
Abstract
The genotype–phenotype correlation of the X-linked Alport syndrome (XLAS) has been well elucidated in males, whereas it remains unclear in females. In this multicenter retrospective study, we analyzed the genotype–phenotype correlation in 216 Korean patients (male:female = 130:86) with XLAS between 2000 and 2021. The patients were divided into three groups according to their genotypes: the non-truncating group, the abnormal splicing group, and the truncating group. In male patients, approximately 60% developed kidney failure at the median age of 25.0 years, and kidney survival showed significant differences between the non-truncating and truncating groups (P < 0.001, hazard ratio (HR) 2.8) and splicing and truncating groups (P = 0.002, HR 3.1). Sensorineural hearing loss was detected in 65.1% of male patients, while hearing survival periods showed a highly significant difference between the non-truncating and truncating groups (P < 0.001, HR 5.1). In female patients, approximately 20% developed kidney failure at the median age of 50.2 years. The kidney survival was significantly different between the non-truncating and truncating groups (P = 0.006, HR 5.7). Our findings support the presence of genotype–phenotype correlation not only in male patients but also in female patients with XLAS.
Similar content being viewed by others

Introduction
Alport syndrome (AS) is a progressive inherited nephropathy that leads to kidney failure and is accompanied by sensorineural hearing loss (SNHL) and ocular abnormalities, such as lenticonus and fleck retinopathy1,2. Although previous studies suggest a prevalence of approximately 1/50,000 for AS, a recent publication reported significantly higher estimated frequencies of predicted pathogenic COL4A4-COL4A5 variants2. While the most common form of AS inheritance is the autosomal dominant form, which affects about 1 in 106 individuals, the X-linked AS (XLAS) form, which is a more severe phenotype, affects approximately 1/2320 of the population2.
In male patients, 90% develop chronic kidney disease (CKD) with a glomerular filtration rate (GFR) category 5 (GFR < 15 ml/min per 1.73 m2 or treatment by kidney replacement) (CKD G5)3 by the age of 40 years, with a median age of 25 years4, and exhibit a very strong genotype–phenotype correlation. For example, truncating variants (rearrangement, nonsense, and frameshift) show a severe phenotype that leads to the earlier development of kidney failure and hearing loss, whereas non-truncating variants (missense and in-frame variants) present a milder phenotype. In the case of splice variants, the overall phenotype was intermediate4,5,6. The development of hearing loss and ocular changes showed a trend similar to that of kidney prognosis4,5,7. In female patients with XLAS, no genotype–phenotype correlations have been observed as they generally present favorable outcomes, with approximately 25% of cases progressing to CKD G5 throughout their lifetime, with a median age of 65 years8,9. However, a recent systematic review article10 reported a correlation between severe genotypes and proteinuria. Missense variants are the most common (60%) variants in male patients, and the overall phenotype is favorable, but the prognosis is variable, ranging from mild to severe11. The majority (about 85%) of missense variants are found in the intermediate collagenous domain, and glycine (Gly) substitutions constitute 95% of these variants (in ARUP, arup.utah.edu/database/)11. Glycine substitution adjacent to a noncollagenous (NC) interruption site is known to often be hypomorphic with a milder phenotype11,12, and other mitigating factors associated with variant location and affected residue have been suggested2. Histologically, the positive expression of the type IV collagen α5 chain in immunofluorescence staining tends to show a milder phenotype12. Here, for the first time, we analyzed the genotype–phenotype correlation in Korean patients of both sexes with XLAS.
Patients and methods
Study participants and definitions
This retrospective study was conducted at 12 medical centers in Korea. A total of 124 probands (85 males and 39 females) diagnosed with XLAS between September 2000 and June 2021 were enrolled. The diagnosis of XLAS was primarily based on the detection of pathogenic COL4A5 variants. Patients with variants classified as having uncertain significance were enrolled if the kidney biopsy findings were consistent with AS. Clinical and laboratory data were obtained through a review of electronic medical records. In addition, some data analyses included the available clinical data of 92 (45 males and 47 females) affected family members with the same COL4A5 variants as the proband. Therefore, the number of analyzed subjects differed for each parameter because of the missing variable data of the affected family members. All patients and their affected family members analyzed in this study were Korean.
The estimated GFR (eGFR) was calculated using the creatinine-cystatin C-based Schwartz 2012 equation13 for patients under 18 years of age and the CKD Epidemiology Collaboration 2021 formula14 for patients aged 18 years and older. CKD stage was determined using the Kidney Disease: Improving Global Outcomes (KDIGO) CKD classification3. Asymptomatic urinary abnormalities (AUA) were defined as the presence of microscopic hematuria and/or proteinuria, detected incidentally during school screening or routine laboratory findings during hospital visits, without any accompanying subjective symptoms. Nephrotic syndrome (NS) was defined as nephrotic-range proteinuria (first morning urine protein to creatinine ratio (UPCR) ≥ 2.0 mg/mg or ≥ 3 + dipstick) and serum albumin < 3.0 g/dL according to the KDIGO 2021 guideline15. The occurrence of hearing impairment was defined as subjective symptoms or according to pure tone audiometry results based on the World Health Organization classification16. Patients with other potential causes of hearing impairment, such as ear infections or medications, were excluded from the analysis. Ocular lesions included maculopathy, lens abnormalities, corneal opacities, cataracts, myopia, keratoconus, and astigmatism7,17,18.
Ethical approval
This study was approved by the institutional ethics committee of each hospital, including the Seoul National University Bundang Hospital, Seoul National University Children’s Hospital, Asan Medical Center, Chonnam National University Hospital, Chungnam National University Hospital, Ewha Womans University Hospital, Inha University Hospital, Korea University Hospital, Kyungpook National University Hospital, Pusan National University Yangsan Hospital, the Catholic University of Korea, Bucheon St. Mary’s Hospital, and Wonju Severance Christian Hospital, which waived the need for informed consent because only retrospective study data was collected. All procedures were performed in accordance with the Declaration of Helsinki.
Mutational analysis
Genetic tests were performed using Sanger sequencing of COL4A5 in most probands, except for four female probands in which targeted exome sequencing (TES) (three patients) and whole-exome sequencing (WES) (one patient) were used. Family members were screened using Sanger sequencing if they presented with clinical symptoms related to AS, such as hematuria, proteinuria, CKD, or hearing loss. The detailed procedures of Sanger sequencing19, TES20, and WES21 have been described in previous literature. Sequences were compared with the reference mRNA sequences (GenBank Accession: NM_000495.4). The interpretation of variants followed the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines, and variants classified as pathogenic or likely pathogenic were considered disease-causing variants (Supplementary Table S1)22. In addition, we included some variants classified as having uncertain significance if the kidney biopsy findings were consistent with AS and a definite family history of kidney disease or hearing loss was present (Supplementary Table S2). The novelty of the variants was determined by referring to the ClinVar, Varsome, LOVD, and HGMD databases.
According to the genotypes, the patients were divided into three groups: Group 1, missense variants or short in-frame deletions, insertions, or duplications; Group 2, abnormal splicing variants; and Group 3, nonsense variants, frame-shifting variants, or large deletions encompassing one or more exons.
Statistical analyses
Statistical analyses were performed using SPSS software, version 20.0 (IBM Corp., Armonk, NY, USA). Descriptive statistics, including the median (interquartile range [IQR]) or frequency counts (percentage), were used to describe the sample population. Categorical variables were analyzed using the Chi-square test or Fisher’s exact test. Continuous variables were compared using the Kruskal–Wallis test or the Mann–Whitney U test. The Kaplan–Meier method and the log-rank statistic were used to present the occurrence of events (age at CKD G5 and age at detection of SNHL). The hazard ratio (HR) was analyzed using the Cox proportional hazards model. Robust statistical tests were used for the comparison between groups and intrafamilial correlations5. A P-value of < 0.05 was considered significant.
Results
Overall manifestations of the patients
Kidney phenotypes
The detailed clinical kidney presentations and outcomes are summarized in Table 1, and the phenotypes and genotypes of the individual patients are listed in Supplemental Table S1. The median age of onset and age at the last visit were 4.4 and 20.8 years, respectively. The most common initial presentation was AUA, followed by macroscopic hematuria, in both males and females. While no female patients presented with NS, approximately 20% of male patients presented with the condition, along with hematuria, at the median onset age of 7.0 (IQR 3.5–10.3) years.
In male patients, UPCR at the initial hospital visit at a median age of 8.9 (IQR 4.1–14.3) years was 1.6 mg/mg (IQR 0.5–4.0 mg/mg), and during follow-up, almost all patients except for one developed overt proteinuria (UPCR > 0.2 mg/mg). More than 90% of female patients developed overt proteinuria during follow-up. All patients exhibited hematuria. Treatment with renin-angiotensin system inhibitors (RASi) was initiated in all male patients at a median age of 9.5 years, except for two patients already in CKD G5 at the initial presentation. Nearly 90% of the female patients started taking RASi at the same median age as the male patients.
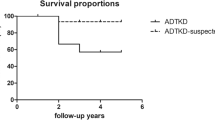
Approximately 60% of male patients had reached CKD G5 at the median age of 25.0 ([95% confidence interval (CI)] 23.5–26.5) years, which was estimated by the Kaplan–Meier method (Fig. 1), and nearly 90% had reached CKD G5 by 39 years. In female patients, approximately 20% progressed to kidney failure at the median age of 50.2 (95% CI 39.0–61.5) years, as estimated by Kaplan–Meier analysis, and approximately 30% and 60% reached CKD G5 by 37 and 55 years, respectively (Fig. 1).

Kidney survival in 188 patients (120 males, 68 females) with X-linked Alport syndrome according to sex.
Kidney biopsy was performed in 52.3% (68 of 130 patients) of male patients at a median age of 10.4 (IQR 7.1–14.3) and 41.9% (36 of 86 patients) of female patients at a median age of 10.6 (IQR 7.0–22.9) years. The pathological diagnosis was AS in 59 (86.8%) male and 20 (55.6%) female patients, while the remaining patients were diagnosed with immunoglobulin A nephropathy, mesangial proliferative glomerulonephritis, or thin basement membrane disease. On the second biopsy, AS was diagnosed in four male and three female patients.
The expression of the type IV collagen α5 chain was examined by immunofluorescence microscopy in 23 male and 17 female patients. In male patients, the expression was completely absent in 17 (73.9%), weak in 3 (13.0%), and normal in 3 (13.0%) patients. In female patients, 8 (47.1%) showed a mosaic pattern, 7 (41.2%) showed a normal pattern, and 2 (11.8%) showed a total loss.
Extrarenal phenotypes
In male patients, 65.1% developed SNHL at the median age (hearing survival age) of 19.9 (95% CI 13.8–26.0) years, while 14.1% of female patients developed SNHL at the median age of 53.4 (95% CI 47.5–59.2) years, which was estimated by Kaplan–Meier analysis (Table 1). A total of 47.9% (34/71; no data available for 25 patients) of male patients and 40% (4/10; no data available for five patients) of female patients with SNHL were recommended for or wore hearing aids at the median ages of 11.3 (IQR 9.9–16.7) and 45.9 (IQR 33.8–50.8) years, respectively. Ocular examination records were available for 63 males and 21 females. Twenty (31.7%) male patients and two (9.5%) female patients showed ocular abnormalities such as anterior lenticonus or fleck retinopathy.
Genotypes
Among the 124 probands, 56 (45.2%) had missense variants, 23 (18.5%) had abnormal splicing, 19 (15.3%) had nonsense variants, 21 (16.9%) had short frame-shifting insertion or deletion variants, and 5 (4.0%) had other types of variants (Table 2 and Supplemental Table S1). Among the 121 different variants, 93 (76.9%) were novel. Two variants (p.Gly400Val and p.Arg1677Ter) were common in three families, and two other variants (c.466-2A>G and c.991-7T>A) were detected in both families. One male proband (P49 in Supplemental Table S1) had two COL4A5 variants, p.Gly1057Glu and p.Gly1442Cys.
Genotype–phenotype correlations
Male patients
Kidney phenotypes
At the time of onset, NS was significantly more prevalent (P = 0.023, odds ratio [OR] 3.3), and AUA was significantly less prevalent (P = 0.043, OR 0.4) in Group 3 patients when compared to Group 1 or 2 patients (Table 3).
When stratified according to variant types, approximately 50% of Group 1 patients and 70% of Group 3 patients developed CKD G5 at the median age (kidney survival periods) of 30 and 20 years, respectively. The kidney survival period showed a significant difference between Group 1 and Group 3 (P < 0.001, HR 2.8) and between Group 2 and Group 3 (P = 0.002, HR 3.1), which was estimated by Kaplan–Meier analysis. However, there was no difference between Groups 1 and 2. The kidney outcomes according to variant type are described in Table 3 and Fig. 2.

Kidney survival in 120 male patients with X-linked Alport syndrome according to the variant types of COL4A5.
Of the 40 different Gly substitution variants, nine were located between exons 1–20, 29 between exons 21–47, and two in the NC domain. The location of Gly substitution variants (exons 1–20 versus 21–47) did not affect the severity of the kidney phenotypes. Furthermore, the difference in the amino acid substitution for Gly did not correlate with the kidney phenotypes (data not shown).
In 15 families, there were at least two (between two and four) male patients who progressed to CKD G5. Intrafamilial homogeneity in CKD progression (less than 15 [0–10.3] years of age at CKD G5) was observed in 13 families. Conversely, intrafamilial heterogeneity (15 or more [between 16 and 22.9] years of age at CKD G5) was observed in the remaining two families.
Extrarenal phenotypes
SNHL was detected more commonly in Group 3 patients when compared to Group 1 patients (P < 0.001, OR 7.6 [95% CI 2.3–24.8]) (Table 3). Kaplan–Meier analyses estimated that the hearing survival period was significantly shorter in Group 3 than in Group 1 (P < 0.001, HR 5.1) and in Group 2 than in Group 1 (P = 0.032, HR 2.2). However, there was no difference between Groups 2 and 3. (Fig. 3) Ocular abnormalities were also more prevalent in Group 3 than in Group 1 (P < 0.001) and in Group 2 than in Group 1 (P < 0.001) (Table 3).

Hearing survival in 89 male patients with X-linked Alport syndrome according to the variant types of COL4A5.
Female patients
Kidney phenotype
The age of onset was lower in Group 3 than in Group 1 (P = 0.029) and Group 2 (P = 0.005), and Group 3 patients presented AUA less frequently than other groups (P = 0.037) (Table 4). Fourteen (20.6%) of the 68 female patients developed CKD G5. Group 3 patients developed CKD G5 significantly earlier than Group 1 (P = 0.006, HR 5.7), as well as Groups 1 and 2 (P = 0.013, HR 2.7) (Table 4 and Fig. 4).

Kidney survival in 68 female patients with X-linked Alport syndrome according to the variant types of COL4A5.
Extrarenal phenotype
The prevalence of SNHL did not differ among the three groups. Ocular phenotypes were not analyzed owing to data limitations.
Discussion
This multicenter study in Korea allowed for the analysis of the genotype–phenotype correlation of male and female patients with XLAS who were diagnosed by genetic testing. Male patients in our study showed a significant difference in kidney survival between those with truncating variants and others, similar to the results of other studies4,5,6,23. In most previously reported studies4,5,6,23, cases with truncating variants developed CKD G5 earlier than cases with non-truncating variants. The kidney prognosis of male patients in our study was similar to that of a European study5. In contrast, the overall median age at CKD G5 in our study was earlier than that in the USA and Japanese cohort studies4,23 (Supplementary Table S3). The proportion of patients with missense variants, known to be associated with better prognosis, in our study (45%) was lower than that in the USA (64%) and Japanese (58%) cohort studies, and the kidney survival of our patients with missense variants (29 years) was shorter than that in the USA (37 years) and Japanese (40 years) studies as well. Although it is difficult to explain these differences, some possible reasons are as follows: Firstly, in our male patients with missense variants, RASi treatment was delayed approximately 6 years after the onset of renal symptoms (at a median age of 10.5 years) due to delayed hospital visits, and they already presented with significant proteinuria (the median UPCR of 1.5 mg/mg) at the first hospital visit. In a considerable number of cases in our study, RASi treatment was initiated when UPCR was ≥ 0.5 mg/mg for two or more consecutive measurements and discontinued when proteinuria improved after treatment. Recent guidelines recommend early and aggressive RASi use because it significantly prolongs kidney survival24. Secondly, our patients may have possessed missense variants resulting in a more severe phenotype. Kamura et al. suggested that poor trimerization or secretion defects could lead to a severe phenotype in missense mutations of the COL4A5 gene25. None of the patients in our study had a genetic variant known as G624D, a common hypomorphic founder variant of European descent, or P628L and G1000V, which have been linked to mild phenotypes in previous studies26,27,28. Thirdly, the presence of modifier gene variants related to podocytes or the glomerular basement membrane may coexist and contribute to early-onset kidney failure29. Our study was limited as most patients were tested only using Sanger sequencing of the COL4A5 gene, which may have resulted in the missed detection of modifier gene variants. Lastly, selection bias may have occurred as patients with a relatively severe phenotype were more likely to have been tested and included in this study.
Our study found that approximately one-fifth of male patients presented with NS as their first manifestation, which, to our knowledge, has not been well studied before. While most male patients (80–100%) present with microscopic hematuria, with or without macroscopic hematuria, proteinuria can be recognized at an early stage of childhood, sometimes leading to nephrotic syndrome30. In this study, we observed that NS was significantly more prevalent as the first manifestation in male patients with truncating variants. The higher incidence of NS at the initial visit could be associated with the higher rate of these variants. Furthermore, recent genetic studies have increasingly reported AS-causing genes in patients with steroid-resistant NS/focal segmental glomerulosclerosis31.
SNHL was detected in 65.1% of male patients in the present study; male patients with missense variants developed hearing problems significantly later than those with other variants, which is consistent with previous reports4,5. More than half of the patients with hearing loss required a hearing aid at the time of diagnosis, which means that the onset of hearing loss might be earlier. Hearing loss can occur even at preschool age in patients with severe variants32. Among female patients, 14.1% were found to have SNHL, which is higher than in the Japanese study (5.5%) but lower than in the European cohort (28%)8,9. The median age of the SNHL was 53.4 years, which is an earlier age than in the European cohort (more than 80 years). Despite these findings, there is limited research on hearing loss in females, so further studies are needed to better understand this condition. Moreover, early detection and proper management of SNHL are required to improve developmental, educational, and cognitive outcomes, particularly in children and adolescents33.
To date, two previous large-scale studies8,9 showed that female patients with XLAS, showed far more benign kidney outcomes compared to male patients: only 12–15% and 30–40% of patients developed CKD G5 by 40 and 60 years of age, respectively, without genotype–phenotype correlations. However, in a smaller study of 24 female patients with XLAS34, patients with non-missense variants showed a higher frequency of early proteinuria and a higher risk of developing CKD. Moreover, a recent systemic review10 provided more supportive evidence for a correlation between severe genotype and proteinuria, with proteinuria likely leading to kidney failure in female patients. The review found that proteinuria detected before 15 years of age progressed rapidly to kidney failure10 (Supplementary Table S4). In our study, more than 90% of the female patients developed overt proteinuria during follow-up, but the exact incidence at disease onset and onset date of proteinuria were unavailable. Despite RASi being used in nearly 90% of female patients at a median age of 9.5 years (approximately 5 years after onset presentation), 20.9% developed CKD G5 with a median age of 50.2 years (youngest age of 16.2 years). Kaplan–Meier analysis showed that over 30% and 60% of the patients progressed to CKD G5 by 40 and 60 years, respectively. In addition, our study showed a significant difference in kidney outcomes between female patients with non-truncating and truncating variants. Although the reasons for the relatively poorer kidney outcomes in our female patients compared to those in other studies are not clear, it is possible that a larger proportion of severe mutations, coexisting digenic variants, combined mutations such as those in other podocyte-related genes, and skewed X chromosome inactivation may have influenced phenotype severity in our patients35,36. Digenic AS, which occurs when variants in both COL4A5 and COL4A3 or COL4A4 genes are present, is known to have worse clinical features than a single variant in females31,35. Our study included more male patients than females, despite the fact that twice as many females as males are affected. It is possible that only women with more severe diseases were referred to the kidney clinic for evaluation, and limited genetic testing of patients with more severe phenotypes may have led to selection bias. Regarding hearing loss in female patients, there was no genotype–phenotype correlation, which is consistent with previous studies8,9.
Our study had several limitations. First, it was a retrospective study; thus, a major limitation was the absence of a unified RASi treatment protocol. Furthermore, there were considerable missing data points, especially for the affected family members. Second, the genetic testing in our study also posed some limitations, given that the test consisted only of Sanger sequencing of the COL4A5 gene in most cases, with digenic and modifier gene variants not tested. Third, other exacerbating factors that might deteriorate kidney function, such as obesity and hypertension, could not be evaluated.
Nevertheless, this is the first multicenter study in Korea to investigate the genotype–phenotype correlation in a sizable number of males and females with XLAS. In particular, this is the first study to confirm the genotype–phenotype relationship between non-truncating and truncating variants in females with XLAS.
Conclusions
In this multicenter retrospective study of Korean patients with XLAS, missense variants (mostly Gly substitutions) were the most common variant in both male (46.2%) and female (46.5%) patients. Despite using RASi in most patients, 57.9% of male and 20.9% of female patients developed CKD G5 at a median age of 25.0 and 50.2 years, respectively. Furthermore, SNHL was detected in 65.1% of male and 14.1% of female patients at a median age of 19.9 and 53.4 years, respectively. Among male patients, those with truncating variants had a shorter kidney survival period and higher frequency of SNHL than those with other types of variants (P < 0.001 in both). Even female patients with truncating variants had significantly poorer kidney survival than patients with other variants. These findings support the presence of genotype–phenotype correlation not only in male patients but also in female patients with XLAS.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Nozu, K. et al. Genetic background, recent advances in molecular biology, and development of novel therapy in Alport syndrome. Kidney Res. Clin. Pract. 39, 402 (2020).
Gibson, J. et al. Prevalence estimates of predicted pathogenic COL4A3-col4a5 variants in a population sequencing database and their implications for Alport syndrome. J. Am. Soc. Nephrol. (2021).
Levey, A. S. et al. Nomenclature for kidney function and disease: Report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int. 97, 1117–1129 (2020).
Bekheirnia, M. R. et al. Genotype–phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 21, 876–883 (2010).
Jais, J. P. et al. X-linked Alport syndrome: Natural history in 195 families and genotype-phenotype correlations in males. J. Am. Soc. Nephrol. 11, 649–657 (2000).
Gross, O., Netzer, K. O., Lambrecht, R., Seibold, S. & Weber, M. Meta-analysis of genotype–phenotype correlation in X-linked Alport syndrome: Impact on clinical counselling. Nephrol. Dial. Transplant. 17, 1218–1227 (2002).
Savige, J. et al. Ocular features in Alport syndrome: Pathogenesis and clinical significance. Clin. J. Am. Soc. Nephrol. 10, 703–709 (2015).
Jais, J. P. et al. X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 14, 2603–2610 (2003).
Yamamura, T. et al. Natural history and genotype–phenotype correlation in female X-linked Alport syndrome. Kidney Int. Rep. 2, 850–855 (2017).
Gibson, J. T., de Gooyer, M., Huang, M. & Savige, J. A Systematic review of pathogenic COL4A5 variants and proteinuria in women and girls with X-linked Alport syndrome. Kidney Int. Rep. 7, 2454–2461 (2022).
Savige, J. et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: Refining the ACMG criteria. Eur. J. Hum. Genet. 29, 1186–1197 (2021).
Hashimura, Y. et al. Milder clinical aspects of X-linked Alport syndrome in men positive for the collagen IV α5 chain. Kidney Int. 85, 1208–1213 (2014).
Schwartz, G. J. et al. Improved equations estimating GFR in children with chronic kidney disease using an immunonephelometric determination of cystatin C. Kidney Int. 82, 445–453 (2012).
Inker, L. A. et al. New creatinine-and cystatin C-based equations to estimate GFR without race. N. Engl. J. Med. 385, 1737–1749 (2021).
Rovin, B. H. et al. KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney Int. 100, S1–S276 (2021).
Olusanya, B. O., Neumann, K. J. & Saunders, J. E. The global burden of disabling hearing impairment: A call to action. Bull. World Health Organ. 92, 367–373 (2014).
Moshirfar, M. et al. Keratoconus in a patient with Alport syndrome: A case report. World J. Clin. Cases 7, 3012 (2019).
Großmann, C. B. J. Characterization of the eye phenotype in children with Alport syndrome. (2022).
Cheong, H. I., Park, H. W., Ha, I. S. & Choi, Y. Mutational analysis of COL4A5 gene in Korean Alport syndrome. Pediatr. Nephrol. 14, 117–121 (2000).
Park, E. et al. Genetic study in Korean pediatric patients with steroid-resistant nephrotic syndrome or focal segmental glomerulosclerosis. J. Clin. Med. 9, 2013 (2020).
Seo, G. H. et al. Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clin. Genet. 98, 562–570 (2020).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015).
Yamamura, T. et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int. 98, 1605–1614 (2020).
Kashtan, C. E. & Gross, O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults—an update for 2020. Pediatr. Nephrol. 36, 711–719 (2021).
Kamura, M. et al. Trimerization and genotype–phenotype correlation of COL4A5 Mutants in Alport SYNDROME. Kidney Int. Rep. 5, 718–726 (2020).
Żurowska, A. M. et al. Mild X-linked Alport syndrome due to the COL4A5 G624D variant originating in the Middle Ages is predominant in Central/East Europe and causes kidney failure in midlife. Kidney Int. 99, 1451–1458 (2021).
Kaneko, K. et al. A family with X-linked benign familial hematuria. Pediatr. Nephrol. 25, 545–548 (2010).
Pierides, A., Voskarides, K., Kkolou, M., Hadjigavriel, M. & Deltas, C. X-linked, COL4A5 hypomorphic Alport mutations such as G624D and P628L may only exhibit thin basement membrane nephropathy with microhematuria and late onset kidney failure. Hippokratia 17, 207 (2013).
Savige, J. et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr. Nephrol. 34, 1175–1189 (2019).
Nozu, K. et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin. Exp. Nephrol. 23, 158–168 (2019).
Savige, J. et al. Guidelines for genetic testing and management of Alport Syndrome. Clin. J. Am. Soc. Nephrol. 17, 143–154 (2022).
Zhang, X. et al. X-linked Alport syndrome: Pathogenic variant features and further auditory genotype–phenotype correlations in males. Orphanet J. Rare Dis. 13, 1–11 (2018).
Yoshinaga-Itano, C., Sedey, A. L., Wiggin, M. & Chung, W. Early hearing detection and vocabulary of children with hearing loss. Pediatrics 140, e20162964 (2017).
Mastrangelo, A. et al. X-Linked Alport syndrome in women: Genotype and clinical course in 24 cases. Front. Med. 7, 580376 (2020).
Savige, J. et al. Digenic Alport syndrome. Clin. J. Am. Soc. Nephrol. 17, 1697–1706 (2022).
Levy Erez, D. & Rheault, M. N. An update on women and girls with Alport syndrome. Curr. Pediatr. Rep. 10, 195–201 (2022).
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1A2C1100974 to H.G.Kang). ※ MSIT: Ministry of Science and ICT.
Author information
Authors and Affiliations
Contributions
J.H.K., Y.H.A. and H.I.C. were primarily responsible for study conception and design, and drafted the article; J.Y.S., M.H.C., H.H., E.M.Y., J.W.L., M.H.C., M.J.P., J.H.L., J.J., K.H.Y., K.M.J., K.S.P., J.S.S., and M.K.N., W.Y.C., S.J.K., E.Y.C., K.M.K., N.H.K., M.K.collected and analyzed data as principal investigators for each center; J.H.P reviewed and analyzed pathologic findings of kidney; J.H.K., S.H.L. and Y.H.A. performed the statistical analysis; J.H.K., H.G.K., Y.H.A. and H.I.C. provided critical revision. The final manuscript has been seen and approved by all the authors, and they have given necessary attention to the manuscript to ensure the integrity of the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, J.H., Lim, S.H., Song, J.Y. et al. Genotype–phenotype correlation of X-linked Alport syndrome observed in both genders: a multicenter study in South Korea. Sci Rep 13, 6827 (2023). https://doi.org/10.1038/s41598-023-34053-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-34053-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
