effect11. In fact, it is now difficult to imagine a world before band-structure-engineered devices and materials.
In current electrical and optical technology, it is highly desirable to be able not only to tune the semiconductor properties, but also to miniaturize the corresponding devices toward nearly atomically thin dimensions. These considerations focused attention to natural or synthesized materials that are formed by stacking individual atomic layers commonly referred to as 2D materials. 2D materials are characterized by a layered crystal structure with strong in-plane bonds, where layers are coupled together by weak van der Waals (vdW) forces. Currently, hundreds of 2D materials are known12,13. Many are natural semiconductors, along with metals and insulators. Because of the weak bonding between layers, numerous heterostructures between 2D materials can also be formed14,15.
Unlike conventional quantum well semiconductors, vdW semiconductors allow for atomic control of their thicknesses. It is well known that thickness variations in semiconductor quantum wells lead to degradation in device performance16. We review the physics of bandgap scaling with layer number and how it differs from the usual effective mass model used in conventional quantum wells, direct-to-indirect bandgap transitions, and the influence of interlayer coupling. One of the most relevant ways of tuning bandgaps in 2D materials originates from the fact that they can be stacked in arbitrary fashion, largely unconstrained by the lattice-matching requirements as in conventional quantum well heterostructures. We review the large degrees of freedom that such an approach can offer in terms of bandgap and band alignment engineering, the influence of stacking, twist angle, interlayer coupling, and experimental growth of these heterostructures. Most importantly, because of the very large surface-to-volume ratios of the 2D layers, their band structure should be very sensitive to external interactions, suggesting that their electronic structure and bandgaps could be modified by external perturbations. We review the tuning of bandgap with the application of an electric field, such as the Franz–Keldysh and Stark effect, the influence of dielectric environment on optical and quasi-particle (QP) gaps, and strain engineering. In all these cases, the bandgap tunability through the above-mentioned external perturbations is about one order of magnitude stronger than in their bulk counterparts. Last, the layered nature of these materials also lends itself to unique chemical approaches to materials engineering. We review the most common schemes, which include alloying, chemical doping, and intercalation of chemical species. These approaches can be very effective, allowing the smooth interpolation of electronic properties, such as through ternary, quaternary, or quinary alloys; tuning of optical gaps and their photoluminescence (PL); and semiconductor-to-metal transitions.
This review is organized as follows: we first provide a survey of the bandgaps of several 2D materials, along with a brief discussion about their most interesting features. We then discuss the influence of the number of layers on the bandgap and the excitonic properties in van der Waals heterostructures (vdWH). Next, we review the latest developments on the use of external electric fields, dielectric environment, and strain engineering for the control of the energies of QP gaps and excitonic peaks in 2D materials. Further, we review the effects of molecular intercala- tion, chemical doping, and alloying on the bandgap. Finally, the bandgap closing and metal–insulator transition in transition-metal dichalcogenides (TMDCs) mediated by structural phase transitions is discussed, followed by our concluding remarks.
Bandgaps in 2D materials family
In Fig. 1, we survey the crystal structures and bandgaps of 2D materials. Currently, research on 2D materials beyond graphene has become very extensive, and is raising expectations for a wide range of applications13. Following similar mechanical exfoliation approaches, monolayers of TMDCs were isolated following graphene12,17. The TMDCs have chemical composition MX2 (M = transition metals, and X = S, Se, and Te) and exhibit various structural phases, such as the 2H, 1T, 1T’, and 1Td. In particular, the semiconducting 2H TMDCs (e.g., MoS2) have bandgaps in the 1–2-eV range and received significant attention due to their interesting circular valley dichroism and exciton physics18. On the other hand, the T phases of TMDCs (e.g., WTe2) are generally metallic to semimetallic19, and are also interesting for their topological properties20. The chalcogenide-based 2D materials also include the semiconducting transition-metal trichalcogenide (e.g., TiS3) whose crystal structure consists of quasi-one-dimensional atomic chains of stacked triangular prism21. There are also group-III-elements 2D chalcogenides, such as GaSe22, which come in different polytypes per their layer-stacking configuration. The group-IV-elements 2D chalcogenides consist of the buckled and puckered monochalcogenides (e.g., SnS and GeSe)23, while their 1T dichalcogenides (e.g., SnS2)24 are mostly semiconducting. The family of 2D transition-metal carbides, nitrides, and carbonitrides are collectively referred to as MXenes25, which exhibits favorable properties of ceramics, such as structural stiffness and good thermal and electrical conductivity of metals. They can have the following chemical formulae: M2X, M3X2, and M4X3, where M is an early-transition metal and X is carbon and/or nitrogen (e.g., Ti3C2). Wide-bandgap III–V 2D semiconductors, such as Ga2N3, were also recently synthesized26.

2D materials are chosen for their experimental significance and demonstration, with depictions of a perspective view of their crystal structures. Arrangement is in accordance with their bandgap, guided by the bottom wavelength/bandgap scale, whereas the bar beneath each structure indicates bandgap range from bulk to monolayer. Typically, the bulk bandgap is smaller than that of its monolayer (black bars), but there are exceptions (red bars). 2D materials on the far left, indicated by a gray box, are zero or near-zero bandgap, metallic, or semimetallic.
Large-bandgap materials such as hexagonal boron nitride (h-BN)27 play a critical role in 2D materials, as its inert and ultraflat nature allows it to serve as a substrate for high-mobility 2D devices. Other large-bandgap materials include the transition-metal oxides (TMO) such as the 2H phase MoO2, 1T phase MnO2, and, more recently, the octahedral α-MoO3, which exhibit hyperbolic optical behavior28, the chromium oxide (e.g., Cr2O3), known for its multiferroic properties29, and mica30.
Other examples of layered materials include perovskites (e.g., SrTiO3), which have been intensively studied as solar cell materials31,32, and topological insulators (e.g., Bi2Se3 and Sb2Se3) known for their topologically protected and spin-momentum-locked electronic transport33. Recent discovery of 2D Cr-based (e.g., CrI3)34 and Fe-based (e.g., Fe3GeTe2)35 ferromagnetic materials, as well as semiconducting ferroelectric materials (e.g., In2Se3)36, introduced an arsenal of functional materials to the existing 2D family. Besides graphene, there are also several notable examples of elemental 2D materials, such as the various phases of phosphorus (e.g., black and blue phosphorus)37, silicene38, germanene39, tellurene40, gallenene41, antimonene42, and borophene43, which can range from metallic to semiconducting. Figure 1 depicts the bandgap range of the above-mentioned 2D materials from monolayer to bulk, showing the span of energy gaps across the electromagnetic spectrum from terahertz, infrared, and visible to ultraviolet.
Bandgap in van der Waals multilayers
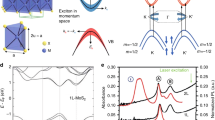
Let us start with the layer dependence of bandgaps in a widely studied elemental semiconductor, namely, black phosphorus (BP). This material is a stable allotrope of phosphorus that can be exfoliated into multilayer puckered honeycomb lattices of phosphorus37, see Fig. 1 for illustration of its crystal structure. The dependence of the absorption spectra of BP on its number of layers N has been experimentally probed by reflectance measurements44,45,46, showing optical gaps ranging from 1.66 eV in monolayers to 0.30 eV in bulk. Typical absorption curves of N-layer black phosphorus (N-BP) are shown in Fig. 2a, showing characteristic peaks that can be traced to transitions between the electron and hole subbands of the same index i47. Figure 2b compiles the energies of these subband transition peaks labeled as Eii in Fig. 2a as a function of the number of layers (symbols). As we will discuss in what follows, the peak features observed in the absorption spectrum of N-BP can be well described within a simple theoretical model, i.e., solid lines in Fig. 2b.

a Experimentally observed absorption spectrum in N-BP, N = 4, 5, 7, 846, where peaks are identified as the Eij transitions labeled in (d). b Positions of experimentally observed (symbols) peaks labeled as E11 (black), E22 (red), E33 (blue), and E44 (green), as a function of the number of layers N. Theoretical predictions (see text) are shown as curves. c Energy bands in monolayer, bilayer, and trilayer BP. d Conduction (Ee) and valence (Eh) band edges, separated by a \({E}_{gap}^{(1)}\) gap in the monolayer case. As the number of layers N increase, more bands appear around Ee(h), with energies Ee(h) ± te(h), for 2-BP, and \({E}_{e(h)}\pm \sqrt{2}{t}_{e(h)}\), for 3-BP. For N-BP, N bands appear around Ee(h), whose energies can be estimated by the eigenvalues of a Toeplitz matrix (see text). Panels a, b reused from Springer Nature/Zhang et al.46, permissible under a CC-BY [4.0/3.0] license.
The number of electron and hole bands in the band structure scales with N. Figure 2c shows the evolution of the band structure of few-layer BP with 1–3 layers, as obtained from tight-binding model and continuum approximation48. At the Γ point, where the QP gap resides, the appearance of these new bands, with lower (higher) energy in the electron (hole) branch, allows the QP gap of black phosphorus to be tuned from 2.2 eV for monolayer to 0.3 eV in bulk. This is also consistent with predictions by density functional theory (DFT) calculations with GW corrections49. The evolution of the bandgap with layer number can be drawn from a very simple model, which we will elaborate below.
If we assume each BP layer as a quantum well for electrons (holes), whose ground state is the band edge Ee(h), a coupled double-well model would predict the splitting of the conduction (valence) band in 2-BP, with energies Ee(h) ± te(h), as sketched in Fig. 2d, where te(h) is the nearest-neighbor interlayer hopping for electrons (holes). In fact, for N-BP, this simple model would consist of a tridiagonal matrix Hamiltonian, \({H}_{e(h)}={E}_{e(h)}\left|i\right\rangle \langle i| -{t}_{e(h)}| i\rangle \langle i+1| -{t}_{e(h)}^{* }| i+1\rangle \langle i| ,{\rm{where}}\ | i\rangle\) represents a state confined to the ith BP layer. Exact diagonalization of such a N × N matrix for N-BP leads to \({E}_{e(h)}^{n}={E}_{e(h)}-2{t}_{e(h)}\cos \left[n\pi /(N+1)\right]\) as the energies of the nth band edge for electrons (holes) at Γ. The peak features in the experimental absorption spectra in Fig. 2a are due to transitions between electron and hole bands with the same index, with energies
where \({E}_{gap}^{(1)}={E}_{e}-{E}_{h}\) is the QP gap of 1-BP. The success of such simple theory is attested by the fair agreement between experimental data (symbols) and predicted transition energies (curves) with layer number N as shown in Fig. 2b.
In contrast to N-BP, the stacking of TMDC layers leads to systems with strikingly different electronic properties. Studies have found that the electronic bandgap, and its band structure in general, depends sensitively on the stacking order, which dictates its interlayer coupling. Indeed, stacking order significantly alters the crystal symmetry and electronic spectrum50, from which second-order harmonics generation51, magnetism35, and superconductivity52 might arise. Nevertheless, for the hexagonal 2H phase of TMDC, these multilayers show weak interlayer interactions at arbitrary fault angles, due to momentum mismatch between the electrons in neighboring layers. This lack of interlayer registry suppresses coherent interlayer motion of electrons, and the conduction and valence band-edge states of each layer remain quantum mechanically decoupled.
Here, we focus our discussion on AB-stacked TMDC, the energetically most favorable atomic configuration in bulk TMDC. As a representative of the TMDC materials, the band structure of bulk and monolayer WS2 is shown in Fig. 3a. Spin–orbit splitting is disregarded here for the sake of simplicity. In the bulk, the valence (conduction) band edge is observed at the Γ (Q) point and the bandgap is therefore indirect. As the number of layers decreases down to bilayer TMDC, both band edges become comparable in energy with those at the K point. In monolayer WS2, the band edges are located at K and the gap becomes direct. The direct transitions at K lead to prominent features in both absorption and PL spectra of monolayer TMDC18,53,54. In the former, these peaks are usually labeled as A and B and assigned to excitonic transitions between different pairs of same-spin states at K. As for the latter, it exhibits an extra lower energy feature, due to the indirect gap transitions, usually labeled as I, as shown in Fig. 3b.

a Band structure of bulk (top) and monolayer (bottom) WS2. High symmetry points are shown on the right side. Band structures of bulk MX2 are qualitatively similar to this, where the lowest energy gap is indirect. This allows for photoluminescence originating from indirect gap transitions, labeled as I, as well as direct gap transitions, labeled as A and B. b Photoluminescence (PL) spectra of monolayer (1ML) to 8-layer (8ML) WS2, measured at temperature T = 5 K, along with optical images of the samples (insets)56. Labels A and I refer to the direct and indirect transitions sketched in (a), respectively. The B transition has higher energy, outside the range of energies in the figure, and is therefore not shown. c Dependence of the energy of the most prominent PL peak on the number of layers for different TMDC58. Panel b used with permission from The Royal Society of Chemistry/Molas et al.56; panel c reprinted by permission from Springer Nature Nature Communications58, Copyright (2014), advance online publication, 06th February 2014 (https://doi.org/10.1038/ncomms4252 Nat. Commun.).
The decrease of the intensity of A and B PL peaks with the number of layers was observed in experiments53,55,56,57, as a result of the transition from direct-to-indirect bandgap. The energies of these peaks, however, exhibit very weak dependence on the number of layers, being just slighly red-shifted (few tens of meV) from monolayer to bulk. This is reasonable, in light of the band structure shown in Fig. 3a, since the energy band is almost flat from K to H (out-of-plane crystallographic direction).
On the other hand, the I peaks observed in PL experiments from bilayer to bulk TMDC, where this peak represents the lowest energy transition, are highly sensitive to the number of MX2 layers, as illustrated in Fig. 3c and reported in refs. 53,55,56,57. Red shifts in this peak are observed in the range of 200–300 meV. Again, this can be understood from the band structure shown in Fig. 3a: in contrast to the energy bands along the K–H direction, the valence band in the Γ–A direction is not flat, which suggests much stronger interlayer hoppings for hole states. Equation (1) would predict the dependence of the I peak energy on the number of layers, as it has been done for WS2 in ref. 56, where the layer dependence of the I peak energy can be fitted with a quantum well-like expression as \({E}_{I}^{(N)}={E}_{0}+{\hslash }^{2}{\pi }^{2}/2{\mu }_{\perp }{L}^{2}\), where E0 = 1.39 eV, μ⊥ = 0.58m0 is the reduced electron–hole effective mass along the vertical direction, m0 is the free electron mass, and the system thickness is approximated as L = Na, where a = 6.16 Å is the thickness of monolayer WS2. The correspondence between such quantum well approximation and the tight-binding model in Eq. (1) is revealed as one takes N → ∞ in this equation, and \({E}_{I}^{bulk}={E}_{gap}^{1}-2({t}_{e}-{t}_{h})\cos (ka)\). This cosine function is then an approximation of the energy dispersion along the vertical direction—its second derivative around Γ (Q) for holes (electrons) leads to electron (hole) effective masses μ⊥ in ref. 56, which are related to te(h) hopping parameters by \({m}_{e(h),\perp }={\hslash }^{2}/2{t}_{e}{a}^{2}\). As one can see in Fig. 1, the bandgap of layered materials is usually reduced as the number of layers increases, with only a few exceptions, such as mica30, a wide-bandgap material with the unique property of exhibiting bandgap narrowing with the decrease in the number of layers.
The behavior of ReS2, on the other hand, is particularly different from the other TMDC, as shown in Fig. 3c. Its lowest energy peak in PL is not strongly affected in either energy or intensity by the number of layers. The band structure of ReS2 is very different from the one of MoS2: as a consequence of a strong decoupling between ReS2 layers, the band structure barely changes from monolayer to bulk58. Similar behavior is also observed for ReSe259. The octahedral 1T phase of TMDC, e.g., for ReS2, undergoes a Peierls distortion resulting in buckled S layers and zigzag Re chains along one of the in-plane lattice vectors, which also lead to electronically and mechanically decoupled monolayers. These materials are thus not suitable for bandgap tuning via the number of layers.
Conversely, there is also TMDC that exhibits an exceptionally strong interlayer coupling energy. This is the case of the noble metal dichalcogenides, such as PdSe2, PtSe2, PdS2, and PtS2, whose bandgap is highly tunable by the number of layers, while their indirect nature is preserved from bulk all the way to the monolayer form. Bandgap in PdSe2, for example, can be tuned from 1.37 eV (monolayer) to 0.5 eV (bulk)60. As for PtSe2, the monolayer gap 1.2 eV drops to only 0.3 eV already in the bilayer, and it becomes metallic for any higher number of layers61. These materials are thus promising candidates for future infrared and mid-infrared photonic and optoelectronic applications.
Bandgap and excitons in vdWH
Vertical stacking of 2D materials into heterostructures allows for the realization of a vastly expanded set of material combinations15,62 that cannot be achieved in 3D materials. In type-I band alignment, conduction band minimum and valence band maximum are in the same material, thus maximizing the electron–hole overlap, which could enable new classes of light-emitting devices63,64. On the other hand, type-II alignment, where conduction band minimum and valence band maximum are in different materials, as well as the semimetallic (zero-bandgap) type-III cases, has potential to create ultrafast photodetectors65, monolayer solar cells66, memory devices67, and tunneling transistors68.
An example of 2D vdWH is illustrated in Fig. 4a, which shows a microscope image of heterobilayer composing of WSe2 monolayer deposited on top of a MoSe2 monolayer69. When different TMDCs are stacked to form vdWH, they typically do not have particular registry, leading to negligible interlayer coupling. Absorption peaks measured in the heterostructure reveal simply a superposition of exciton and trion peaks in each separate TMDC, except for small energy shifts. PL measurements, on the other hand, consistently exhibit an extra low-energy peak. This is associated with an interlayer exciton70, i.e., an exciton where electrons and holes lie in different layers69, as sketched in the inset of Fig. 4b. An example of such PL spectrum is shown in Fig. 4b, where neutral X\({}_{Mo(W)}^{0}\) and negatively charged X\({}_{Mo(W)}^{-}\) exciton peaks of Mo(W)Se2 are seen at energies greater than ≈1.65 eV, along with an interlayer XIL exciton peak at ≈1.38 eV69. Indeed, the XIL feature is present only in the PL of the vdWH region of the sample in Fig. 4a, whereas measurements at the monolayer Mo(W)Se2 region exhibit only X\({}_{Mo(W)}^{0}\) and X\({}_{Mo(W)}^{-}\). Hence, vdWH figures as an efficient way to control optical bandgaps in TMDC.

a Microscope image of a MoSe2/WSe2 van der Waals heterostructure. b PL spectrum (at T = 20 K) of the sample shown in (a)69. The inset sketches intralayer (red and green) excitons in the Mo(W)Se2 layer, associated with the features labeled as X\({}_{Mo(W)}^{0}\) in the PL spectrum, and the interlayer (yellow) exciton, associated with the XIL peak. X\({}_{Mo(W)}^{-}\) refers to intralayer trions. c Top panels: band structures of single-layer MoS2 (green) and WS2 (red) superimposed, and band structure of a MoS2/WS2 heterostructure. Colors in the latter represent probability-density projections in each material. Bottom panels: the same as top, but for GeS (green) and SiTe (red) layers in the buckled form. d Survey of intralayer (triangles) and IL (circles) exciton peaks, experimentally observed in PL measurements of different combinations of TMDC as vdWH. Background colors delimit the range of energies observed for exciton absorption in single-layer experiments with MoS2 (green), MoSe2 (red), WS2 (blue), and WSe2 (yellow)69,75,76,77,78,79,80,81,82,83,84,85,86,87. Colors of triangles are chosen as to match those corresponding to each material involved in the observed intralayer exciton peak. Panels a, b reprinted by permission from Springer Nature Nature Communications69, Copyright (2015), advance online publication, 24th February 2015 (https://doi.org/10.1038/ncomms7242 Nat.Commun).
For rotation angles where the crystal lattices of the stacked TMDC layers are commensurate, namely, 0∘ (AA stacking) and 60° (AA’ stacking), the conduction (valence) band minimum (maximum) is found at the K point of the Brillouin zone. The band-edge states are mostly composed of d orbitals of the transition-metal atoms, which are buried in-between the chalcogens. These orbitals from transition metals of different layers do not interact strongly, i.e., its bandgap at K should not differ much from a simple superposition of the bandgaps of its constituent materials71, as one can verify in the top panels of Fig. 4c, obtained from first-principles DFT calculations for a WS2/MoS2 heterostructure. In the case of separate monolayers, the band structure of Mo(W)S2 is shown as green (red) lines, whereas in the vdWH, green (red) refers to the projection of the states on the Mo(W)S2 layer. At K, the conduction (valence) band-edge state of WS2/MoS2 vdWH is practically fully confined within the Mo(W)S2 layer, leading to negligible electron–hole overlap. As a consequence, despite the direct gap nature of the K-point electron–hole state, light absorption via valence-to-conduction transition at K point is much suppressed, since the optical susceptibility, as given, e.g., by Elliot formula72, depends not only on the dipole-matrix element of the bands states, but also on the electron–hole overlap of the exciton state. This explains the lack of an IL exciton peak in the absorption spectra of TMDC vdWH. In PL measurements, however, light emission from IL states is made possible as electrons and holes excited by light in the same material may drift toward different layers seeking for lower energy band edges, thus producing a charge-separated IL exciton.
This relaxation process is shown to be faster than recombination of intralayer excitons73,74. Recent observations of IL excitons in PL spectra of MoSe2/WSe2 vdWH75,76,77 suggest that these states may be indirect not only in real space, but also in reciprocal space. In refs. 75,76, two IL exciton peaks are observed at ≈1.4 and ≈1.42 eV and interpreted as reciprocal space indirect excitons where the hole is at the K point, while the electron lies in a spin-split Q valley (see Fig. 4c, top). In ref. 77, an IL exciton peak is observed at 1.33 eV and assigned to a similar reciprocal space indirect exciton, but the second IL exciton peak, at 1.38 eV, is attributed to a K–K direct transition. In a MoS2/WSe2 vdWH, evidence from the dependence of the lowest energy PL peak on the twisting angle between layers points toward an interpretation of this peak as being due to a reciprocal space indirect Γ-to-K IL exciton78. These Γ–K and K–Q indirect excitons share the characteristic of having one of the quasi-particles spread among both TMDC layers, i.e., the particles are only partially separate in real space, which helps to enhance the light emission efficiency of these states.
Figure 4d surveys energies of experimentally observed IL exciton peaks (black circles) in vdWH: MoS2/MoSe279,80, MoS2/WSe278,81, MoSe2/WSe269,75,76,77,82,83, MoS2/h-BN/MoSe284, and MoSe2/WS285,86,87. A simple theoretical model can be made to predict the energy of such IL excitons: (i) once the band edges involved in the IL transition are identified, the effective masses of electrons and holes in those band edges are obtained, (ii) a tight-binding model for the stacked layers similar to the one proposed earlier is constructed for electron and hole states, from where IL hopping parameters te and th are obtained, (iii) electron–hole binding energies Eij are calculated assuming an electron in layer i and a hole in layer j, using an appropriate interaction potential88,89, and (iv) the system Hamiltonian is approximated to be \({H}_{\rm{exc}}={E}_{ij}\left|ij\right\rangle \langle ij| -[{t}_{e}| ij\rangle \langle i+1\,j| -{t}_{h}| ij\rangle \left\langle i\,j+1\right|+c.c.]\) and properly diagonalized, leading to the exciton energy states. Similar recipes were followed, e.g., in refs. 71,90, as well as in ref. 78 for a MoS2/WSe2 vdWH, which helped to understand the apparently high IL exciton-binding energy and peak intensity as being due to a partially charge-separated Γ–K IL exciton. K–K IL exciton transitions have also been observed in PL91 and electroluminescence92 experiments with MoS2/WSe2 vdWH. In this case, the IL peak is observed with lower energy (≈1.1–1.3 eV) as compared with the Γ–K one (≈1.58 eV, see Fig. 4d), as a consequence of the smaller IL gap at K point, as compared with the one at Γ (see, e.g., Supplemental Material of ref. 78).
Intralayer (direct) exciton transitions are also featured in PL measurements of vdWH. Figure 4d shows the energies of these transitions as triangles. The vdWH surveyed in Fig. 4d are those that were experimentally realized to date, and where IL excitons were observed, but possible combinations of TMDC into vdWH are potentially vast. In order to investigate this further, Fig. 5a surveys the bandgaps theoretically obtained for stacked TMDC in the limit of zero interlayer coupling for different TMDC. In this limit, the bandgap is obtained by the difference between the minimum conduction and maximum valence band-edge energies of the constituent monolayers (Anderson model). These TMDC heterostructures exhibit bandgaps in the terahertz-to-infrared range, with different band alignments, namely, of types I–III.

a Quasi-particle bandgap values and types for various 2D vertical heterostructures composed of TMDC, obtained using DFT–PBE calculations and displayed as a heatmap. Monolayer bandgap values are given in the left column, while heterostructure bandgap values, as obtained by the Anderson rule, are shown in the upper triangular half. Type-I, -II, and -III heterostructures are represented by green-, red-, and blue-checkered boxes, respectively, in the bottom triangular region. Top and side views of crystal structure for the different TMDC phases, namely 2H, 1T, and 1Td, are displayed on the far left. b The same as a, but for MMC heterostructures. Bandgap values for monolayer IV chalcogenide in puckered (buckled) phase are shown on the left (right) panel. In general, they do not obey the Anderson’s rule, due to strong interlayer coupling. Monolayer values are shown in the first column of each panel. Top and side views of crystal structures for puckered and buckled crystal groups are displayed as the inset. For crystal structure labels, refer to Fig. 1a.
The metal (group-IV) monochalcogenides (MMC) are also layered semiconductors that have recently been synthesized, and are theoretically predicted to form mechanically rigid heterostructures23,93,94,95,96,97,98. Their monolayers are stable buckled and puckered structures, as depicted in the inset of Fig. 5b. The interlayer coupling for the puckered phase is generally stronger than the buckled counterpart. As a result, the energy gaps for the puckered heterostructures are smaller, mostly in the infrared. On the other hand, the gap in buckled heterostructures mostly spans from near-infrared to red. Figure 5b summarizes the bandgaps of group-IV-chalcogenide heterostructures, obtained from DFT calculations. Due to the strong interlayer coupling, the electronic wavefunctions are highly delocalized across the two layers93, in contrast to the case of TMDC vdWH previously discussed. Hence, their bandgaps do not follow Anderson rule99, as one can verify by comparing the band structures of, e.g., monolayer GeS (green) and SiTe (red) with those of a SiTe/GeS vdWH, which are shown in the bottom panels of Fig. 4c. Thus, from the point of view of electronic structure, a MMC stack should be viewed as a new material, instead of a heterostructure.
The vast majority of reports on 2D material-based heterostructures and devices have used flakes exfoliated and transferred from naturally occuring crystals or crystals grown by chemical vapor transport (CVT). However, those flakes have high levels of extrinsic impurities and intrinsic point defects, which lead to degraded material and device performance with high variability100,101. Furthermore, it is well established that heterostructures that are created through exfoliation and even the “cleanest” subsequent transfer processes102, still result in residual contamination at interfaces and surfaces, which again degrades the heterostructure properties103.
Chemical vapor deposition (CVD) of TMDC has been shown to be quite promising104,105. In particular, CVD films have been demonstrated with very large grains106 and reduced impurity107 and structural defect levels. Transport in CVD-grown TMDC is also quite competitive with reported field-effect mobilities of 20–90 cm2/Vs106,108,109,110,111 for MoS2 and 30 cm2/Vs (true MOCVD)8,112 and 95 cm2/Vs (powder vaporization)113,114 for WSe2 that is approaching the values reported for flakes exfoliated from geologic crystals. Heterostructures using MOCVD have also been reported. Successful heterostructures include MoS2/WSe2115,116, WSe2/MoSe2117, WTe2/WSe2118, GaSe/MoSe2119, and SnS2/WSe2120. However, CVD typically requires growth temperatures in excess of 700 °C that is often much too high to be compatible in heterostructures with underlying chalcogenide layers121. The elevated growth temperatures can result in point defect and vacancy formation and undesired reactions at interfaces116.
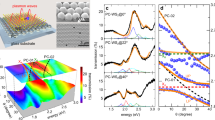
Molecular beam epitaxy (MBE) can oftentimes solve these challenges enabling the growth of 2D materials and heterostructures with ultraclean interfaces, as illustrated in Fig. 6. Enhanced quality films and interfaces with very low-impurity concentrations are enabled in MBE through the utilization of elemental sources of high purity coupled with the cleanliness of ultrahigh vacuum. Moreover, the lower growth temperature capability of MBE minimizes issues associated with vertical heterostructure growth, including vacancy formation, layer intermixing, and interface chemical reactions. MBE, therefore, enables a variety of novel materials, including 2D-layered oxides, nitrides, arsenides, and the wide variety of 2D chalcogenides122,123,124,125,126,127,128. TMDC materials, such as WSe2129, MnSe2130, MnTe2 and VSe2131, VTe2 and HfSe2132, SnSe2125, MoSe2133, MoTe2134, and WTe2135, have all been grown by MBE. In addition, TMDC alloys with mixed metals or mixed chalcogens have been advanced lately, further expanding the library of potential TMDC and heterostructures136,137,138,139,140,141.

a HfSe2 grown on MoS2132, b WSe2 grown on Bi2Se3277, c WTe2 grown on MoS2135, and d helical Te grown on graphite278. In each case, the interfaces between the two materials are atomically sharp with no misfit dislocations due to the lack of strain in the covalent-free interface interactions. Heterostructures can therefore be designed and synthesized with minimal regard for lattice matching. Panels reproduced with permission from a reprinted with permission from132 ACS Nano 2015, 9, 1, 474–480 Publication Date: December 11, (2014) https://doi.org/10.1021/nn5056496 Copyright (2014) American Chemical Society; panel b reused from IOP Publishing/Yue et al.277 permissible under a CC-BY [4.0/3.0] licence; panel d used with permission from John Wiley and Sons/Zhou et al.278.
In TMDC and other van der Waals materials, the layer-to-layer or layer-to-substrate interactions are much weaker than the intralayer bond strength. Coupled with the surface termination of van der Waals crystals, layer-by-layer growth without islanding can be achieved, although determining the growth parameter window that enables that this can be a challenge142. Experimental results of epitaxially grown TMDC have demonstrated that TMDC grown on other inert, hexagonal substrates (other TMDC, graphite, etc.) grows unstrained, with its own lattice constant, and does not form any misfit dislocations143. Utilizing this so-called van der Waals epitaxy enables144 vertical heterostructures with constituent materials chosen almost exclusively for their electronic properties with signifcantly relaxed criteria for crystal lattice matching. Recent findings have also indicated that through controlled growth conditions, the stacking sequence of thin films of TMDC can be altered15. In a high chalcogen background environment in conjunction with a relatively fast growth rate, for example, one can promote differences in edge dimerization and growth rates that result in AA stacking in TMDC that normally has AB stacking in equilibrium (like WSe2 or MoS2). Atomic heterostructuring is an exciting approach whose full potential is yet to be explored.
External electric field effect
In the presence of an external electric field F, energy band edges in a semiconductor are distorted in real space, so that electrons and holes are pushed toward opposite directions. As a consequence, their QP envelope wave functions are no longer sine waves, but rather Airy functions. These functions, however, still exhibit a tail that makes the electron–hole overlap nonnegligible, thus allowing for a finite probability of interband transition mediated by light absorption. As illustrated in Fig. 7a, neglecting electron–hole interactions and excitonic effects, the QP-state transition has energy EFK, which is smaller than the zero-field gap Eg. This phenomenon, also known as the Franz–Keldysh effect, is responsible for a tail in the absorption spectrum of semiconductors at energies below the zero-field QP gap of the material, which suggests the use of an electric field to actively tune optical gap properties of semiconductors.

a Sketch of the quantum-confined Franz–Keldysh effect (top): in the presence of an applied field (blue, F ≠ 0), the otherwise constant bands (black, F = 0) are tilted, which pushes electrons and holes toward opposite sides of the system. Electron and hole wave functions are given by Airy functions (red) with tails that slightly overlap, thus allowing for a light absorption-induced transition with energy EFK lower than the original quasi-particle gap Eg of the system. Bottom panels show an experimental observation of the Franz–Keldysh effect in light absorption for few-layer (9-nm-thick) BP, with several values of applied bias Vg (in volts) and light polarized along the armchair (AC) and zigzag (ZZ) directions of the material145. b Experimental observation of Franz–Keldysh effect as a subgap shoulder (FK) in the absorption spectrum, which emerges among A exciton peaks as the gate voltage VB increases in monolayer WSe2 at temperature T = 30 K151. c Band structure of 2-BP, as obtained with tight-binding (red dashed) and continuum (blue solid) models, in the presence of 1.5-eV (top) and 2-eV (bottom) bias158. d Experimental verification of the bandgap closing and consequent formation of a Dirac cone in few-layer BP due to an effective bias across the sample155. Experimental data and theoretical simulations are shown together, for comparison. e Stark shift of the PL peak (at T = 4.2 K) due to IL excitons in a MoSe2/WSe2 vdWH, as a function of bias induced by top and bottom gates, set at potentials VTG and VBG, respectively. Two energy peaks, labeled as IX1 and IX2 are consistently observed76. Panel a reprinted with permission from145 Nano Lett. 2017, 17, 10, 6315–6320 Publication Date: September 27, 2017 https://doi.org/10.1021/acs.nanolett.7b03050, Copyright (2017) American Chemical Society; panel b reused from Springer Nature/Massicotte et al.151, permissible under a CC-BY [4.0/3.0] license; panel d used with permission from AAAS/Kim et al.155; panel e reprinted by permission from Springer Nature Nature Photonics76, Ciarrocchi, A., Unuchek, D., Avsar, A. et al. Polarization switching and electrical control of interlayer excitons in two-dimensional van der Waals heterostructures. Nature Photon. 13, 131–136 (2019), Copyright (2019) advance online publication, 31st December 2018 (https://doi.org/10.1038/s41566-018-0325-y Nature Photon.).
The 2D nature adds another feature to this scenario: due to the small thickness of the material, electrons and holes pushed toward the top and bottom layers become strongly confined, in what is called the quantum-confined Franz–Keldysh (QCFK) effect. The consequence of this effect is clearly seen in the modulation of the absorption spectrum of 9-nm-thick few-layer BP in the bottom panels of Fig. 7a145. When light is polarized along the zigzag (ZZ) direction of the N-BP crystal lattice, no feature is observed, as expected, due to particular selection rules in BP13. For polarization along the armchair (AC) direction, however, very clear features are observed, such as the presence of absorption peaks and a modulation of the absorption coefficient. Although the Franz–Keldysh effect alone tunes the bandgap only within the range of a few tens of meV, much broader bandgap tuning is achieved as stronger electric fields are applied perpendicularly to multilayer BP146,147 and TMDC148.
For monolayer TMDC, the excitonic Stark shifts are predicted to be small, of the order of a few meV149,150. This has been confirmed in a recent experiment with monolayer WSe2151. On the other hand, a clear subbandgap shoulder related to the Franz–Keldysh effect in this case is observed ≈70 meV below the QP bandgap of this material, amid excitonic state peaks. This is illustrated in Fig. 7b, where the shoulder is seen to emerge as the gate potential VB increases from −0.2 V up to 2.5 V, while the few meV Stark shift of the A exciton peak is not visible in the plot.
Theoretical works146,152,153,154 have suggested that applying an electric field perpendicular to N-BP lowers its QP gap and may close it for high enough fields155. In fact, a variation of the bandgap of few-layer BP as large as 200 meV has been observed for a 2 V/nm displacement field, due to quantum-confined Stark effect, in a SiO2/BP/BN-sandwiched structure156. Such gate-tunable giant Stark effect in few-layer BP was confirmed by a direct measurement, using low-temperature scanning tunneling microscopy157. Further increase in the electric field then produces a linear band crossing along the AC direction, as illustrated in Fig. 7c158. This perculiar band structure was subsequently experimentally observed in ARPES measurements155, as illustrated in Fig. 7d, where experimental data and theoretical simulations are shown side by side for comparison. In this experiment, the strong electric field accross the N-BP layers is obtained by doping the uppermost layer with potassium atoms. Similar surface-doping technique has also been used to tune the bandgap of multilayer TMDC up to hundreds of meV159.
Bandgap modulation induced by a vertical electric field has also been observed in electrical experiments156,160,161,162,163, with significant impact on the transport properties of 2D materials as well. For example, a room-temperature hole Hall mobility of few-layer BP as high as 5000 cm2/Vs is reported in BN/BP/BN van der Waals quantum well160, which exceeds the theoretical limit161 without consideration of QCFK156,162,163.
Electric field tuning of electronic properties of multilayer TMDC is also relevant in the context of vdWH composed of these materials71. As previously discussed, vdWH exhibits a low-energy PL peak related to the IL exciton state. Due to the intrinsic electric dipole across the layers, application of a perpendicular electric field yields a predominantly linear Stark shift69. Figure 7e shows an example of such linear Stark effect, experimentally observed in a MoSe2/WSe2 vdWH, where two interlayer exciton states, labeled IX1 and IX2, are consistently observed and interpreted as being formed by indirect and direct transitions in the reciprocal space, respectively76. In this case, a 138-meV tuning of this lowest-transition energy peak is obtained. Similar tuning is also obtained for IL exciton peaks in MoS2/WSe2 vdWH in ref. 92.
Excitons trapped in defects in monolayer TMDC have been observed as intense features in the PL spectra with energy slightly lower than the optical gap. These optically active defects have attracted interest as possible quantum emitters at low temperature. The energy of these peaks can also be tuned by an external applied field, via quantum-confined Stark effect164. In this case, trapped exciton states can exhibit either linear or quadratic Stark shifts, depending on the character of the defect. The linear contribution is dominant, provided there is an intrinsic electric dipole moment, as a result of a defect that lacks of a center of symmetry. Linear Stark shifts of a few tens of meV were experimentally observed for optically active defects in monolayer WSe2 encapsulated in h-BN164.
Dielectric screening and many-body effects
It is clear that in a 2D semiconductor, the combination of dielectric screening by the material layer itself and that provided by its surrounding environment plays a major role in defining not only exciton-binding energies, but also the QP gaps18,165,166. However, it is known that the dielectric screening-induced shifts of these two effects are opposite; hence, their isolated effects are difficult to discern experimentally. Indeed, early attempts to understand optical gaps under the influence of the surrounding dielectric environment were inconclusive165,167,168,169,170,171,172,173,174,175,176. In these reports, either fused silica or oxides, such as SiO2, MgO, and sapphire, are used as substrates and/or capping layers. Only recently, experimental studies demonstrated that encapsulation of 2D semiconductors with different layered materials, such as h-BN and graphene166,167, produced significant effects in the optical bandgap consistent with theory. This ushered in a new approach of optical gap engineering via encapsulation techniques and dielectric engineering.
A clear experimental evidence of this effect has been reported in ref. 166, where the excitonic ground and excited states in a WS2 monolayer in the presence of a few-layer graphene capping are carefully investigated by differential reflectivity measurements. On the one hand, additional screening from the graphene layers should reduce electron–hole interactions and lead to a blueshift of the excitonic peak. On the other hand, it should also reduce electron–electron interactions, which leads to a redshift in the QP gap renormalization177,178. Overall, the latter was found to dominate, and a red-shift in the optical gap was observed.
Figure 8a surveys experimentally obtained optical gaps under different dielectric environment for various TMDC, with optical gap tuning in the range of a few hundred of meV165,166,167,176,179,180. As for BP, it is commonly covered by capping layers of different materials, such as h-BN and sapphire. A recent theoretical study shows that such encapsulation causes significant changes (≈120 meV for 1-BP) on the optical gap173. This has been experimentally verified in 2-BP and 3-BP173,181, by comparing the results for N-BP supported on SiO2, PDMS45,46, and h-BN substrates44, with and without further coating with sapphire.

a Survey of recent experimentally observed optical gaps of 2D materials in different dielectric environments. For TMDC, lowest energies correspond to samples encapsulated in h-BN. In general, energies increase as the substrate is changed from h-BN to SiO2, CYTOP, MgO, Al2O3, and LaAlO3, respectively167. For N-BP, lower energies correspond to samples on sapphire covered by h-BN173, whereas uncovered samples on Si/SiO2 substrate181 exhibit higher energy. b Difference between optical bandgaps (top) of the suspended monolayer (ε = 1) and those in a medium with an effective dielectric constant \({\varepsilon }_{m}=\frac{{\varepsilon }_{1}+{\varepsilon }_{2}}{2}\) for four TMDC, as calculated by solving SBE for the gapped Dirac Hamiltonian72. The parameter r0 was fitted for each TMDC as to obtain the experimental optical bandgap: 1.91 eV for suspended MoS2279 using r0 = 108 Å, 1.66 eV for MoSe2 deposited on SiO2280 with r0 = 90 Å, 1.74 eV for WSe2 deposited on SiO2178 with r0 = 47 Å, and 2.04 eV for WS2 deposited on SiO2281 with r0 = 23 Å. The other parameters were obtained from the GGA calculations of ref. 282. The effect on the QP bandgap, as calculated by the method explained in the Supplemental Material, is shown in the bottom panel.
Understanding the physics behind the dielectric screening effect on the optical gap is fundamental to provide control of its optoelectronic properties. Let us provide a glimpse of how such a model could be constructed. The simplest way to see the dependence of effective electron–electron interaction in 2D materials on the screening by the environment is to consider a thin slab with width d cladded between two different dielectrics with permittivities ε1 and ε2. In the d → 0 limit, the eletrostatic potential due to a charge bound to the slab is given by the Rytova–Keldysh (RK) potential182,183
where r0/2π is the 2D polarizability of the thin slab and εm = (ε1 + ε2)/2 is the mean value of the dielectric constants of the environment. The corresponding static dielectric function is linear in q, given by ε2D(q) = VRK(q)/V0(q) = εm(1 + r0q), with V0(q) = e2/(2εq) being the 2D Fourier transform of the vacuum Coulomb potential. Therefore, in contrast to bulk materials, where the dielectric constant is an intrinsic property, for a 2D material, the dielectric constant depends on extrinsic properties (εm) and intrinsic properties (r0). The inverse Fourier transform of the potential Eq. (2) is
where H0 and Y0 are the Struve function and the Bessel function of the second kind, respectively. This potential can be further generalized to include finite thickness89, anisotropy184, and a spacer between the 2D material and the substrate168.
Equation (2) can also be obtained from a microscopic viewpoint, as the long-wavelength limit of the dielectric function in the random-phase approximation88. This provides an ab initio way to calculate the parameter r0 from the dielectric constant185. In computational codes for ab initio calculations, it is necessary to put an interlayer distance Lc large enough to avoid interaction between layers. It can be shown that the effective dielectric constant scales with the interlayer distance Lc as88
The dependence of the screening of the carrier–carrier interaction on the environment, as shown by Eq. (2), opens avenues to engineer the desired electronic and optical properties of 2D materials167,169,186.
The optical bandgap is effectively defined as the QP bandgap corrected by the exciton-binding energy. The latter can be obtained by solving the Wannier equation, namely, a hydrogen-like equation describing an exciton187
where r is the relative coordinate between the electron and hole, Eg is the QP bandgap, μ = memh/(me + mh) is the reduced mass, ψnℓ(r) is the exciton envelope wave function, and n (ℓ) is the principal (angular) quantum number. When the RK potential Eq. (3) is used, this equation only has, as input, the external dielectric constant εm, the 2D polarizability r0, and the effective masses. Eq. (5) can be straightforwardly generalized to account for the anisotropic mass and screened potential in, e.g., N-BP184,188. This provides a good aproximation for the first energy level in the exciton Rydberg series187.
While Eq. (5) accounts for the effect of dielectric environment on the exciton-binding energy, it does not account for the QP bandgap renormalization that results from it. On the other hand, both effects are naturally included in the semiconductor Bloch equations (SBE)189, which has been already succesfully applied for 2D materials72,190. Here, we use the SBE to study the effects of the environment on the exciton properties and the QP bandgap. Details of these calculations are shown in the Supplemental Material.
Since the results for N-BP were already previously discussed173, let us now focus on TMDC. Exciton energies and QP bandgaps for TMDC, as obtained by the above-mentioned theoretical procedure, are presented as a function of the effective dielectric constant of the environment in Fig. 8b, which depicts the changes both in the optical ΔEopt = E0(εm) − E0(εm = 1) and QP \(\Delta {E}_{{\rm{Q}}P}={\Sigma }_{{\bf{0}},+}^{{\rm{xc}}}({\varepsilon }_{m})-{\Sigma }_{{\bf{0}},+}^{{\rm{xc}}}({\varepsilon }_{m}=1)\) gaps. Here, E0 is the ground-state exciton energy, and \({\Sigma }_{{\bf{0}},+}^{{\rm{xc}}}\) accounts for the exchange-correlation function191. More details are presented in the Supplemental Material. One clearly sees that the optical bandgap has a weak dependence on the substrate, in accordance with previous calculations192 and experiments179,193. However, the QP bandgap alone exhibits stronger dependence on the environment, also in accordance with experiments170 and theory172,175. It is also clear from Fig. 8b that tungsten-based materials are more susceptible to optical gap modulation by the dielectric environment, as compared with the molybdenum-based ones, which is in accordance with the experimental data shown in Fig. 8a. The results thus far suggest that more is yet to be done in this area in order to exploit the full capability of optical bandgap modulation through dielectric engineering.
Strain engineering effects
Strain engineering aims at modifying the bandgap or the electronic properties of materials by the application of strain. Traditionally, this technique has been developed in the context of conventional semiconductors (such as silicon) toward the enhancement of electronic transport properties194. Its potential use to break the inversion symmetry of the crystal lattice in graphene, thus opening a bandgap in its otherwise gapless band structure, is among the first applications of strain in the context of 2D materials195,196,197. According to theoretical calculations, in order to open a technologically relevant bandgap in graphene of a few hundred meV, a large strain ε ≈ 15–30% is needed198.
Contrary to graphene, TMDC and N-BP are 2D semiconductors with bandgap energies in the range between 0.3 and 2.0 eV, which can be further engineered with strain. Theoretical calculations based on DFT199,200 suggest that the direct QP gap energy in MoS2 decreases by ≈0.8 eV with 9% of biaxial strain. The indirect (K–Q) gap energy undergoes an even stronger decrease; consequently, a direct-to-indirect bandgap transition is observed as biaxial strain is applied in MoS2 and the same is predicted for other TMDCs. Conversely, in monolayer BP, an increase of ≈0.65 (0.4) eV is observed in the optical gap for 4.5% uniaxial strain applied in the AC (ZZ) direction201. Such substantial modifications of the bandgap with the application of moderate strain levels in both cases demonstrate the feasibility of strain-based bandgap engineering of 2D semiconductors.
The tuning of the bandgap in TMDCs by the application of uniaxial and biaxial strain has been experimentally demonstrated in various works202,203,204,205,206. Contrary to dielectric environment engineering, uniaxial strain is demonstrated to lead to large QP gap modulation, but with very weak effect on the exciton-binding energy206. The latter results from slight modifications of the effective masses with strain.
Typically, uniaxial strain is applied by bending or stretching the substrate underneath the 2D material, or by inducing ripples in the material itself. For example, uniaxially-strained monolayer MoS2 can be achieved by bending its polycarbonate substrate in a four-point bending apparatus207. The top panel of Fig. 9a shows the PL of the unstrained and strained (up to a value of 1.8%) monolayer MoS2. The A exciton PL peak, related to the direct bandgap of MoS2, shifts to lower energies when a tensile uniaxial strain is applied, indicating a closing of the bandgap204,207,208. Uniaxial strain tuning of the bandgap has been also demonstrated for other TMDCs, like MoSe2209 and WSe2210, and it has been shown to have a strong impact on the exciton–phonon coupling211. The bottom panel of Fig 9a shows a different experiment in which biaxial strain of monolayer MoS2 was achieved by exploiting the thermal expansion or contraction of the polypropylene substrate carrying the flake212. Alternatively to the use of thermal expansion, biaxial strain has also been demonstrated by transferring MoS2 onto a piezoelectric substrate203, or by applying a pressure difference across suspended MoS2 membranes205. The A exciton peak in the differential reflectance spectra shifts to lower energies when applying biaxial tensile strain (up to 0.9%) and to higher energies for a compressive strain (up to 0.1%). The reported bandgap tunabilities with strain (or gauge factors) are −45 meV/% and −25 meV/%, respectively, for uniaxial and biaxial strain, in good agreement with theoretical predictions206. A 100- (125) meV redshift of the optical (quasi-particle) gap has also been observed in monolayer WSe2 under 2.1% uniaxial strain213. Strain engineering also has the potential to modify the interlayer interaction in multilayered 2D materials. This has been recently employed to modify the interlayer interaction in few-layer black phosphorus214, and to tune the energy of the interlayer excitons in bilayer MoS2215,216 flakes.

a Bandgap tuning in uniaxially207 (top) and biaxially (bottom)212 strained monolayer MoS2. b Superlattice built by introducing a local and periodic strain in a monolayer MoS2 by patterning nanocones in the substrates221. c Exciton funneling and inverse funneling effect in 2D semiconductors201. The bottom panels show an experimental realization of inverse funneling in a locally strained HfS2 photodetector225. Panel a reprinted with permission from207 Nano Lett. 2013, 13, 8, 3626–3630. Publication Date: July 2, 2013 https://doi.org/10.1021/nl4014748 Copyright (2013) American Chemical Society; b reused from Springer Nature/Li et al.221, permissible under a CC-BY [4.0/3.0] license; c reused with permission from APS/San Jose et al.201, and Springer Nature/Sanctis et al.225, permissible under a CC-BY [4.0/3.0] license.
Strain engineering has been recently used to fabricate straintronic devices based on 2D semiconductors. For example, the thermal expansion-based biaxial straining has been recently employed to tune photodetectors based on single-layer MoS2. By controlling the level of strain, it was shown that one can tune the photoresponsivity (by 2–3 orders of magnitude), the response time (from <80 ms to 1.5 s), and the spectral bandwidth (with a gauge factor of 135 meV/% or 58 nm/%) of the device217. Uniaxial strain engineering has also been exploited to modify the in-plane anisotropy of MoS2-based photodetectors. Photodetectors with strong linear polarization-sensitive response from visible to near-infrared range have been shown for monolayer MoS2 devices fabricated onto polydimethylsiloxane, and the polarization anisotropic ratio can reach over 2.0 under 4.5% stretching of the polymeric substrate218.
While the experiments discussed above deal with global and homogeneous strain, many interesting physical effects can be achieved with local strain distributions, such as pseudomagnetic fields197, the creation of wrinkles219, or even “artificial atoms” single-photon emitters, by the exciton-funneling effect220,221. The latter has been proposed as a way to modulate the intrinsic bandgap of monolayer MoS2 and thus improve its photovoltaic and photodetecting properties199. The diagram in the top panel of Fig. 9b sketches an example of local strain distributions induced by patterning nanocones in the substrate, where the bandgap of MoS2 is modulated in such a way that the locations of maximum strain mimic the Coulomb potential around ions in a crystal. A similar approach, exploiting the local induced strain reduction of the bandgap, has been employed to deterministically define single-photon emitters222,223. In the bottom panel of Fig. 9b, a tilted false-color scanning electron microscopy (SEM) image of the strained MoS2 crystal defined by such nanocones array is shown.
A local strain center in a 2D semiconductor leads to exciton funneling, which is a way of controlling exciton motion by means of inhomogeneous strains. The top panel of Fig. 9c shows a representation of exciton funneling in MoS2 (left)224 and inverse exciton funneling in black phosphorous (right)201 together with a schematic band diagram of the process. An experimental realization of the inverse funneling effect was recently demonstrated225, where the photoresponse of a HfS2-based device is tuned by introducing a local strain center through laser-assisted oxidation of HfS2 to HfO2. Other alternative local straining approaches are based on indentation of the 2D material with an AFM tip, that produces strong strain localization right under itself226,227,228.
Strain also plays an important role in lateral heterojunctions between 2D materials with different lattice parameters. The fabrication of seamless lateral heterojunctions has been pursued over the past few years as a path toward monolayer diodes and transistors, see refs. 231,232,233,234. for reviews on their synthesis, optoelectronic properties, and device applications. In this case, a spatial modulation of both the energy and intensity of PL peaks has been experimentally observed229 in the MoS2 region nearby a MoS2–WSe2 lateral heterojunction, where energies were found between 1.79 and 1.91 eV. This modulation has been addressed to the effects on the band structure due to the strain distribution caused by the lattice mismatch between these two materials. Strain engineering in such monolayer lateral junctions has been proven a challenging task, but it would allow for control of the alignment between energy bands of the materials in the heterojunction. Theoretical predictions also suggest that the band structure modification due to tensile strain across a monolayer MoS2/WS2 lateral heterojuction would lead to charge carrier confinement at the interface, thus forming a one-dimensional channel that brings prospects for applications in high-electron-mobility transistors and low-power switching devices230,231,233.
Molecular intercalation and chemical doping effects
Intercalation of 2D materials by organic molecules leads to organic–inorganic hybrid structures, such as p–n junctions235, heterojunctions, and superlattices236,237. This opens a new avenue to modulate and manage materials electronic and optical properties, thermoelectric performances, and doping effects by tailoring their molecular structures, sizes, and symmetries. Molecular intercalation techniques thus build a bridge between traditional integrated circuits and atomically layered materials. In contrast to previous organic–inorganic hybrid systems, organic molecule-intercalated hybrid structures exhibit unique features, such as intrinsic interfaces, atomically seamless junctions, high yield, and reproducibility235,236,237. Recent experiments involving intercalation of layered semiconductors, such as bulk TMDCs and BP with organic molecules237, forming stable superlattices of alternating monolayer atomic crystals with molecular layers, could effectively remove the interlayer coupling and thus allow access to tune the bandgap of bulk layered materials. In situ measurement of PL during the process that intercalates organic molecules (such as cetyl-trimethylammonium bromide, CTAB) into bulk BP by electrochemical approach reveals the gradual modulation of PL peak position from no apparent emission in bulk to remarkable emission at ≈1.38, ≈1.75, and ≈2.26 eV as the number of layers in the intercalation process increases (Fig. 10a). These three values roughly correspond to band-edge transition of trilayer, bilayer, and monolayer BP, respectively, but the latter is slightly more than that of ideal monolayer BP. This may be due to the bandgap expansion and orbital symmetry breaking due to the intercalation.

a PL signals of different stages of intercalation of monolayer BP molecular superlattices (MPMS)237. The simulated atomic structure of MPMS is shown in the side panel. b Band structure of intrinsic bilayer CVD SnS2 and Cu-intercalated SnS2. The inset in the left panel shows the lattice structure of bilayer SnS2 with AA stacking. The inset in the right panel shows seamless integration of n-type SnS2, p-type Cu–SnS2, and metallic Co–SnS2 within a single flake. Valence band V1 and conduction bands C1 and C2 are marked by yellow, orange, and blue, respectively. The calculated Fermi level is set to be zero235. c PL spectra for undoped and F4TCNQ-doped monolayer MoS2 (bottom-left panel). The atomic structure of the latter is shown in the top-left panel. The right panel illustrates relative potentials of monolayer MoS2 and n- and p-type dopants. d PL images of monolayer MoS2 before and after TFSI treatment. Insets show optical micrographs. Panel a reprinted by permission from Springer Nature237 Wang, C., He, Q., Halim, U. et al. Monolayer atomic crystal molecular superlattices. Nature 555, 231–236 (2018), Copyright (2018), advance online publication, 8th March 2018 (doi: 10.1038/nature25774 Nature); panel b reprinted by permission from Springer Nature Nature Nanotechnology235, Gong, Y., Yuan, H., Wu, C. et al. Spatially controlled doping of two-dimensional SnS2 through intercalation for electronics. Nature Nanotech 13, 294–299 (2018), Copyright (2018), advance online publication, 26th February 2018 (https://doi.org/10.1038/s41565-018-0069-3 Nature Nanotech.).
Furthermore, intercalating metal atoms into CVD SnS2 flakes was found to change its charge-conductive states235. Naturally grown SnS2 flakes have n-type charge transport behavior, resulting from S vacancy. However, after those flakes are intercalated by copper and cobalt atoms, they display p-type semiconductor properties, with record hole mobility, and highly conductive metal properties. DFT calculations provide an understanding of these changes. In Fig. 10b, we label the valence and conduction bands as V1, C1, and C2, respectively. For intrinsic bilayer SnS2 [left panel in Fig. 10b], the bandgap arises from the difference between V1 bands and C1 bands, ≈1.5 eV. When intercalating Cu atoms into SnS2, the s-orbital electrons of Cu atoms extend and partly fill into C1 bands due to the coupling among intercalated atoms and SnS2 layers. The bandgap that at this point comes from the difference C1–C2 (Δ12) decreases by 0.9 eV due to the increase in the C1 bandwidth [right panel in Fig. 10b], compared with that of pristine SnS2. Therefore, the Cu-intercalated SnS2 behaves like a p-type semiconductor. For Co-intercalated SnS2, the strong coupling hybridization among d orbitals of Co atoms, s orbitals of Sn atoms, and p orbitals of S atoms imports plenty of hybridized states around the Fermi level, leading to excellent metal conductivity. The intercalation technique has been used to demonstrate the seamless integration of n-type SnS2, p-type Cu–SnS2, and metallic Co–SnS2 within a single flake. Therefore, such intercalation strategy can be regarded as an effective way to tailor the electro-optical properties of parent materials.
The traditional dopant diffusion or ion-injection scheme is not suitable for doping 2D-layered materials, due to their ultrathin nature. Therefore, many efforts have been devoted to develop new chemical doping techniques to modify their electrical and optical properties, such as absorbing NO2 molecules238 or potassium (K) on their surface239,240,241, covering them with organic molecular films239,240,241, soaking them into organic solutions242, and treating their surface with organic superacids243. Although these chemical doping methods do not usually modify the intrinsic band-edge transition energy, they can effectively dope 2D-layered materials and manipulate electron and exciton transport to enhance their electrical244 and optical240,243 performances. Organic molecules, such as 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) and 7,7,8,8-tetracyanoquinodimethane (TCNQ), have been used as p-type dopants, while nicotinamide adenine dinucleotide (NADH) is used as n-type dopant to dope monolayer MoS2240. With increasing F4TCNQ doping step, the PL intensity (peak A) continuously increases until saturation, and its peak energy undergoes a blueshift to 1.88 eV (left panel in Fig. 10c). It has been claimed240 that such shift in PL spectra does not originate from the modification of band-edge energy, but rather derives from the suppression of negative trion (X−) exciton recombination. For undoped monolayer MoS2, X− is still formed due to unintentional electron doping, and its PL peak (≈1.84 eV) is stronger than that of the intrinsic exciton (X) peak (at ≈1.88 eV). Hence, the low PL-intensity peak at 1.84 eV is observed. For p-type doping samples, monolayer MoS2 samples, F4TCNQ, or TCNQ, electron acceptors consume excess electrons and drastically inhibit the X− trion formation [right panel in Fig. 10c]. These flakes present intrinsic exciton properties and, for n-type doping of a MoS2 flake using NADH as electron donor, excess electrons and more negative trions are formed, which decreases the PL intensity [right panel in Fig. 10c]. Similarly, the defects or unintentional doping can give rise to nonradiative recombination, thus resulting in low PL quantum yield (QY) in monolayer MoS2 flake. A method of chemical treatment by organic superacid to remove the above defect-mediated nonradiative recombination was also reported243. After bis(trifluoro methane) sulfonimide (TFSI) treatment, the luminescence QY of monolayer MoS2 flake can reach near-unity, and PL intensity enhances by two orders of magnitude (Fig. 10d).
Alloying effects
Historically, the alloying strategy allows one to achieve continuously tunable bandgap and electronic structures for various applications in optoelectronics. Due to their low dimensionality, 2D alloys are comparatively more difficult to realize. Theoretical calculations indicate that monolayer TMDC alloys can exhibit continuously tunable gaps245 spanning across those of its constituents. So far, mechanical exfoliation139,246,247, as well as physical and CVDs (PVD138 and CVD136,137,248) have been explored to synthesize 2D- layered TMDC alloys. Although mechanically exfoliated 2D-layered alloys139,246,247 exhibit long stability247 without phase separation139,247, they have small size, poor thickness control, and low yield. This, along with the ultralong growth time of their parent bulk crystals, limits them to fundamental researches, in which the bulk crystals can be directly synthesized by CVT technique247,249. Their PVD counterparts, on the other hand, seem to have a great potential for producing large-area continuous films; however, 2D PVD alloys to date are hard to synthesize at high dopant content, due to the component decomposition resulting from high temperature, so that only 2D MoS2(1−x)Se2x alloys with 20-μm domain size have been achieved250. Conversely, the CVD approach shows some outstanding advantages, so that it enables not only the mass production of single-crystal domains, but also the fabrication of large-area continuous films with a lower growth temperature than the PVD approach [see left panel in Fig. 11a], thus being preferred for practical applications. So far, numerous 2D TMDC alloys, including ternary (such as MoS2(1−x)Se2x, WS2xSe2(1−x), and WSe2(1−x)Te2x), quaternary (MoxNb(1−x)S2ySe2(1−y)), and quinary (VxWyMo1−x−yS2zSe2(1−z)), have successfully been synthesized251 via sulfurization, selenization, and tellurization of transition metals or transition- metal compounds by CVD technique.

a Typical optical images of monolayer and single-crystal domains and continuous MoS2(1−x)Se2x (x = 0.1) films (left panel), scanning transmission electron microscope (STEM) image of Se-doped MoS2 with ≈12% local Se concentration (middle panel), and composition-dependent PL emission peak position of 0% Se-doped MoS2 (monolayer pristine MoS2,blue), 30% Se-doped MoS2 (green), 50% Se-doped MoS2 (purple), 75% Se-doped MoS2 (orange), and 100% Se-doped MoS2 (monolayer pristine MoSe2, red), respectively137. b Composition-dependent optical bandgap of 2D-layered alloys. c STEM image of quinary VxWyMo1−x−yS2zSe2(1−z) alloy (left panel)251, quaternary MoxW1−xS2ySe2(1−y) alloy (middle panel), and DFT-calculated bandgap as a function of composition parameters (right panel). In the latter, experimental points are indicated as white (650 °C), blue (700 °C), and green (750 °C) dots255. Panel a reprinted with permission from137 Nano Lett. 2014, 14, 2, 442–449. Publication Date: December 24, 2013 https://doi.org/10.1021/nl4032296, Copyright (2013) American Chemical Society; panel c reprinted by permission from Springer Nature251 Zhou, J., Lin, J., Huang, X. et al. A library of atomically thin metal chalcogenides. Nature 556, 355–359 (2018), advance online publication, 18th April 2018 (https://doi.org/10.1038/s41565-018-0069-310.1038/s41586-018-0008-3 Nature) and from John Wiley and Sons/Susarla et al.255.
Alloying of 2D monolayer TMDCs, such as MoS2(1−x)Se2x and Mo1−xWxSe2, has robust potential to continuously modulate the band-edge transitions, by changing their constituent stoichiometries, while maintaining direct gap character and thermodynamical stability. A MoS2(1−x)Se2x monolayer film with well-controlled Se concentration from 0 to ≈75% by CVD enables its bandgap modulation over 10% and features one dominant peak in PL spectroscopy, as demonstrated in ref. 137. These observations suggest that the distributions of S and Se atoms in MoS2(1−x)Se2x alloy are random. This is consistent with the result of scanning transmission electron microscope (STEM) in Fig. 11a, which shows clear separation between doped- and undoped domains in Se-doped MoS2 with ≈12% Se concentration, where each type of atom can be easily distinguished by quantitative image-intensity analysis. Noticeably, a redshift of peak position in PL spectrum with increasing Se concentration (Fig. 11a) suggests that the band-edge transition energy of monolayer MoS2(1−x)Se2x alloy can be continuously fine-tuned from 1.85 eV (for pure monolayer MoS2) to 1.54 eV (for pure monolayer MoSe2), where the band-edge transition energy exhibits a linear dependence on the Se concentration [see cyan open right triangles in Fig. 11b]. Such arrangement of constituent atoms and composition-dependent band-edge transition energy is usually a common feature in other TMDC alloys. Furthermore, monolayer WSe2(1−x)Te2x alloy exhibits a phase transition [magenta open circles in Fig. 11b], so that the optical bandgap is continuously red-shifted from 1.67 to 1.44 eV with increasing Te atom concentration from 0 to 60%, and suddenly jumps to 0 eV when the dopant concentration is over 60%140. This indicates that the monolayer WSe2(1−x)Te2x alloy transforms from semiconducting 2H phase to metallic 1Td phase. This transition will be discussed in more detail in the next section.
In general, the bandgap of 2D ternary TMDC alloys MX2(1−x)Y2x, can be modeled with252
where EPL,MXY, EPL,MX, EPL,MY, and b are, respectively, the optical bandgap of the alloy, pure MX2, pure MY2, and bowing parameter. The latter characterizes the bowing effect of the bandgap of 2D TMDC alloys, which is a joint effect of alloy volume deformation, strengthened structure relaxation by low dimensionality, and chemical difference between two constituent compounds. Usually, a larger-size mismatch and chemical potential difference will lead to a higher bowing parameter such as b(S,Te) > b(Se,Te) > b(S,Se), and then result in an obvious bandgap bowing effect as shown for Mo1−xWxS2 and Mo1−xWxSe2 in Fig. 11b. Taking Mo1−xWxS2 as an example246, the large bowing effect of the bandgap (b ≈ 0.28 eV) comes from the bowing of the LUMO energy level. Notice that the bowing parameter can also be negligible; in this case, the optical bandgap shows a linear modulation trend as a function of dopant concentrations (see WS2xSe2(1−x) and MoS2xSe2(1−x) in Fig. 11b).
2D-layered materials of group-V elements can tune their bandgap to mid-infrared regime. A clear example is BP (As), whose bandgap goes from 0.3 (0.3) eV in bulk to 2.2 (1.0) eV in monolayer form253,254. So, black arsenic–phosphorus ternary alloy (b-AsxP(1−x)) can be regarded as a promising candidate for application in industrial thermal imaging, infrared tracking, and free-space optical communication. Unlike 2D TMDC alloys, direct synthesis of 2D AsxP(1−x) alloys has not been reported. Most of such alloys have been obtained by mechanical exfoliation from their parent crystals. These can be a mixture of red phosphorus and gray arsenic with a desired ratio, which are then placed, along mineralizer additives, into evacuated silica grass ampoules to perform phase formation and crystal growth via the vapor transport method137,254. Different composition alloys may require different mineralizer additives and growth temperatures and times. Dark-yellow diamonds in Fig. 11b show the composition-dependent bandgap of b-AsxP(1−x) alloy with flake thickness over 30 nm, in which the bandgap can be fully modulated from 0.3 to 0.15 eV with increasing As concentration254. The observed fluctuation of bandgap at each arsenic concentration originates from the polarization-sensitive optical properties of these materials47.
Besides the binary and ternary alloys, multielement alloys have drawn intensive attention as they can also widely modify the bandgap and greatly enrich the physical properties. Recently, several groups251,255 have successfully synthesized multiple monolayer, quaternary, and quinary alloys using the CVD method. For instance, a molten-salt-assisted CVD process, which can grow a wide variety of TMD alloys, was developed251. This includes 11 ternary, one quaternary, and one quinary (such as MoSxTe2−x, MoxNb1−xS2ySe2(1−y), and VxWyMo1−x−yS2zSe2(1−z)), where mixing NaCl or KI together with metal or metal oxides served as precursors to decrease the melting point of the reactants and facilitate the formation of intermediate products. A facile CVD method to successfully synthesize monolayer quaternary MoxW1−xS2ySe2(1−y) alloy was also developed255, where the content of metals and chalcogens can be tuned by controlling the growth temperature during the growth process. For these quaternary and quinary alloys, STEM image characterization (Fig. 11c) shows that each atom was mostly uniformly distributed throughout the alloy samples. CVD-synthesized monolayer quaternary MoxW1−xS2ySe2(1−y) alloy exhibits a wide tunable range of bandgap, from 1.61 to 1.85 eV255. The DFT-calculated gap of such quaternary alloy presents good variability with values ranging from 1.60 to 2.03 eV. The right panel in Fig. 11c shows such variation of bandgap with composition. In summary, these alloys show the possibility to modulate the conductivity type (such as n-type, p-type, and semimetal), and provide a feasibile scheme for tunable material bandgap and electronic structure.
Bandgap closing: metal–insulator transition
An important topic on its own, the metal–insulator transition (MIT) of 2D semiconductor materials can also be considered as a rather extreme version of bandgap engineering. While the structural phase transition of bulk MoS2 under alkali metal intercalation256,257 and α- to β-phase transition of MoTe2258,259 under heating had been reported since the 1970s, the isolation of atomically thin layers of TMDC has rekindled the research interest. Owing to its polymorphism, the MIT in 2D TMDC may be either a pure electronic phase transition, or also accompanies a structural phase transition.
In a nutshell, the general driving force of the structural phase transition is attributed to the change in the oxidation number of the central transition-metal cation and its electron configuration. In trigonal prismatic (2H) phase, the typical oxidation number of the transition-metal cation is +4. Under 2H coordination, the d-orbital splitting has one lowest state at d\({}_{{z}^{2}}\), which can host two d-orbital electrons. However, the reduction of central transition-metal atom requires an additional electron to occupy a high-energy state. Thus, it becomes energetically favorable for the crystal to assume the octahedral (1T/1T’) phase, with three degenerate low-energy states. Hence, alkali metal intercalation has shown to be an effective method to trigger a phase transition in TMDC19,256,257 (for a more detailed discussion, refer to ref. 260). In this regard, triangular prism phase (H) MoTe2 has been the model case of phase transition owing to the small energy difference between semiconducting trigonal prismatic (2H) phase and metallic distorted octahedral phase (1T’). Theory predicts that the smallest stimulus for triggering structural phase transition is in 2H-MoTe2261. To understand what makes MoTe2 more susceptible to phase transition compared with its counterpart TMDC, an intuitive picture is presented to link the ionic radii of atoms and the preferred phases of different TMDCs262,263. First, because of the aforementioned d-orbital splitting, the preferred coordination is trigonal prismatic. However, if we view the crystal as a close-packed layer of a large spherical anion (r−) and metal cation (r+), the close-packed spheres cannot be arranged in a trigonal prismatic phase when the radius ratio between r+/r− is too small. From such a simple geometrical picture, the ratio limit for 2H phase is 0.527. This simple picture can lead to a clean separation of phases by the ratio of the cation radius and the anion radius r− by a straight line with a slope of 0.49. In addition to this simple picture, the ionic nature of the bond between metal and chalcogen plays an important role: if the bonding is ionic, the ionic radii of chacogen anions are larger, resulting in a stronger anion–anion repulsion. Taking the bond ionicity and the ratio r+/r− together, a separation of phases in TMDC is possible as illustrated in Fig. 12a (reproduced from ref. 263). As one can see, MoTe2 is right on the boundary where a transition to the octahedral phase is feasible. According to this intuitive picture, any stimuli that increase the anion–anion repulsion (whether by reduction of anions or by tensile strain) would increase the repulsion between the Te atoms. This becomes an additional driving force for phase transition, as the repulsion can be relaxed by adopting the octahedral phase, where the distance between anions is greater263 (see Fig. 12b). Consequently, the structural phase transition in MoTe2 can be triggered by a relatively small change in the crystal geometry, which is not the case for other TMDCs. Evidently, such structural phase transition (accompanied by the semiconductor-to-metal transition) of MoTe2 has been demonstrated to be triggered by heating264, tensile strain265, electron doping266, and electrostatic gating267 (see Fig. 12c).

a Ratio of the single-bond covalent radii of the metal atom and chalcogen plotted against Paulings resonating bond ionicity (reproduced from ref. 263). b Intuitive picture of phase transition in MoTe2 with the anion–anion repulsion. c Structural phase transition of monolayer 2H-MoTe2 with ionic gating267. d Asymmetric critical exponent T0 versus δn for monolayer and multilayer MoS2, showing the co-existence of percolation-like and quantum-phase transition in the same sample269. Panel a used with permission from Elsevier B.V./Madhukar263; c reprinted by permission from Springer Nature267 Wang, Y., Xiao, J., Zhu, H. et al. Structural phase transition in monolayer MoTe2 driven by electrostatic doping. Nature 550, 487–491 (2017), Copyright (2017), advance online publication, 11th October 2017 (https://doi.org/10.1038/nature24043 Nature); d reused from Springer Nature/Moon et al.269, permissible under a CC-BY [4.0/3.0] license.
MIT of 2D materials, especially in TMDC, that do not involve structural phase transition, has been an active research field as well. Both percolation-like transition through disorder268,269 and quantum-phase transition270,271 have been attributed as the underlying mechanism. It should be noted that although the device geometry employed maybe similar to that typical of bandgap engineering through an external electric field, the electrostatic gate-induced MIT in TMDC essentially differs from the excitonic Stark effect discussed in previous sections. The distinction between the disorder and quantum mechanisms is based on the different scaling behaviors: \(\sigma =A{(n-{n}_{c})}^{\delta }\) for percolation-like transition, while the quantum-phase transition follows the conductivity scaling of σ(δn, T) = σc(T)F[T/T0(δn)]. Naturally, the quality of the sample appears to play a critical role in determining the origin of the MIT, as both the percolation-like transition and quantum-phase transition were observed in the same sample in multilayer MoS2269. With the recent progress in obtaining high-quality samples, more intricate physical phenomena are being revealed. It is intriguing that the critical carrier density is typically in the order of few 1012 cm−2, which is easily achievable for a monolayer (and even for few-layer TMDC) through electrostatic gating. From a technological point of view, the MIT offers complementary advantages to the bandgap engineering, whether it originates from structural phase transition or from other mechanisms, as it provides a route to improve the contacts as heterophased devices19,264. Moreover, MIT enables optoelectric applications, such as optoelectrical switches, infrared detection, and more271.
Conclusions
The emergence of 2D semiconductors offers a new platform to explore band structure engineering effects. Here, we reviewed the state-of-the-art bandgap engineering approaches, but their applications in materials science, large-scale growth, and devices are still in early stages. Outstanding issues include the realization of heterostructures with tailored band alignment for devices, such as tunnel field effect transistors, light-emitting diodes, solar cells, electrocatalysis, and exciton condensate devices, among many others. Another area where 2D materials depart from most conventional semiconductors is in their polymorphic nature, which allows them to display vastly different band structure and electronic properties in different phases. Such phase engineering can be used to improve transistor Ohmic contacts19 for the study of exotic quantum behavior, such as spin-valley and Weyl fermion physics, and realize memristive devices with potential applications in reconfigurable RF272 and neuromorphic computing applications273,274. In addition, 2D materials can also exhibit ferromagnetism34,35, ferroelectricity275, topological effects276, as well as superconductivity52, which offers a multitude of other opportunities for a wide range of novel functional devices, made possible by the ability to manipulate the band structure in entirely new ways compared with conventional 3D semiconductors.
Data availability
Data sharing was not applicable to this paper, as no original datasets were generated or analyzed for this review.
References
Kroemer, H. Nobel lecture: quasielectric fields and band offsets: teaching electrons new tricks. Rev. Mod. Phys. 73, 783–793 (2001).
Esaki, L. & Tsu, R. Superlattice and negative differential conductivity in semiconductors. IBM J. Res. Dev. 14, 61–65 (1970).
Lee, M. L., Fitzgerald, E. A., Bulsara, M. T., Currie, M. T. & Lochtefeld, A. Strained si, sige, and ge channels for high-mobility metal-oxide-semiconductor field-effect transistors. J. Appl. Phys. 97, 011101 (2005).
Ning, C.-Z., Dou, L. & Yang, P. Bandgap engineering in semiconductor alloy nanomaterials with widely tunable compositions. Nat. Rev. Mater. 2, 17070 (2017).
Chu, M., Sun, Y., Aghoram, U. & Thompson, S. E. Strain: a solution for higher carrier mobility in nanoscale MOSFETs. Annu. Rev. Mater. Res. 39, 203–229 (2009).
Thijs, J., Tiemeijer, L. F., Kuindersma, P., Binsma, J. & Van Dongen, T. High-performance 1.5 μ m wavelength InGaAs-InGaAsP strained quantum well lasers and amplifiers. IEEE J. Quantum Electron. 27, 1426–1439 (1991).
Mimura, T., Hiyamizu, S., Fujii, T. & Nanbu, K. A new field-effect transistor with selectively doped GaAs/n-alx ga1−x as heterojunctions. Jpn. J. Appl. Phys. 19, L225–L227 (1980).
Kroemer, H. A proposed class of hetero-junction injection lasers. Proc. IEEE 51, 1782–1783 (1963).
Faist, J. et al. Quantum cascade laser. Science 264, 553–556 (1994).
Chang, L. L. & Esaki, L. Semiconductor quantum heterostructures. Phys. Today 45, 36–43 (1992).
Tsui, D. C., Stormer, H. L. & Gossard, A. C. Two-dimensional magnetotransport in the extreme quantum limit. Phys. Rev. Lett. 48, 1559–1562 (1982).
Novoselov, K. S. et al. Two-dimensional atomic crystals. Proc. Natl Acad. Sci. USA 102, 10451–10453 (2005).
Avouris, P., Heinz, T.F & Low, T. 2D Materials. (Cambridge University Press, 2017).
Geim, A. K. & Grigorieva, I. V. van der Waals heterostructures. Nature 499, 419 (2013).
Novoselov, K. S., Mishchenko, A., Carvalho, A. & Castro Neto, A. H. 2D materials and van der Waals heterostructures. Science 353, aac9439 (2016).
Redaelli, L. et al. Effect of the quantum well thickness on the performance of ingan photovoltaic cells. Appl. Phys. Lett. 105, 131105 (2014).
Wang, Q. H., Kalantar-Zadeh, K., Kis, A., Coleman, J. N. & Strano, M. S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 7, 699 (2012).
Wang, G. et al. Colloquium: excitons in atomically thin transition metal dichalcogenides. Rev. Mod. Phys. 90, 021001 (2018).
Kappera, R. et al. Phase-engineered low-resistance contacts for ultrathin MoS2 transistors. Nat. Mater. 13, 1128 (2014).
Fei, Z. et al. Edge conduction in monolayer WTe2. Nat. Phys. 13, 677 (2017).
Island, J. O. et al. Ultrahigh photoresponse of few-layer TiS3 nanoribbon transistors. Adv. Opt. Mater. 2, 641–645 (2014).
Lei, S. et al. Synthesis and photoresponse of large gase atomic layers. Nano Lett. 13, 2777–2781 (2013).
Li, L. et al. Single-layer single-crystalline SnSe nanosheets. J. Am. Chem. Soc. 135, 1213–1216 (2013).
Huang, Y. & Sutter, P., SnS2: an emerging layered metal dichalcogenide semiconductor. APS March Meeting Abstracts (2015).
Anasori, B., Lukatskaya, M. R. & Gogotsi, Y. 2D metal carbides and nitrides (MXenes) for energy storage. Nat. Rev. Mater. 2, 16098 (2017).
AlBalushi, Z. Y. et al. Two-dimensional gallium nitride realized via graphene encapsulation. Nat. Mater. 15, 1166 (2016).
Dean, C. R. et al. Boron nitride substrates for high-quality graphene electronics. Nat. Nanotechnol. 5, 722 (2010).
Ma, W. et al. In-plane anisotropic and ultra-low-loss polaritons in a natural van der Waals crystal. Nature 562, 557 (2018).
Lodesani, A. et al. Graphene as an Ideal Buffer Layer for the Growth of High-Quality Ultrathin Cr2 O3 Layers on Ni (111). ACS Nano 13, 4361–4367 (2019).
Lui, C. H., Liu, L., Mak, K. F., Flynn, G. W. & Heinz, T. F. Ultraflat graphene. Nature 462, 339 (2009).
Hodes, G. Perovskite-based solar cells. Science 342, 317–318 (2013).
Liu, M., Johnston, M. B. & Snaith, H. J. Efficient planar heterojunction perovskite solar cells by vapour deposition. Nature 501, 395 (2013).
Fu, L. & Kane, C. L. Superconducting proximity effect and majorana fermions at the surface of a topological insulator. Phys. Rev. Lett. 100, 096407 (2008).
Huang, B. et al. Layer-dependent ferromagnetism in a van der Waals crystal down to the monolayer limit. Nature 546, 270 (2017).
Gong, C. et al. Discovery of intrinsic ferromagnetism in two-dimensional van der Waals crystals. Nature 546, 265 (2017).
Ding, W. et al. Prediction of intrinsic two-dimensional ferroelectrics in In2 Se3 and other III2 -VI3 van der Waals materials. Nat. Commun. 8, 14956 (2017).
Liu, H. et al. Phosphorene: an unexplored 2D semiconductor with a high hole mobility. ACS Nano 8, 4033–4041 (2014).
Vogt, P. et al. Silicene: compelling experimental evidence for graphenelike two-dimensional silicon. Phys. Rev. Lett. 108, 155501 (2012).
Li, L. et al. Buckled germanene formation on pt (111). Adv. Mater. 26, 4820–4824 (2014).
Zhu, Z. et al. Multivalency-driven formation of te-based monolayer materials: a combined first-principles and experimental study. Phys. Rev. Lett. 119, 106101 (2017).
Kochat, V. et al. Atomically thin gallium layers from solid-melt exfoliation. Sci. Adv. 4, e1701373 (2018).
Ji, J. et al. Two-dimensional antimonene single crystals grown by van der Waals epitaxy. Nat. Commun. 7, 13352 (2016).
Wu, R. et al. Large-area single-crystal sheets of borophene on cu (111) surfaces. Nat. Nanotechnol. 14, 44 (2019).
Li, L. et al. Direct observation of the layer-dependent electronic structure in phosphorene. Nat. Nanotechnol. 12, 21 (2017).
Zhang, G. et al. Determination of layer-dependent exciton binding energies in few-layer black phosphorus. Sci. Adv. 4, eaap9977 (2018).
Zhang, G. et al. Infrared fingerprints of few-layer black phosphorus. Nat. Commun. 8, 14071 (2017).
Low, T. et al. Tunable optical properties of multilayer black phosphorus thin films. Phys. Rev. B 90, 075434 (2014).
de Sousa, D. J. P., de Castro, L. V., daCosta, D. R., Pereira, J. M. & Low, T. Multilayered black phosphorus: From a tight-binding to a continuum description. Phys. Rev. B 96, 155427 (2017).
Rudenko, A. N. & Katsnelson, M. I. Quasiparticle band structure and tight-binding model for single-and bilayer black phosphorus. Phys. Rev. B 89, 201408 (2014).
He, J., Hummer, K. & Franchini, C. Stacking effects on the electronic and optical properties of bilayer transition metal dichalcogenides MoS2, MoSe2, WS2, and WSe2. Phys. Rev. B 89, 075409 (2014).
Li, Y. et al. Probing symmetry properties of few-layer MoS2 and h-bn by optical second-harmonic generation. Nano Lett. 13, 3329–3333 (2013).
Xi, X. et al. Ising pairing in superconducting NbSe2 atomic layers. Nat. Phys. 12, 139 (2016).
Mak, K. F., Lee, C., Hone, J., Shan, J. & Heinz, T. F. Atomically thin MoS2: a new direct-gap semiconductor. Phys. Rev. Lett. 105, 136805 (2010).
Splendiani, A. et al. Emerging photoluminescence in monolayer MoS2. Nano Lett. 10, 1271–1275 (2010).
Arora, A., Nogajewski, K., Molas, M., Koperski, M. & Potemski, M. Exciton band structure in layered MoSe2: from a monolayer to the bulk limit. Nanoscale 7, 20769–20775 (2015).
Molas, M. R. et al. The optical response of monolayer, few-layer and bulk tungsten disulfide. Nanoscale 9, 13128–13141 (2017).
Zhao, W. et al. Evolution of electronic structure in atomically thin sheets of WS2 and WSe2. ACS Nano 7, 791–797 (2013).
Tongay, S. et al. Monolayer behaviour in bulk ReS2 due to electronic and vibrational decoupling. Nat. Commun. 5, 3252 (2014).
Jariwala, B. et al. Synthesis and characterization of ReS2 and ReSe2 layered chalcogenide single crystals. Chem. Mater. 28, 3352–3359 (2016).
Zhang, G. et al. Optical and electrical properties of two-dimensional palladium diselenide. Appl. Phys. Lett. 114, 253102 (2019).
Yu, X. et al. Atomically thin noble metal dichalcogenide: a broadband mid-infrared semiconductor. Nat. Commun. 9, 1545 (2018).
Özçelik, V. O., Azadani, J. G., Yang, C., Koester, S. J. & Low, T. Band alignment of two-dimensional semiconductors for designing heterostructures with momentum space matching. Phys. Rev. B 94, 035125 (2016).
Bellus, M. Z. et al. type-I van der Waals heterostructure formed by MoS2 and ReS2 monolayers. Nanoscale Horiz. 2, 31–36 (2017).
Nakamura, S., Senoh, M., Iwasa, N. & Nagahama, S.-I. High-brightness ingan blue, green and yellow light-emitting diodes with quantum well structures. Jpn. J. Appl. Phys. 34, L797 (1995).
Hong, X. et al. Ultrafast charge transfer in atomically thin MoS2 /WS2 heterostructures. Nat. Nanotechnol. 9, 682 (2014).
Bernardi, M., Palummo, M. & Grossman, J. C. Extraordinary sunlight absorption and one nanometer thick photovoltaics using two-dimensional monolayer materials. Nano Lett. 13, 3664–3670 (2013).
Cheng, R. et al. High-performance, multifunctional devices based on asymmetric van der Waals heterostructures. Nat. Electron. 1, 356 (2018).
Li, M. O., Esseni, D., Nahas, J. J., Jena, D. & Xing, H. G. Two-dimensional heterojunction interlayer tunneling field effect transistors (thin-TFETs). IEEE J. Electron Devices Soc. 3, 200–207 (2015).
Rivera, P. et al. Observation of long-lived interlayer excitons in monolayer MoSe2 -WSe2 heterostructures. Nat. Commun. 6, 6242 (2015).
Rivera, P. et al. Interlayer valley excitons in heterobilayers of transition metal dichalcogenides. Nat. Nanotechnol. 13, 1004–1015 (2018).
Chaves, A., Azadani, J. G., Özçelik, V. O., Grassi, R. & Low, T. Electrical control of excitons in van der Waals heterostructures with type-II band alignment. Phys. Rev. B 98, 121302 (2018).
Chaves, A. J., Ribeiro, R. M., Frederico, T. & Peres, N. M. R. Excitonic effects in the optical properties of 2d materials:an equation of motion approach. 2D Mater. 4, 025086 (2017).
Nayak, K. et al. Probing evolution of twist-angle-dependent interlayer excitons in MoSe2 /WSe2 van der Waals heterostructures. ACS Nano 11, 4041–4050 (2017).
Yu, Y. et al. Equally efficient interlayer exciton relaxation and improved absorption in epitaxial and nonepitaxial MoS2 /WS2 heterostructures. Nano Lett. 15, 486–491 (2015).
Hanbicki, A. T. et al. Double indirect interlayer exciton in a MoSe2 /WSe2 van der Waals heterostructure. ACS Nano 12, 4719–4726 (2018).
Ciarrocchi, A. et al. Polarization switching and electrical control of interlayer excitons in two-dimensional van der Waals heterostructures. Nat. Photonics 13, 131 (2019).
Miller, B. et al. Long-lived direct and indirect interlayer excitons in van der Waals heterostructures. Nano Lett. 17, 5229–5237 (2017).
Kunstmann, J. et al. Momentum-space indirect interlayer excitons in transition-metal dichalcogenide van der Waals heterostructures. Nat. Phys. 14, 801 (2018).
Zhang, N. et al. A. Castellanos-Gomez and Plochocka, Moiré intralayer excitons in a MoSe2/MoS2 heterostructure. Nano Lett. 18, 7651–7657 (2018).
Mouri, S. et al. Thermal dissociation of inter-layer excitons in MoS2/MoSe2 hetero-bilayers. Nanoscale 9, 6674–6679 (2017).
Fang, H. et al. Strong interlayer coupling in van der Waals heterostructures built from single-layer chalcogenides. Proc. Natl. Acad. Sci. USA 111, 6198–6202 (2014).
Seyler, K. L. et al. Signatures of moiré-trapped valley excitons in MoSe2/WSe2 heterobilayers. Nature 567, 66 (2019).
Nagler, G. et al. Interlayer exciton dynamics in a dichalcogenide monolayer heterostructure. 2D Mater. 4, 025112 (2017).
Calman, E. et al. Indirect excitons in van der Waals heterostructures at room temperature. Nat. Commun. 9, 1895 (2018).
Kozawa, D. et al. Evidence for fast interlayer energy transfer in MoSe2 /WS2 heterostructures. Nano Lett. 16, 4087–4093 (2016).
Ceballos, F., Bellus, M. Z., Chiu, H.-Y. & Zhao, H. Probing charge transfer excitons in a MoSe2-WS2 van der Waals heterostructure. Nanoscale 7, 17523–17528 (2015).
Bellus, M. Z., Ceballos, F., Chiu, H.-Y. & Zhao, H. Tightly bound trions in transition metal dichalcogenide heterostructures. ACS Nano 9, 6459–6464 (2015).
Thygesen, K. S. Calculating excitons, plasmons, and quasiparticles in 2D materials and van der Waals heterostructures. 2D Mater. 4, 022004 (2017).
Cavalcante, L., Chaves, A., Van Duppen, B., Peeters, F. & Reichman, D. Electrostatics of electron-hole interactions in van der Waals heterostructures. Phys. Rev. B 97, 125427 (2018).
Gao, S., Yang, L. & Spataru, C. D. Interlayer coupling and gate-tunable excitons in transition metal dichalcogenide heterostructures. Nano Lett. 17, 7809–7813 (2017).
Huang, Z. et al. Robust room temperature valley hall effect of interlayer excitons. Nano Lett. 20, 1345–1351 (2019).
Binder, J. et al. Upconverted electroluminescence via auger scattering of interlayer excitons in van der Waals heterostructures. Nat. Commun. 10, 2335 (2019).
Özçelik, V. O., Fathi, M., Azadani, J. G. & Low, T. Tin monochalcogenide heterostructures as mechanically rigid infrared band gap semiconductors. Phys. Rev. Mater. 2, 051003 (2018).
Tian, Z., Guo, C., Zhao, M., Li, R. & Xue, J. Two-dimensional SnS: a phosphorene analogue with strong in-plane electronic anisotropy. ACS Nano 11, 2219–2226 (2017).
Huang, L., Wu, F. & Li, J. Structural anisotropy results in strain-tunable electronic and optical properties in monolayer gex and snx (x= s, se, te). J. Chem. Phys. 144, 114708 (2016).
Sa, B., Sun, Z. & Wu, B. The development of two dimensional group iv chalcogenides, blocks for van der Waals heterostructures. Nanoscale 8, 1169–1178 (2016).
Brent, J. R. et al. Tin (ii) sulfide (SnS) nanosheets by liquid-phase exfoliation of herzenbergite: Iv-vi main group two-dimensional atomic crystals. J. Am. Chem. Soc. 137, 12689–12696 (2015).
Patel, M., Kim, J. & Kim, Y. K. Growth of large-area SnS films with oriented 2D SnS layers for energy-efficient broadband optoelectronics. Adv. Funct. Mater. 28, 1804737 (2018).
Sun, H., Wang, Z. & Wang, Y. Band alignment of two-dimensional metal monochalcogenides MXs (M=Ga,In; X=S,Se,Te). AIP Adv. 7, 095120 (2017).
McDonnell, S., Addou, R., Buie, C., Wallace, R. M. & Hinkle, C. L. Defect-dominated doping and contact resistance in MoS2. ACS Nano 8, 2880–2888 (2014).
Addou, R. et al. Impurities and electronic property variations of natural MoS2 crystal surfaces. ACS Nano 9, 9124–9133 (2015).
Shim, J. et al. Controlled crack propagation for atomic precision handling of wafer-scale two-dimensional materials. Science 362, 665–670 (2018).
Gong, C. et al. Rapid selective etching of pmma residues from transferred graphene by carbon dioxide. J. Phys. Chem. C 117, 23000–23008 (2013).
Eichfeld, S. M. et al. Highly scalable, atomically thin WSe2 grown via metal-organic chemical vapor deposition. ACS Nano 9, 2080–2087 (2015).
Eichfeld, S. M., Colon, V. O., Nie, Y., Cho, K. & Robinson, J. A. Controlling nucleation of monolayer WSe2 during metal-organic chemical vapor deposition growth. 2D Mater. 3, 025015 (2016).
Kang, K. et al. High-mobility three-atom-thick semiconducting films with wafer-scale homogeneity. Nature 520, 656 (2015).
Lin, Y.-C. et al. Realizing large-scale, electronic-grade two-dimensional semiconductors. ACS Nano 12, 965–975 (2018).
Huo, N. et al. High carrier mobility in monolayer cvd-grown MoS2 through phonon suppression. Nanoscale 10, 15071–15077 (2018).
Chen, W. et al. Oxygen-assisted chemical vapor deposition growth of large single-crystal and high-quality monolayer MoS2. J. Am. Chem. Soc. 137, 15632–15635 (2015).
Amani, M. et al. Electrical performance of monolayer MoS2 field-effect transistors prepared by chemical vapor deposition. Appl. Phys. Lett. 102, 193107 (2013).
Schmidt, H. et al. Transport properties of monolayer MoS2 grown by chemical vapor deposition. Nano Lett. 14, 1909–1913 (2014).
Gong, Y., Zhang, X., Redwing, J. M. & Jackson, T. N. Thin film transistors using wafer-scale low-temperature mocvd WSe2. J. Electron. Mater. 45, 6280–6284 (2016).
Huang, J.-K. et al. Large-area synthesis of highly crystalline WSe2 monolayers and device applications. ACS Nano 8, 923–930 (2013).
Huang, J. et al. Large-area synthesis of monolayer WSe2 on a SiO2/Si substrate and its device applications. Nanoscale 7, 4193–4198 (2015).
Zhang, C. et al. Interlayer couplings, moiré patterns, and 2D electronic superlattices in MoS2 /WSe2 hetero-bilayers. Sci. Adv. 3, e1601459 (2017).
Lin, Y.-C. et al. Atomically thin resonant tunnel diodes built from synthetic van der Waals heterostructures. Nat. Commun. 6, 7311 (2015).
Gong, Y. et al. Two-step growth of two-dimensional WSe2 /MoSe2 heterostructures. Nano Lett. 15, 6135–6141 (2015).
Lee, C.-S. et al. Epitaxial van der Waals contacts between transition-metal dichalcogenide monolayer polymorphs. Nano Lett. 19, 1814–1820 (2019).
Li, X. et al. Two-dimensional gase/MoSe2 misfit bilayer heterojunctions by van der Waals epitaxy. Sci. Adv. 2, e1501882 (2016).
Zribi, J. et al. Strong interlayer hybridization in the aligned SnS2/WSe2 hetero-bilayer structure. npj 2D Mater. Appl. 3, 27 (2019).
An, V., Irtegov, Y. & Izarra, C. D. Study of tribological properties of nanolamellar WS2 and MoS2 as additives to lubricants. J. Nanomater. 2014, 188 (2014).
Koma, A., Sunouchi, K. & Miyajima, T. Fabrication and characterization of heterostructures with subnanometer thickness. Microelectron. Eng. 2, 129–136 (1984).
Aminalragia-Giamini, S., Marquez-Velasco, J., Tsipas, P., Tsoutsou, D. & Renaud, G. Molecular beam epitaxy of thin HfTe2 semimetal films. 2D Mater. 4, 015001 (2016).
Liu, H. et al. Molecular-beam epitaxy of monolayer and bilayer WSe2: a scanning tunneling microscopy/spectroscopy study and deduction of exciton binding energy. 2D Mater. 2, 034004 (2015).
Diaz, H. C., Chaghi, R., Ma, Y. & Batzill, M. Molecular beam epitaxy of the van der Waals heterostructure MoTe2 on MoS2: phase, thermal, and chemical stability. 2D Mater. 2, 044010 (2015).
Park, Y. W. et al. Molecular beam epitaxy of large-area SnSe2 with monolayer thickness fluctuation. 2D Mater. 4, 014006 (2016).
Yan, M. et al. High quality atomically thin ptse2 films grown by molecular beam epitaxy. 2D Mater. 4, 045015 (2017).
Xenogiannopoulou, E. et al. High-quality, large-area MoSe2 and MoSe2/Bi2 Se3 heterostructures on AlN (0001)/Si (111) substrates by molecular beam epitaxy. Nanoscale 7, 7896–7905 (2015).
Zhang, Y. et al. Electronic structure, surface doping, and optical response in epitaxial WSe2 thin films. Nano Lett. 16, 2485–2491 (2016).
O’Hara, D. J. et al. Room temperature intrinsic ferromagnetism in epitaxial manganese selenide films in the monolayer limit. Nano Lett. 18, 3125–3131 (2018).
Bonilla, M. et al. Strong room-temperature ferromagnetism in vse2 monolayers on van der Waals substrates. Nat. Nanotechnol. 13, 289 (2018).
Yue, R. et al. HfSe2 thin films: 2D transition metal dichalcogenides grown by molecular beam epitaxy. ACS Nano 9, 474–480 (2014).
Vishwanath, S. et al. Comprehensive structural and optical characterization of mbe grown MoSe2 on graphite, caf2 and graphene. 2D Mater. 2, 024007 (2015).
Chen, J. et al. Quantum effects and phase tuning in epitaxial hexagonal and monoclinic MoTe2 monolayers. ACS Nano 11, 3282–3288 (2017).
Walsh, L. A. et al. WTe2 thin films grown by beam-interrupted molecular beam epitaxy. 2D Mater. 4, 025044 (2017).
Li, H. et al. Growth of alloy MoS2x Se2(1−x) nanosheets with fully tunable chemical compositions and optical properties. J. Am. Chem. Soc. 136, 3756–3759 (2014).
Gong, Y. et al. Band gap engineering and layer-by-layer mapping of selenium-doped molybdenum disulfide. Nano Lett. 14, 442–449 (2013).
Feng, Q. et al. Growth of large-area 2D MoS2(1−x) Se2x semiconductor alloys. Adv. Mater. 26, 2648–2653 (2014).
Zhang, M. et al. Two-dimensional molybdenum tungsten diselenide alloys: photoluminescence, raman scattering, and electrical transport. ACS Nano 8, 7130–7137 (2014).
Yu, J. et al. Metal-semiconductor phase-transition in WSe2(1−x) Te2x monolayer. Adv. Mater. 29, 1603991 (2017).
Barton, A. et al. WSe2−x tex alloys grown by molecular beam epitaxy. 2D Mater. (2019).
Nie, Y. et al. Dislocation driven spiral and non-spiral growth in layered chalcogenides. Nanoscale 10, 15023–15034 (2018).
Walsh, L. A., Addou, R., Wallace, R. M. & Hinkle, C. L. Molecular beam epitaxy of transition metal dichalcogenides. In Mohamed H. (ed.), Molecular Beam Epitaxy. 515–531 (Elsevier, 2018).
Walsh, L. A. & Hinkle, C. L. Van der Waals epitaxy: 2D materials and topological insulators. Appl. Mater. Today 9, 504–515 (2017).
Peng, R. et al. Midinfrared electro-optic modulation in few-layer black phosphorus. Nano Lett. 17, 6315–6320 (2017).
Pereira, J. M. & Katsnelson, M. I. Landau levels of single-layer and bilayer phosphorene. Phys. Rev. B 92, 075437 (2015).
Lin, C., Grassi, R., Low, T. & Helmy, A. S. Multilayer black phosphorus as a versatile mid-infrared electro-optic material. Nano Lett. 16, 1683–1689 (2016).
Lu, X. & Yang, L. Stark effect of doped two-dimensional transition metal dichalcogenides. Appl. Phys. Lett. 111, 193104 (2017).
Scharf, B. et al. Excitonic stark effect in MoS2 monolayers. Phys. Rev. B 94, 245434 (2016).
Cavalcante, L. S. R., da Costa, D. R., Farias, G. A., Reichman, D. R. & Chaves, A. Stark shift of excitons and trions in two-dimensional materials. Phys. Rev. B 98, 245309 (2018).
Massicotte, M. et al. Dissociation of two-dimensional excitons in monolayer WSe2. Nat. Commun. 9, 1633 (2018).
Dolui, K. & Quek, S. Y. Quantum-confinement and structural anisotropy result in electrically-tunable dirac cone in few-layer black phosphorous. Sci. Rep. 5, 11699 (2015).
Baik, S. S., Kim, K. S., Yi, Y. & Choi, H. J. Emergence of two-dimensional massless dirac fermions, chiral pseudospins, and berry’s phase in potassium doped few-layer black phosphorus. Nano Lett. 15, 7788–7793 (2015).
Li, L., Partoens, B. & Peeters, F. Tuning the electronic properties of gated multilayer phosphorene: a self-consistent tight-binding study. Phys. Rev. B 97, 155424 (2018).
Kim, J. et al. Observation of tunable band gap and anisotropic dirac semimetal state in black phosphorus. Science 349, 723–726 (2015).
Yang, L., Lin, Y.-M., Tsai, W. & Peide, D. Y. Experimental demonstration of electrically-tunable bandgap on 2D black phosphorus by quantum confined stark effect. In 2017 Symposium on VLSI Technology. T48–T49 (Organizers/Chairs: S. Yamakawa, W. Rachmady, and C.-P. Chang, IEEE, 2017).
Liu, Y. et al. Gate-tunable giant stark effect in few-layer black phosphorus. Nano Lett. 17, 1970–1977 (2017).
de Sousa, D., Cavalcante, L., Chaves, A., Pereira Jr, J.M. & Low, T. Plasmons in bias-induced topological phase transition in black phosphorus. Preprint at https://arxiv.org/abs/1808.08869 (2018).
Kang, M. et al. Universal mechanism of band-gap engineering in transition-metal dichalcogenides. Nano Lett. 17, 1610–1615 (2017).
Long, G. et al. Achieving ultrahigh carrier mobility in two-dimensional hole gas of black phosphorus. Nano Lett. 16, 7768–7773 (2016).
Rudenko, A., Brener, S. & Katsnelson, M. Intrinsic charge carrier mobility in single-layer black phosphorus. Phys. Rev. Lett. 116, 246401 (2016).
Deng, B. et al. Efficient electrical control of thin-film black phosphorus bandgap. Nat. Commun. 8, 14474 (2017).
Li, D. et al. Tunable bandgap in few-layer black phosphorus by electrical field. 2D Mater. 4, 031009 (2017).
Chakraborty, C. et al. Quantum-confined stark effect of individual defects in a van der Waals heterostructure. Nano Lett. 17, 2253–2258 (2017).
Martín-Sánchez, J. et al. Effects of dielectric stoichiometry on the photoluminescence properties of encapsulated WSe2 monolayers. Nano Res. 11, 1399–1414 (2018).
Raja, A. et al. Coulomb engineering of the bandgap and excitons in two-dimensional materials. Nat. Commun. 8, 15251 (2017).
Borghardt, S. et al. Engineering of optical and electronic band gaps in transition metal dichalcogenide monolayers through external dielectric screening. Phys. Rev. Mater. 1, 054001 (2017).
Florian, M. et al. The dielectric impact of layer distances on exciton and trion binding energies in van der Waals heterostructures. Nano Lett. 18, 2725–2732 (2018).
Merkl, Steinleitner. et al. Dielectric engineering of electronic correlations in a van der Waals heterostructure. Nano Lett. 18, 1402–1409 (2018).
Drüppel, M., Deilmann, T., Krüger, P. & Rohlfing, M. Diversity of trion states and substrate effects in the optical properties of an MoS2 monolayer. Nat. Commun. 8, 2117 (2017).
Stier, A. V., Wilson, N. P., Clark, G., Xu, X. & Crooker, S. A. Probing the influence of dielectric environment on excitons in monolayer WSe2 : insight from high magnetic fields. Nano Lett. 16, 7054–7060 (2016).
Ryou, J., Kim, Y.-S., Santosh, K. & Cho, K. Monolayer MoS2 bandgap modulation by dielectric environments and tunable bandgap transistors. Sci. Rep. 6, 29184 (2016).
Qiu, D. Y., da Jornada, F. H. & Louie, S. G. Environmental screening effects in 2D materials: renormalization of the bandgap, electronic structure, and optical spectra of few-layer black phosphorus. Nano Lett. 17, 4706–4712 (2017).
Gerber, I. C. & Marie, X. Dependence of band structure and exciton properties of encapsulated WSe2 monolayers on the hbn-layer thickness. Phys. Rev. B 98, 245126 (2018).
Naik, M. H. & Jain, M. Substrate screening effects on the quasiparticle band gap and defect charge transition levels in MoS2. Phys. Rev. Mater. 2, 084002 (2018).
Park, S. et al. Direct determination of monolayer MoS2 and WSe2 exciton binding energies on insulating and metallic substrates. 2D Mater. 5, 025003 (2018).
Ugeda, M. M. et al. Giant bandgap renormalization and excitonic effects in a monolayer transition metal dichalcogenide semiconductor. Nat. Mater. 13, 1091 (2014).
Hanbicki, A., Currie, M., Kioseoglou, G., Friedman, A. & Jonker, B. Measurement of high exciton binding energy in the monolayer transition-metal dichalcogenides WS2 and WSe2. Solid State Commun. 203, 16–20 (2015).
Lin, Y. et al. Dielectric screening of excitons and trions in single-layer MoS2. Nano Lett. 14, 5569–5576 (2014).
Stier, A. V. et al. Magnetooptics of exciton rydberg states in a monolayer semiconductor. Phys. Rev. Lett. 120, 057405 (2018).
Yang, J. et al. Optical tuning of exciton and trion emissions in monolayer phosphorene. Light 4, e312 (2015).
Rytova, N. S. Screened potential of a point charge in a thin film. Proc. MSU, Phys., Astron. 3, 30 (1967). Preprint at https://arxiv.org/abs/1806.00976 (2018).
Keldysh, L. Coulomb interaction in thin semiconductor and semimetal films. Sov. J. Exp. Theor. Phys. Lett. 29, 658 (1979).
Rodin, A., Carvalho, A. & Neto, A. C. Excitons in anisotropic two-dimensional semiconducting crystals. Phys. Rev. B 90, 075429 (2014).
Berkelbach, T. C., Hybertsen, M. S. & Reichman, D. R. Theory of neutral and charged excitons in monolayer transition metal dichalcogenides. Phys. Rev. B 88, 045318 (2013).
Yu, Y. et al. Engineering substrate interactions for high luminescence efficiency of transition-metal dichalcogenide monolayers. Adv. Funct. Mater. 26, 4733–4739 (2016).
Chernikov, A. et al. Exciton binding energy and nonhydrogenic rydberg series in monolayer WS2. Phys. Rev. Lett. 113, 076802 (2014).
Xia, F., Wang, H. & Jia, Y. Rediscovering black phosphorus as an anisotropic layered material for optoelectronics and electronics. Nat. Commun. 5, 4458 (2014).
Lindberg, M. & Koch, S. W. Effective bloch equations for semiconductors. Phys. Rev. B 38, 3342–3350 (1988).
Berghäuser, G. & Malic, E. Analytical approach to excitonic properties of mos2. Phys. Rev. B 89, 125309 (2014).
Henriques, J. C. G. et al. Optical absorption of single-layer hexagonal boron nitride in the ultraviolet. J. Phys.: Condens. Matter. 32, 025304 (2020).
Komsa, H.-P. & Krasheninnikov, A. V. Effects of confinement and environment on the electronic structure and exciton binding energy of mos2 from first principles. Phys. Rev. B 86, 241201 (2012).
Ye, Z. et al. Probing excitonic dark states in single-layer tungsten disulphide. Nature 513, 214 (2014).
Sun, Y., Thompson, S.E. & Nishida, T. Strain Effect in Semiconductors: Theory and Device Applications. (Springer Science & Business Media, 2009).
Ni, Z. H. et al. Uniaxial strain on graphene: Raman spectroscopy study and band-gap opening. ACS Nano 2, 2301–2305 (2008).
Sun, L. et al. Strain effect on electronic structures of graphene nanoribbons: a first-principles study. J. Chem. Phys. 129, 074704 (2008).
Guinea, F., Katsnelson, M. & Geim, A. Energy gaps and a zero-field quantum hall effect in graphene by strain engineering. Nat. Phys. 6, 30 (2010).
Si, C., Sun, Z. & Liu, F. Strain engineering of graphene: a review. Nanoscale 8, 3207–3217 (2016).
Feng, J., Qian, X., Huang, C.-W. & Li, J. Strain-engineered artificial atom as a broad-spectrum solar energy funnel. Nat. Photonics 6, 866 (2012).
Van Der Zande, A. & Hone, J. Optical materials: inspired by strain. Nat. Photonics 6, 804 (2012).
San-Jose, V., Parente, F., Guinea, R., Roldán, P. & Prada, E. Inverse funnel effect of excitons in strained black phosphorus. Phys. Rev. X 6, 031046 (2016).
Roldán, R., Castellanos-Gomez, A., Cappelluti, E. & Guinea, F. Strain engineering in semiconducting two-dimensional crystals. J. Phys. 27, 313201 (2015).
Hui, Y. Y. et al. Exceptional tunability of band energy in a compressively strained trilayer MoS2 sheet. ACS Nano 7, 7126–7131 (2013).
He, K., Poole, C., Mak, K. F. & Shan, J. Experimental demonstration of continuous electronic structure tuning via strain in atomically thin MoS2. Nano Lett. 13, 2931–2936 (2013).
Lloyd, D. et al. Band gap engineering with ultralarge biaxial strains in suspended monolayer MoS2. Nano Lett. 16, 5836–5841 (2016).
Feierabend, M., Morlet, A., Berghäuser, G. & Malic, E. Impact of strain on the optical fingerprint of monolayer transition-metal dichalcogenides. Phys. Rev. B 96, 045425 (2017).
Conley, H. J., Wang, B., Ziegler, J. I., HaglundJr, R. F., Pantelides, S. T. & Bolotin, K. I. Bandgap engineering of strained monolayer and bilayer MoS2. Nano Lett. 13, 3626–3630 (2013).
Zhu, C. et al. Strain tuning of optical emission energy and polarization in monolayer and bilayer MoS2. Phys. Rev. B 88, 121301 (2013).
Island, J. O. et al. Precise and reversible band gap tuning in single-layer MoSe2 by uniaxial strain. Nanoscale 8, 2589–2593 (2016).
Schmidt, R. et al. Reversible uniaxial strain tuning in atomically thin WSe2. 2D Mater. 3, 021011 (2016).
Niehues, I. et al. Strain control of exciton-phonon coupling in atomically thin semiconductors. Nano Lett. 18, 1751–1757 (2018).
Frisenda, R. et al. Biaxial strain tuning of the optical properties of single-layer transition metal dichalcogenides. npj 2D Mater. Appl. 1, 10 (2017).
Aslan, O. B., Deng, M. & Heinz, T. F. Strain tuning of excitons in monolayer WSe2. Phys. Rev. B 98, 115308 (2018).
Huang, S. et al. Strain-tunable van der Waals interactions in few-layer black phosphorus. Nat. Commun. 10, 2447 (2019).
Niehues, I., Blob, A., Stiehm, T., de Vasconcellos, S. M. & Bratschitsch, R. Interlayer excitons in bilayer MoS2 under uniaxial tensile strain. Nanoscale 11, 12788–12792 (2019).
Carrascoso, F., Lin, D.-Y., Frisenda, R. & Castellanos-Gomez, A. Biaxial strain tuning of interlayer excitons in bilayer MoS2. J. Phys. Mater. 3, 015003 (2020).
Gant, P., Huang, P., de Lara, D. P., Guo, D. & Frisenda, R. A strain tunable single-layer MoS2 photodetector. Materials Today 27, 8–13 (2019).
Tong, L. et al. Artificial control of in-plane anisotropic photoelectricity in monolayer MoS2. Appl. Mater. Today 15, 203–211 (2019).
Yang, S. et al. Tuning the optical, magnetic, and electrical properties of ReSe2 by nanoscale strain engineering. Nano Lett. 15, 1660–1666 (2015).
Quereda, J. et al. Castellanos-Gomez, Strong modulation of optical properties in black phosphorus through strain-engineered rippling. Nano Lett. 16, 2931–2937 (2016).
Li, H. et al. Optoelectronic crystal of artificial atoms in strain-textured molybdenum disulphide. Nat. Commun. 6, 7381 (2015).
Branny, A., Kumar, S., Proux, R. & Gerardot, B. D. Deterministic strain-induced arrays of quantum emitters in a two-dimensional semiconductor. Nat. Commun. 8, 15053 (2017).
Palacios-Berraquero, C. et al. Large-scale quantum-emitter arrays in atomically thin semiconductors. Nat. Commun. 8, 15093 (2017).
Castellanos-Gomez, A. et al. Local strain engineering in atomically thin MoS2. Nano Lett. 13, 5361–5366 (2013).
De Sanctis, A., Amit, I., Hepplestone, S. P., Craciun, M. F. & Russo, S. Strain-engineered inverse charge-funnelling in layered semiconductors. Nat. Commun. 9, 1652 (2018).
Manzeli, S., Allain, A., Ghadimi, A. & Kis, A. Piezoresistivity and strain-induced band gap tuning in atomically thin MoS2. Nano Lett. 15, 5330–5335 (2015).
Benimetskiy, F. et al. Measurement of local optomechanical properties of a direct bandgap 2D semiconductor. APL Materials 7, 101126 (2019).
Rosenberger, M. R. et al. Quantum calligraphy: Writing single-photon emitters in a two-dimensional materials platform. ACS Nano 13, 904–912 (2019).
Li, M.-Y. et al. Epitaxial growth of a monolayer wse2-mos2 lateral pn junction with an atomically sharp interface. Science 349, 524–528 (2015).
Rubel, O. One-dimensional electron gas in strained lateral heterostructures of single layer materials. Sci. Rep. 7, 1–9 (2017).
Ávalos-Ovando, O., Mastrogiuseppe, D. & Ulloa, S. E. Lateral heterostructures and one-dimensional interfaces in 2D transition metal dichalcogenides. J. Phys. 31, 213001 (2019).
Li, M.-Y., Chen, C.-H., Shi, Y. & Li, L.-J. Heterostructures based on two-dimensional layered materials and their potential applications. Mater. Today 19, 322–335 (2016).
Huang, T., Wei, W., Chen, X. & Dai, N. Strained 2D layered materials and heterojunctions. Ann. Phys. 531, 1800465 (2019).
Taghinejad, H., Eftekhar, A. A. & Adibi, A. Lateral and vertical heterostructures in two-dimensional transition-metal dichalcogenides. Opt. Mater. Express 9, 1590–1607 (2019).
Gong, Y. et al. Spatially controlled doping of two-dimensional SnS2 through intercalation for electronics. Nat. Nanotechnol. 13, 294 (2018).
Wan, C. et al. Flexible n-type thermoelectric materials by organic intercalation of layered transition metal dichalcogenide TiS2. Nat. Mater. 14, 622 (2015).
Wang, C. et al. Monolayer atomic crystal molecular superlattices. Nature 555, 231 (2018).
Fang, H. et al. High-performance single layered WSe2 p-FETs with chemically doped contacts. Nano Lett. 12, 3788–3792 (2012).
He, D. et al. High-performance black phosphorus field-effect transistors with long-term air stability. Nano Lett. 19, 331–337 (2018).
Mouri, S., Miyauchi, Y. & Matsuda, K. Tunable photoluminescence of monolayer MoS2 via chemical doping. Nano Lett. 13, 5944–5948 (2013).
Fang, H. et al. Degenerate n-doping of few-layer transition metal dichalcogenides by potassium. Nano Lett. 13, 1991–1995 (2013).
Yang, L. et al. Chloride molecular doping technique on 2D materials: WS2 and MoS2. Nano Lett. 14, 6275–6280 (2014).
Amani, M. et al. Near-unity photoluminescence quantum yield in MoS2. Science 350, 1065–1068 (2015).
Du, Y., Yang, L., Zhou, H. & Peide, D. Y. Performance enhancement of black phosphorus field-effect transistors by chemical doping. IEEE Electron Device Lett. 37, 429–432 (2016).
Komsa, H.-P. & Krasheninnikov, A. V. Two-dimensional transition metal dichalcogenide alloys: stability and electronic properties. J. Phys. Chem. Lett. 3, 3652–3656 (2012).
Chen, Y. et al. Tunable band gap photoluminescence from atomically thin transition-metal dichalcogenide alloys. Acs Nano 7, 4610–4616 (2013).
Tongay, S. et al. Two-dimensional semiconductor alloys: Monolayer Mo1−x Wx Se2. Appl. Phys. Lett. 104, 012101 (2014).
Mann, J. et al. 2-dimensional transition metal dichalcogenides with tunable direct band gaps: MoS2(1−x) Se2x monolayers. Adv. Mater. 26, 1399–1404 (2014).
Dumcenco, D., Chen, K., Wang, Y., Huang, Y. & Tiong, K. Raman study of 2H-Mo1−x Wx S2 layered mixed crystals. J. Alloy. Compd. 506, 940–943 (2010).
Feng, Q. et al. Growth of MoS2(1−x) Se2x (x= 0.41-1.00) monolayer alloys with controlled morphology by physical vapor deposition. ACS nano 9, 7450–7455 (2015).
Zhou, J. et al. A library of atomically thin metal chalcogenides. Nature 556, 355 (2018).
Kang, J., Tongay, S., Li, J. & Wu, J. Monolayer semiconducting transition metal dichalcogenide alloys: stability and band bowing. J. Appl. Phys. 113, 143703 (2013).
Zhang, Z. et al. Manifestation of unexpected semiconducting properties in few-layer orthorhombic arsenene. Appl. Phys. Express 8, 055201 (2015).
Liu, B. et al. Black arsenic-phosphorus: layered anisotropic infrared semiconductors with highly tunable compositions and properties. Adv. Mater. 27, 4423–4429 (2015).
Susarla, S. et al. Quaternary 2D transition metal dichalcogenides (TMD s) with tunable bandgap. Adv. Mater. 29, 1702457 (2017).
Somoano, R., Hadek, V. & Rembaum, A. Alkali metal intercalates of molybdenum disulfide. J. Chem. Phys. 58, 697–701 (1973).
Woollam, J. A. & Somoano, R. B. Physics and chemistry of MoS2 intercalation compounds. Mater. Sci. Eng. 31, 289–295 (1977).
Hughes, H. & Friend, R. Electrical resistivity anomaly in β -MoTe2 (metallic behaviour). J. Phys. C 11, L103 (1978).
Dawson, W. & Bullett, D. Electronic structure and crystallography of MoTe2 and WTe2. J. Phys. C 20, 6159 (1987).
Yang, H., Kim, S. W., Chhowalla, M. & Lee, Y. H. Structural and quantum-state phase transitions in van der Waals layered materials. Nat. Phys. 13, 931 (2017).
Li, Y., Duerloo, K.-A. N., Wauson, K. & Reed, E. J. Structural semiconductor-to-semimetal phase transition in two-dimensional materials induced by electrostatic gating. Nat. Commun. 7, 10671 (2016).
Gamble, F. Ionicity, atomic radii, and structure in the layered dichalcogenides of group IVb, Vb, and VIb transition metals. J. Solid State Chem. 9, 358–367 (1974).
Madhukar, A. Structural classification of layered dichalcogenides of group iv b, vb and vi b transition metals. Solid State Commun. 16, 383–388 (1975).
Cho, S. et al. Phase patterning for ohmic homojunction contact in MoTe2. Science 349, 625–628 (2015).
Song, S. et al. Room temperature semiconductor-metal transition of MoTe2 thin films engineered by strain. Nano Lett. 16, 188–193 (2015).
Kim, S. et al. Long-range lattice engineering of MoTe2 by a 2D electride. Nano Lett. 17, 3363–3368 (2017).
Wang, Y. et al. Structural phase transition in monolayer MoTe2 driven by electrostatic doping. Nature 550, 487 (2017).
Chen, X. et al. Probing the electron states and metal-insulator transition mechanisms in molybdenum disulphide vertical heterostructures. Nat. Commun. 6, 6088 (2015).
Moon, B. H. et al. Soft coulomb gap and asymmetric scaling towards metal-insulator quantum criticality in multilayer MoS2. Nat. Commun. 9, 2052 (2018).
Pradhan, N. R. et al. Metal to insulator quantum-phase transition in few-layered ReS2. Nano Lett. 15, 8377–8384 (2015).
Radisavljevic, B. & Kis, A. Mobility engineering and a metal-insulator transition in monolayer MoS2. Nat. Mater. 12, 815 (2013).
Ge, R. et al. Atomristor: nonvolatile resistance switching in atomic sheets of transition metal dichalcogenides. Nano Lett. 18, 434–441 (2018).
Zhu, X., Li, D., Liang, X. & Lu, W. D. Ionic modulation and ionic coupling effects in MoS2 devices for neuromorphic computing. Nat. Mater. 18, 141 (2019).
Zhang, F. et al. Electric-field induced structural transition in vertical MoTe2 -and Mo1−x Wx Te2-based resistive memories. Nat. Mater. 18, 55 (2019).
Chang, K. et al. Discovery of robust in-plane ferroelectricity in atomic-thick snte. Science 353, 274–278 (2016).
Fatemi, V. et al. Electrically tunable low-density superconductivity in a monolayer topological insulator. Science 362, 926–929 (2018).
Yue, R. et al. Nucleation and growth of WSe2: enabling large grain transition metal dichalcogenides. 2D Mater. 4, 045019 (2017).
Zhou, G. et al. High-mobility helical tellurium field-effect transistors enabled by transfer-free, low-temperature direct growth. Adv. Mater. 30, 1803109 (2018).
Klots, A. et al. Probing excitonic states in suspended two-dimensional semiconductors by photocurrent spectroscopy. Sci. Rep. 4, 6608 (2014).
Ross, J. S. et al. Electrical control of neutral and charged excitons in a monolayer semiconductor. Nat. Commun. 4, 1474 (2013).
Zhu, C. et al. Exciton valley dynamics probed by Kerr rotation in WSe2 monolayers. Phys. Rev. B 90, 161302 (2014).
Kormányos, A. et al. k ⋅ p theory for two-dimensional transition metal dichalcogenide semiconductors. 2D Mater. 2, 022001 (2015).
Acknowledgements
Discussions and interactions with D.R. Reichman, F. Tavazza, N.M.R. Peres, and K. Choudhary are gratefully acknowledged. A.C. acknowledges financial support by CNPq, through the PRONEX/FUNCAP and PQ programs. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No. 755655, ERC-StG 2017 project 2D-TOPSENSE). Computational support from the Minnesota Supercomputing Institute (MSI) and EU Graphene Flagship funding (Grant Graphene Core 2, 785219) is acknowledged. R.F. acknowledges support from the Netherlands Organization for Scientific Research (NWO) through the research program Rubicon with project number 680-50-1515. D.H., J.Z., and X.W. acknowledge support by National Natural Science Foundation of China 61734003, 61521001, 61704073, 51861145202, and 61851401, and National Key Basic Research Program of China 2015CB921600 and 2018YFB2200500. J.Z. and Z.L. acknowledge support by RG7/18, MOE2017-T2-2-136, MOE2018-T3-1-002, and A*Star QTE program. S.H.S. and Y.H.L. acknowledge the support from IBS-R011-D1. Y.D.K. is supported by Samsung Research and Incubation Funding Center of Samsung Electronics under Project Number SRFC-TB1803-04. S.J.K acknowledges financial support by the National Science Foundation (NSF), under award DMR-1921629. T.L. and J.G.A. acknowledge funding support from NSF/DMREF under Grant Agreement No. 1921629. S.-H.O. acknowledges support from the U.S. National Science Foundation (NSF ECCS 1809723) and Samsung Global Research Outreach (GRO) project.
Author information
Authors and Affiliations
Contributions
P.A., S.J.K., and Y.D.K. contributed with the introduction and concluding remarks. J.G.A. and H.A. contributed with the sections “Bandgaps in 2D materials family and Bandgap and excitons in van der Waals heterostructures”. A.C. and T.L. contributed with the sections “Bandgap in van der Waals multilayers, Bandgap and excitons in van der Waals heterostructures, External electric field effect, and with the theoretical models therein”. P.D.Y. contributed with the section “External electric field effect”. C.L.H. contributed with the section Bandgap and excitons in van der Waals heterostructures. A.J.C. contributed with the section “Dielectric screening and many-body effects”. R.F. and A.C.G. contributed with the section “Strain engineering effects”. D.H., J.Z., Z.L., and X.W. contributed with the sections “Molecular intercalation and chemical doping effects and Alloying effects”. S.H.S. and Y.H.L. contributed with the section “Bandgap closing: metal–insulator transition”. A.C., S.H.O., D.R.C., T.L., Y.D.K., F.M.P., and P.A. edited the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chaves, A., Azadani, J.G., Alsalman, H. et al. Bandgap engineering of two-dimensional semiconductor materials. npj 2D Mater Appl 4, 29 (2020). https://doi.org/10.1038/s41699-020-00162-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-020-00162-4
This article is cited by
-
Critical assessment of G0W0 calculations for 2D materials: the example of monolayer MoS2
npj Computational Materials (2024)
-
Prediction of highly stable 2D carbon allotropes based on azulenoid kekulene
Nature Communications (2024)
-
Broadband miniaturized spectrometers with a van der Waals tunnel diode
Nature Communications (2024)
-
Microwave graphitic nitrogen/boron ultradoping of graphene
npj 2D Materials and Applications (2024)
-
Synthesis of 2D layered transition metal (Ni, Co) hydroxides via edge-on condensation
Scientific Reports (2024)
