
Abstract
This study uses wastewater-based epidemiology (WBE) to rapidly and, through targeted surveillance, track the geographical distribution of SARS-CoV-2 variants of concern (Alpha, Beta and Delta) within 24 wastewater treatment plants (WWTPs) in the Western Cape of South Africa. Information obtained was used to identify the circulating variant of concern (VOC) within a population and retrospectively trace when the predominant variant was introduced. Genotyping analysis of SARS-CoV-2 showed that 50% of wastewater samples harbored signature mutations linked to the Beta variant before the third wave, with the Delta variant absent within the population. Over time, the prevalence of the beta variant decreased steadily. The onset of the third wave resulted in the Delta variant becoming the predominant variant, with a 100% prevalence supporting the theory that the Delta variant was driving the third wave. In silico molecular docking analysis showed that the signature mutations of the Delta variant increased binding to host proteins, suggesting a possible molecular mechanism that increased viral infectivity of the Delta variant.
Similar content being viewed by others


Introduction
Since December 2019, the deadly severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has caused social and economic disruption and has had a major impact on public healthcare systems globally. In an attempt to control the pandemic, COVID-19 vaccines have been developed at an extraordinary pace. Vaccination programs have become vital due to the emergence of immune-evading Variants of Concern (VOCs) as a result of high mutation rates within the SARS-CoV-2 genome. In this context, a VOC can be classified as a variant of which there is evidence of an increase transmissibility, disease severity and decrease vaccine-induced protection1.
Through the course of the pandemic, several rapidly spreading SARS-CoV-2 VOCs have arisen in countries, including the United Kingdom (UK)2, South Africa (SA)3, Brazil (BZ)4, India5 and the United States of America (USA)5,6,7,8. These VOCs include B.1.1.7 (Alpha), B.1.351 (Beta), P.1 (Gamma), B.1.617 (Kappa/Delta) and, more recently, C.1.2. lineage6,9,10. The identified lineages are termed VOCs depending on the propensity to infect and transmit due to the acquisition of multiple signature mutations in the viral spike (S) glycoprotein, a key target for vaccine development, potentially affecting viral infectivity and antigenicity6,11. The S- protein is a 180–200 kDa protein that is found as a glycosylated trimer surrounding the virion surface. Each monomeric structure consists of an S1 and S2 subunit, which is further characterized by an N-terminal domain (NTD), a receptor-binding domain (RBD), a fusion peptide (FP), heptapeptide repeat sequences 1 and 2 (HR1/2), a transmembrane domain and a cytoplasmic domain12. During viral infection, the RBD domains of the trimeric structure confer an “open state” confirmation, which initiates entry into host cells by binding to angiotensin-converting enzyme 2 (ACE 2)13. Once bound, the S1 and S2 subunits of the protein are subjected to proteolysis by host proteases. The S1/S2 boundary site residues proline-arginine-arginine-alanine (PRRA) are cleaved by furin convertase, detaching the two subunits, while transmembrane protein 2 (TMPRSS2) cleaves the S2 site (residues KPSK) to expose the fusion peptide. During this time, the HR1/2 peptides undergo conformational modifications to form a “6-helix bundle fusion core” complex that leads to the linking and fusion of the viral and host cell membranes. Once fusion occurs, the viral RNA is released into the host cell for further replication14,15. Given the necessity of the spike protein in SARS-CoV-2 replication through viral binding, it is of no surprise that the viral particle has rapidly evolved in an attempt to evade prophylactic and therapeutic regimens. The phenotypic modifications observed in the VOCs are populated with several S-protein mutations and deletions, as elucidated below.
The B.1.1.7 The alpha variant was first identified in the UK and has since disseminated to more than 110 countries, with the most prominent mutations being the N501Y (role in viral entry into human cells), P681H, H69/V70 and Y144/145 deletions, with the latter two deletions altering the shape of the spike, allowing the virus to evade an immune response2. First identified in South Africa, in December 2020, the B.1.351 The Beta variant escalated to one of the most predominant VOCs, harboring the characteristic N501Y, K417N and E484K mutations. The Beta variant has since spread to 68 countries3 and shares two common mutations (N501Y and E484K) with the P.1 Gamma variant, first identified in Japan in four travelers from Manaus, Brazil16. Since its detection, the Gamma variant has spread to more than 37 countries with signature mutations, including N501Y, K417N, and E484K, which may aid the virus in evading an immune response. The B.1.617 variant appeared in India in October 2020. This variant continued to evolve into B.1.617.1, known as the Kappa variant, and B.1.617.2, known as Delta variant, which is now the most common variant in India, having two prominent mutations, E484Q and L452R. Last, the B.1.427/429 Epsilon lineage appeared for the first time in California and carried the L452R mutation, which has been reported to be highly contagious17. Taken together, the mentioned mutations within VOCs are of functional importance, thus augmenting public health concerns surrounding potential detrimental clinical implications, including infection volatility and vaccine efficacy17,18,19,20. Key VOC mutations may also result in diagnostic test failures and could potentially lead to a decrease in the affinity of antiviral candidates for SARS-CoV-2, thus necessitating the rollout of recurring SARS-CoV-2 vaccines.
In the last year, wastewater surveillance has been used across the globe to monitor for the presence of COVID-19 infections within a community and to track infection trends21,22,23. To date, limited studies have utilized wastewater to routinely screen for circulating variants11,24,25,26. Furthermore, most studies used next-generation sequencing (NGS) to gather information about the diversity of SARS-CoV-2 variants at the community level27,28. While NGS sequencing is informative to identify new variants, it is costly to perform routine screening of sewage samples to identify and profile known mutations associated with VOCs. As such, this study aimed to conduct weekly wastewater surveillance using a robust TaqMan SARS-CoV-2 mutation panel and report on key VOCs at the community level within 24 WWTPs in Cape Town, South Africa. This study also identified and determined the emergence of the VOC that drove South Africa’s third wave and elaborated on the key structural implications of VOC mutations on viral infectivity.
Results and discussion
In the current study, routine surveillance was conducted on 272 wastewater samples within 24 WWTPs in the Cape Town Municipality, South Africa. At the start of the experiments, the epidemiological limit of detection (LOD) was determined and set at 700 g.c./mL. The latter results were higher than the LOD of 50 g.c/mL reported by Randazzo et al.29. Subsequently, RT-qPCR analysis was used to confirm the presence and quantify the SARS-CoV-2 viral load within the 24 WWTP between April and July 2021. Over the three months, the average viral load was 12 798 g.c./mL, and only sites with a viral load > 1500 g.c/mL were used for subsequent variant testing (Fig. 1).

Temporal analysis of SARS-Cov-2 viral load in wastewater against diagnosed COVID-19 cases in Western Cape.
This study reports on the emergence of a highly fit SARS-CoV-2 lineage with several S-protein mutations that decrease vaccine efficacy while preluding possible reinfection. Wastewater contains a mixture of inactivated SARS-CoV-2 nucleic acid fragments shed from multiple infected individuals. Genotyping these fragments can identify the circulating variant and its geographic distribution within a given catchment area with much less effort than clinical surveillance30. This is especially important with the current lineage of SARS-CoV-2 that is constantly mutating, affecting vaccine efficacy and viral transmission. Understanding the dynamics of a highly virulent circulating strain will allow for prompt response and informed decision making to control and restrain viral spread. Likewise, recent scientific evidence highlighted the importance of wastewater surveillance in tracking disease prevalence in the general population and, more recently, tracking signature mutations in VOC. Hence, we report a robust TaqMan SARS-CoV-2 genotyping method to monitor wastewater for signature mutations linked to VOCs at the population level. Additionally, this information will identify spatial hotspots while tracking temporal trends of VOCs, including their prevalence and frequency over time.
As the Western Cape, South Africa approached the third wave (early June 2021 week 24), a steady increase in SARS-CoV-2 viral load was observed, with an average of 12 798 genome copies (g.c.) /mL recorded over a 3-month period. The orange line graph represents the average viral load across all wastewater treatment plant, whilst the black line graph indicates diagnosed cumulative COVID-19 cases within the Western Cape (Fig. 1).
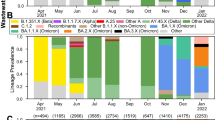
Next, the mutational frequency, which refers to the proportional of wastewater samples that were positive for a mutation divided by the total number of samples, where detected for all VOC. Figure 2 displays the mutational frequency for the Alpha (N501Y, 69/70DEL and P681H), Beta (K417N, N501Y and E484K) and Delta (L452R and P681R) variants of SARS-CoV-2 found in sewage water over a period of 3 months. According to Abdool Karim et al.6 and later Tegally et al.3, the Beta variant was the predominant circulating variant within the Western Cape. Our results confirmed previous reports showing that the Beta variant was indeed the predominant variant between 26 April 2021 and 24 May 2021 (Epidemiological weeks 17 and 21, respectively, Fig. 3). This study also showed that the Beta variant harbored a combination of key mutations (K417N, N501Y and E484K) that define this lineage at a stable frequency of > 60% (Fig. 2A). The presence of these mutations was also confirmed by whole genome sequencing of a subset of samples collected from selected WWTPs within the Cape town municipalities (Supplementary data). However, the frequency for mutations of the Alpha variant was very low, corresponding to a prevalence ≤ 40%, while the Delta variant was not introduced during the respective period (Figs. 2B,C and 3). Temporal analysis showed that from 7 June to 26 July 2021, the combination of K417N, N501Y and E484K mutations for the beta variant decreased at a mean frequency of < 40% (Figs. 2 and 3). The surge in viral load and decreased occurrence of various mutations of the Beta variant were followed by a simultaneous rise in the frequency of mutations corresponding to the Delta variant. In the current study, genotyping of SARS-CoV-2 RNA for key mutations of the Delta variant (L452R and P681R) and whole genome sequencing (Supplementary data) confirmed the presence of this variant within wastewater by mid-May 2021, corresponding to epiweek 20 (Figs. 2C and 3).

Mutation frequency linked to key Variants of Concern (VOC). Frequency of viral RNA showing spike protein mutation linked to the (A) Beta variant with three key substitutions in the RBD (K417N, E484K and N501Y), (B) the Alpha variant with the N501Y, P681H and 69/70 Deletion and, (C) the Delta variant harbouring mutations for P681R and L452R.

Temporal analysis of SARS-CoV-2 variants of concern compared to diagnosed COVID-19 cases. Data suggests that the Beta variant predominance decreases with dominance of Delta variant. Data is represented as a stacked bar graph. Each colour represents the average frequency of a variant type that occurs across all WWTPs, and the proportion of that specific variant compared to other variants is given by week.
Our data also revealed the cooccurrences of key Delta mutations (L452R and P681R) with the P681H mutation corresponding to the Alpha variant (Fig. 2). Happi et al. (2020) reported that the P681H mutation was first observed in March 2020 in Nigeria and Hawaii, whereafter it was detected in New York and Israel and is now characterized as part of the global spreading B.1.1.7 VOC (35, 36). In this study, the P681H mutation was introduced as early as April, even when the viral load was low (Fig. 1B). An erratic pattern in the frequency of the P681H mutation was observed, with the lowest frequency of 25% across most of the sites. Thereafter, the P681H mutation became static across various sample sites, showing a frequency of 100% throughout the peak of the third wave (Figs. 2B, 3 and 4B). According to Shang et al. (37) and Zukerman et al. (35), the P681H mutation is a critical mutation found in the proximity of the furin cleavage site. Although it has been postulated that the P681H mutations play a role in increased furin cleavage, allowing virus entry into the cell, they do not decrease vaccine efficacy (35, 38, 39). Nonetheless, the P681H mutations are still being monitored globally (35).
Furthermore, apart from the P681H mutation, a 69/70 deletion, a key mutation for the Alpha variant, was observed only in two sample sites, namely, the Mitchells Plain and Zandvliet WWTP. It could be speculated that this variant could have been endemic to those catchment areas but not transmissible, as the viral load remained high throughout May 2021 (frequency > 50%) without offsetting any resurgence (Figs. 2 and 4).
Delta variant predominant in the third wave
This study evaluated whether the Delta variant was the dominant variant during the third wave, identifying when it emerged and elaborating on the protein structure of key mutations that may affect cellular infectivity. The Delta variant was first detected in India and became the leading variant globally31,32,33. The Delta variant is 60% more transmissible than its ancestry SARS-CoV-2 strain, meaning infection is more likely to occur in those exposed to the strain34.
The spatiotemporal analysis showed that the Delta variant was initially detected in the Gordons Bay and Melkbosstrand WWTP, whereafter it spread to the remaining 15 catchment areas within the City of Cape Town (Figs. 4 and 5). Figures 3 and 4 confirmed that as the Western Cape approached the third wave (early June 2021), a steady increase in SARS-CoV-2 viral load was observed, which was concomitant with the observed increase in the Delta variant. Spatial mapping showed that between 6 June and 21 July 2021, the Delta variant was detected in more than 95% of the WWTPs with a viral load ranging between 2000 and 50 000 g.c/mL (Figs. 3, 4 and 5). This co-occurred with a decline in the Beta and Alpha variants (Figs. 3 and 4), suggesting that this heightened prevalence observed during the third wave could have been attributed to the circulating Delta variant. Similar findings were reported by Scheepers et al. (2021), with the Delta variant being predominant by the end of June (317/422, 75% in week 24; 203/230, 88% in week 25), with the first mutations being identified within the Western Cape in Epidemiological 2110. The latter finding confirmed our study results; however, using wastewater, the Delta variant was detected as early as Epidemiological week 20 (Fig. 5). Furthermore, it can be speculated that the rapid emergence and spread of the Delta variant could have been introduced during travel-related importations, whereafter it continued to spread within the population, resulting in possible community transmission. However, evidence related to community transmission remains inconclusive. Wastewater surveillance and the use thereof to track, discriminate, and quantitate SARS-CoV-2 variants using targeted surveillance, such as NGS sequencing and the highly specific and discriminatory VOC RT-qPCR mutation panel, has become a commonly adopted method. However, despite allowing for the fast and cost-effective detection of signature mutations linked to VOC, it should be noted that wastewater samples contain a mixture of different isolates. Therefore, the method can only be utilized to detect the circulating variant in the community and cannot discriminate that the mutations linked to the VOC are present on the same virus genome.

Temporal analysis of the City of Cape Town municipality WWTP. The frequency of the spike protein mutations during the epidemiological weeks seeks to explain patterns of the shift of key mutations linked to 3 variants of concern; (A) Beta, (B) Alpha, and (C) Delta variant detected over time. The mutational frequency on the y axis is represented by a stacked bar graph. Each colour represents the average mutation frequency of a variant type, which refers to the proportion of wastewater samples that were positive for a mutation divided by the total number of samples.

Spatial analysis of the rapidly evolving Delta variant over three months. The Delta variant was initially detected in the Melkbosstrand WWTP, where after it spread to the remaining catchment areas. Spatial mapping showed that the Delta variant was detected in more than 95% of the WWTP. Red-Beta variant, Green-Delta and Black indicates the presence of the P681H mutation. All maps were produced using ArcGIS 10.6.1 (https://www.arcgis.com/).
Nonetheless, collectively, our results suggest that wastewater can be used as a proxy for prompt detection and informed decision making that will allow health officials to introduce rapid nonpharmaceutical interventions to curb the spread of this highly transmissible strain.
Structural Insights into SARS-CoV-2 Spike Glycoprotein VOCs on Cellular Infectivity
To further elucidate the understanding and consequences of these VOCs within the SARS-CoV-2 spike protein, in silico modeling techniques were utilized. This was done by comparing the wild-type versus mutant RBD domain when bound to the ACE2 receptor, as well as the affinity of the furin enzyme to the S1/S2 boundary site when mutated.
Infectivity
Binding of the spike protein RBD to ACE2 is critical to viral replication. After RBD-ACE2 binding, furin binds to the S1/S2 boundary site; thus, mutations seen at both of these domains can have detrimental implications on patients who lack neutralizing antibodies. Numerous studies have eluded the chemical inferences of mutations within the spike protein, with most in silico studies concluding that amino acid alterations, specifically at the RBD domain, increase ACE2-spike interactions (2,5–7). This study chose to assess two vital binding regions of the spike protein by comparing mutants from four of the major VOCs assessed in wastewater RT-QPCR surveillance. The mutations/deletions of concern were computed as per VOC, followed by molecular docking and interaction analysis to identify and compare the binding landscape between the viral and host proteins. Table 1 demonstrates the wild-type (WT) and VOCs complexed with ACE2 and Furin (Fig. 6).

Schematic representation of SARS-CoV-2 spike glycoprotein: Schematic representation of SARS-CoV-2 spike glycoprotein: (A) Surface area representation of trimeric spike protein conferring an “open conformation” RBD domain (S1 domain depicted in blue and the S2 domain represented in purple) (B) Atomistic representation of characterized subdomains within the monomeric spike protein and the spatial mapping of defined protein binding regions. The identified VOCs within the RBD, NTD and S1/S2 boundary site are also demonstrated. The Alpha variant mutation and deletion are represented in green, the Beta variant in red and the Delta variant in orange.
It was evident from the docked complexes that direct binding site mutations did not alter the binding affinity of either ACE2 or furin (Table 1). Increased binding affinity was noted when ACE2 bound to the Delta variant compared to the WT. This was evidenced by an increase of 7 interacting residues and a surface interaction area of 399 Å2. The alpha variant docked to ACE2 with a suboptimal binding affinity. However, interaction analysis revealed 4 additional binding site residues, which led to an increased surface interaction area of 77 Å2 when compared to the Delta complex.
The Beta-ACE2 complex demonstrated the best docked structure, indicative of the increased interaction surface area of both proteins. The alteration from LYS to ASN at amino acid 417 resulted in the formation of a hydrogen bond with LYS31 of ACE2 (Supplementary). Compared to the other VOC docked structures, 4 additional hydrogen bonds and approximately double the hydrophobic interactions were noted. Interestingly, these common functional amino acids identified in the spike and ACE2 proteins have previously been reported to upregulate spike-ACE2 binding35. It is evident that Beta variant mutations induce stabilizing intermolecular forces, thus increasing the affinity of ACE2 to the altered RBD and potentially increasing infectivity.
An additional integral viral entry-point complex was assessed to further rationalize the increased dissemination of the viral mutants. Furin was docked to the “PRRA” S1/S2 boundary site of the spike protein. Docking results indicated the most favorable binding affinity between furin and the Delta variant spike protein, characterized by a P681R alteration at the S1/S2 boundary site. The Beta variant and WT proteins demonstrated identical docking scores, while the Alpha-Furin complex displayed the least optimal binding affinity. A similar trend was noted in the interaction analysis, with the Beta-Furin complex resulting in the lowest number of intermolecular interactions. The Delta-Furin complex constituted 41 interacting residues, with an interaction surface area of 2007 Å2. Of particular interest to the infectivity rate observed with the Delta variant is forming the triple ARG cleavage site. The positively charged ARG mutation at residue 681 resulted in a salt bridge with the negatively charged ASP191 of furin. This attractive force and the increased overall positive charge would increase the affinity of furin to the spike protein. This results in a unique Delta variant, as this additional salt bridge is reliant on the mutation. A characteristic feature of the PRO amino acid is the lack of a hydrogen located on the alpha-amino group. This hinders the amino acid from protonating secondary structures of the protein, thus resulting in the formation of protein loops or turns25. The HIS amino acid is positively charged and is usually characterized by its ability to allow for protein folding, stability and function36. As a result of the above information, it is not surprising that the alpha-furin complex contained the least optimal binding landscape. The PRO-HIS alteration at amino acid 681 could lead to a more stable protein structure at the S1/S2 binding site, resulting in “energy-heavy” furin cleavage.
For this study, only molecular docking was assessed to confer a general understanding of the structural mechanics. To gain a more comprehensive and reliable dataset, molecular dynamic simulations are required. This will evaluate the reactivity of all the mutations in a VOC by allowing for flexibility within the complex. This is especially important when elucidating the consequences of the Alpha variant H69/V70 deletion on structural loop dynamics and spike protein stability.
It is evident from the binding of ACE2 and furin to the spike VOCs that the Delta variant results in increased binding of both host proteins and thus suggests a possible molecular mechanism of the current excessive infectivity rates.
Materials and methods
Wastewater sample concentration and RNA extraction
Raw sewage samples were collected weekly from 24 WWTPs in Cape Town, South Africa, between 19 May-31 July 2021 (16 weeks). For RNA extraction, 100 mL of grab sample was mixed and centrifuged at 2500 xg for 20 min, whereafter 2.5 mL of the resultant pellet was used for total RNA extraction using a previously described protocol of Johnson et al. (2021). Total RNA was subjected to spectrophotometry using the NanoDrop ND-8000 (Thermo Scientific, USA). Only samples with a A260/280 ratio between 1.8–2 and a A260/230 ratio between 1.8–2.1 was used for subsequent After isolation, the consequent RNA (70 µL) was aliquoted and stored at –80 °C until required for molecular analysis.
Detection of spiked inactivated SARS-CoV-2 in wastewater sample as a positive extraction control
Inactivated SARS-CoV-2 was cultured from a nasopharyngeal swab for both wild type (E6.2) and variant (501Y. V2) was previously isolated and fully characterized and was a kind gift from Prof Wolfgang Preiser (National Health Laboratory Services, University of Stellenbosch, Tygerberg, Cape Town)3. The viral titer of inactivated SARS-CoV-2 was calculated and found to be 1 × 106 g.c./µL. Wastewater matrix samples, which previously were negative for SARS-CoV-2, were spiked with and without the inactivated wild-type and mutant virus at a concentration of 2 × 104 g.c/mL. RNA was extracted and used as an extraction positive control in all subsequent real-time quantitative polymerase chain reactions (RT-qPCR) reactions.
Real-time quantitative polymerase chain reaction (RT-qPCR) analysis
For quality assurance and control (QC/QA) the combined primer/probe mix (Integrated DNA Technologies, Coralville, IA, USA) (Table 2) as well as PCR positive controls were resuspended, aliquot and stored at − 20 ºC to limit freeze thaw cycles. For each PCR run, (i) a cultured inactivated nasopharyngeal swab SARS-CoV-2 spike extraction positive control, (ii) a extraction negative control (wastewater sample with absence of SARS-CoV-2), (iii) a PCR non-template control (NTC; nuclease free water, Cat no. 11–04-02–01) (Integrated DNA Technologies, Coralville, IA, USA), as well as a (iv) Synthetic SARS-CoV-2 DNA plasmid at 200 g.c/µL (2019-nCoV_N Positive; Cat no. 10006625) (Integrated DNA Technologies, Coralville, IA, USA) was included in each PCR run. As per our in-house SOP, failure of any one of these controls invalidated the test.
For all PCR reactions, the Minimum Information for Publication of Quantitative Real-Time PCR experiments (MIQC) guidelines was followed to ensure reliability and data integrity37. For each run, the following RT-qPCR MIQC detail was adhere to; (i) the threshold was set at 0.02 which was in the linear phase of the amplification plot, (ii) PCR efficiency was considered acceptable if in the range of 90–100%, (iii) the slope was acceptable if between − 3.55 and − 3.30, (iv) all R2 values was acceptable if > 0.99, (v) a minimum of 5 points for the dilution series was used to generate the standard curve and (vi) all samples was run in duplicate and a minimum SD range of < 0.5 was required for both standards and samples. Next, the presence of SARS-CoV-2 was determined and quantified using the Centre for Disease Control and Prevention (CDC) approved 2019-nCoV CDC EUA Combined primer/probe kit (Cat no. 10006670) (Integrated DNA Technologies, Coralville, IA, USA) using N1 and N2 as a target (Table 2). For quantitative analysis, the standard curve method was employed using the 2019-nCoV_N Positive Control (Cat no. 10006625) (Integrated DNA Technologies, Coralville, IA, USA) to construct the serial diluted calibrator/ standard curve matric of 200 000- 20 g.c./µL , whilst the threshold was set in the linear phase at 0.02 to ensure that all samples amplified above the threshold with a correlation coeffient > 0.99 and a PCR efficiency between 90–100% (slope − 3.30 to − 3.55). SARS-CoV-2 viral RNA from both sewage and spiked control samples was reversed, transcribed and quantified using the Bio-Rad iTaq Universal Probes One-Step Kit (Cat no. 1725141, Bio-Rad Laboratories, Hercules, CA) according to the manufacturer’s instructions. Extracted RNA was standardized at a concentration of 0.2 µg/µL, and RT-qPCR was performed as previously described by Johnson et al.38. Briefly, 1 µL of 0.2 µg/mL RNA was added to 2.5 µL iTaq Universal Probe One Step mix (Bio-Rad, Hercules, CA), in a final volume of 10 µL containing 0.5 µL of the IDT 2019-nCoV CDC approved nucleocapsid (N) primer/probe set (Cat no. RV202001/2, Integrated DNA Technologies, Coralville, IA, USA) and 3.25 µL nuclease free water (Cat no. 11-04-02-01, Integrated DNA Technologies, Coralville, IA, USA). PCR was conducted on an Applied Biosystems™ QuantStudio™ 7 Flex Real-Time PCR System (Thermo Fisher Scientific, Waltham, USA) under the following cycling conditions: Reverse transcription and RT-qPCR reaction was done at 50° for 10 min; followed by a 95° for 3 min and amplification was done at 95° for 15 s and 60° for 1 min (40 cycles) (Table 2). All reactions were performed in duplicate, and a plasmid SARS-CoV-2 positive control at a viral titer of 200 g.c./µL was included as a RT-qPCR positive control.
All Subsequently, samples positive for SARS-CoV-2 were used to detect S-protein signature mutations for the Alpha (N501Y, DelH69V70 and P681H)39, Beta (N501Y, E484K, and K417N)3 and Delta (L452R, and P681R)40 variants using the TaqMan SARS-CoV-2 Mutation Panel as per manufacturer’s instructions (Thermo Fisher Scientific, Waltham, USA) (Table 2).
Calibration curves and limit of detection determination
Given the complexity of the wastewater matrix, the detection limit was used to determine the lowest viral load the assay will detect in g.c./µL. Briefly, a tenfold serial dilution from 200 000–1 g.c./µL of RNA was run in duplicate using the 2019-nCoV_N plasmid control as the calibrator as well as the spike wastewater sample to confirm the limit of detection (LOD) using the N-gene. Linear regression was performed between the log copy number and the Ct values from the RT-qPCR results on all calibration curves that resulted in a good fit and allowed for the determination of the detection limit.
Genotyping analysis to detect mutations within Variant of Concern (VOCs) in wastewater
To perform variant detection, allele-specific RT-qPCR was performed in duplicate using the TaqPath™ RT-qPCR assay (Table 2, Thermo Fisher Scientific, Waltham, USA) according to the manufacturer’s instructions. Briefly, a SNP genotyping reaction was set up in duplicate with the addition of 4 µL of RNA with a viral titer > 1500 g.c./mL, 2.5 µL of TaqPath one-step RT-qPCR master mix and 0.25 µL of TaqMan™ SARS-CoV-2 Mutation Panel (Table 2) and this reaction was made up with nuclease free water to a final volume of 10 µL. The following variants of interest were tested for each assay: Alpha, Beta, and Delta with mutations N501Y (Cat no. A51812), E484K (Cat no. A51813), K417N (Cat no. A51814), DelH69V70 (Cat no.: A51808), P681H (Cat no. A51816), L452R (Cat no. A51819), and P681R (Cat no. A51822) (Table 2).
Next, the Genotyping PCR was conducted as specified in Table 2. Briefly, PCR cycling was performed on an Applied Biosystems™ QuantStudio™ 7 Flex Real-Time PCR System instrument (Thermo Fisher Scientific, Waltham, USA) using the following conditions: reverse transcription was performed at 60 °C for 30 s followed by 50 °C for 10 min, Taq polymerase activation at 95 °C for 2 min, followed by 45 cycles of denaturation at 95 °C for 3 s, and annealing of 30 s at 60 °C with a final Postread for 30 s at 60 °C. All RT-qPCR experiments included a NTC, a wild-type AcroMetrix Coronavirus 2019 (COVID-19) RNA control (RUO) (Cat no. 954519, (Thermo Fisher Scientific, Waltham, USA) supplied with the assay, and the extracted nucleic acids from wild-type and variant nasopharyngeal swabs positive for SARS-CoV-2 PCR.
Whole-genome sequencing and genome assembly
Library preparation and sequencing
SARS-CoV-2 RNA libraries were produced using the ATOPlex platform, which is a multiplexed amplicon-based sequencing workflow that is tailor-made for detecting SARS-CoV-2 fragments (~ 500 bp). Following reverse transcription of 10 µL RNA, a two-step PCR with 13 and 27 cycles for target enrichment and sequence adapter with dual-barcode primer (Cat No: 1000021626) addition, respectively, was carried out in the first and second PCRs. The amplified PCR products were purified by magnetic bead-based techniques followed by quantity and quality checks using the Qubit dsDNA high sensitivity assay kit (Life Technologies, USA) and gel-based electrophoreses, respectively. Libraries were equally pooled, and single strand circularized DNA (ssDNA) was synthesized using the MGIEasy Dual Barcode Circularization kit (Cat No:1000020570). DNA nanoballs (DNBs) were constructed for both the ssDNA sample library and a dual barcoded balance library (Cat No: 1000022270) in parallel, which was then mixed 3:1 prior to sequencing on the DNBSEQ-G50RS instrument at SAMRC’s Genomics Centre. Data analysis was conducted using a sweet of bioinformatics tools (https://github.com/MGI-tech-bioinformatics/SARS-CoV-2_Multi-PCR_v1.0).
In silico characterization of the binding landscape of mutations found within VOCs of the SARS-CoV-2 spike protein
Most available SARS-CoV-2 spike glycoprotein crystal structures either lack the resolved structure of the S1/S2 boundary site or demonstrate an already cleaved spike protein. To assess the binding landscape of the spike protein, all fully formed quaternary structures of the protein must be available. To accommodate this requirement, the S1/S2 boundary site and any other missing residues were homology modeled using the SwissModel webserver41. The monomeric model was established from template structures including PDB codes: 7mkm, 6wpt and 7a94. Once modeled, all mutations within the VOCs were computed using UCSF Chimera visualization software and structurally minimized using the AMBER20 molecular dynamics package42. Host proteins furin and ACE2 were retrieved from the Protein Data Bank using the PDB codes 4z2a and 7a94, respectively. The proteins were then prepared on UCSF Chimera, in which all nonstandard residues were deleted, hydrogen atoms were added and proteins charged. Patchdock43 and Firedock44 online webservers were utilized to assess docking at the spike protein. These tools utilize rigid-body docking algorithms to determine the geometric fit within the binding region of the target molecule. This is assessed according to the surface shape of the target molecule. To identify the best docking pose of the VOC complexes, the protein structures were uploaded to the server, and the identified binding regions were defined. Once scored, the best docked complexes were subjected to the firedock webserver to refine and filter the best structures. The best 10 structures were assessed individually on UCSF Chimera software to disregard any complexes that yielded atomistic clashes or steric hindrance. The final structures were then ranked based on docking scores, and the best complex was chosen for further analysis. The identified complexes were then subjected to interaction analysis using the EMBL-EBI PDBsum online webserver26. This server provides critical information on the 3D structure of a complex, including the secondary structure, molecules within the complex and schematic diagrams of the interactions within the complex. Intermolecular interactions identified between amino acids in the complex, including salt bridges, hydrogen bonds and other noncontact interactions, are also described. The information provided in PDBsum was subsequently utilized to elucidate the potential molecular mechanisms of increased VOC infectivity.
Statistical analysis
Descriptive statistics for continuous variable days were calculated with the mean, standard deviation, median, and interquartile range.
Spatial data
Suburb shapefiles were obtained from the City of Cape Town open data portal. Coordinates for each WWTP were collected using a handheld GPS and verified using Google Earth. All maps were produced using ArcGIS 10.6.1. The WWTP catchment areas (comprising 753 suburbs) were joined to the corresponding WWTPs.
Conclusion
It is imperative in the battle against COVID-19 to identify hotspots and monitor circulating strain in real time, allowing mitigation strategies to curb further spread of a highly transmissible strain. PCR genotyping provides real-time tracking and detection of circulating variants. This study acknowledges that RT-qPCR genotyping has various limitations compared to the gold standard NGS sequencing that can detect both known and unknown variants. However, PCR genotyping can be routinely performed; it allows for real-time monitoring of VOCs, the technology is readily available in most laboratories, and more importantly, it can provide information on the circulating variant at the population level. Therefore, early detection through WBE can give public health officials an additional and much needed metric to rapidly triangulate the prevalence of circulating variants within a community, making this type of surveillance essential for current and future outbreaks.
References
Mishra, S. et al. Changing composition of SARS-CoV-2 lineages and rise of Delta variant in England. EClinicalMedicine 39, 101064 (2021).
Grint, D. J. et al. Case fatality risk of the SARS-CoV-2 variant of concern B. 1.1. 7 in England, 16 November to 5 February. Eurosurveillance 26(11), 2100256 (2021).
Tegally, H. et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 592(7854), 438–443 (2021).
Faria, N. R. et al. Genomics and epidemiology of the P. 1 SARS-CoV-2 lineage in Manaus, Brazil. Science 372(6544), 815–821 (2021).
Singh, J. et al. Structure-function analyses of new SARS-CoV-2 variants B. 1.1. 7, B. 1.351 and B. 1.1. 28.1: clinical, diagnostic, therapeutic and public health implications. Viruses 13(3), 439 (2021).
Abdool, K. S. S. & De Oliveira, T. New SARS-CoV-2 variants—clinical, public health, and vaccine implications. New Engl. J. Med. 384(19), 1866–1868 (2021).
Deng, X. et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. MedRxiv 3, 1079 (2021).
Zhang, W. et al. Emergence of a novel SARS-CoV-2 variant in Southern California. JAMA 325(13), 1324–1326 (2021).
Parums, D. V. Revised World Health Organization (WHO) terminology for variants of concern and variants of interest of SARS-CoV-2. Med. Sci. Monit. 27, e933622 (2021).
Scheepers, C. et al. The continuous evolution of SARS-CoV-2 in South Africa: a new lineage with rapid accumulation of mutations of concern and global detection. medRxiv 2, 1071 (2021).
La Rosa, G. et al. Rapid screening for SARS-CoV-2 variants of concern in clinical and environmental samples using nested RT-PCR assays targeting key mutations of the spike protein. Water Res. 197, 117104 (2021).
Huang, Y. et al. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 41(9), 1141–1149 (2020).
Alai, S. et al. Pan-India novel coronavirus SARS-CoV-2 genomics and global diversity analysis in spike protein. Heliyon 7(3), e06564 (2021).
Xia, S. et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 17(7), 765–767 (2020).
Zhang, J. et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 372(6541), 525–530 (2021).
de Souza, A. S. et al. Molecular dynamics analysis of fast-spreading severe acute respiratory syndrome coronavirus 2 variants and their effects in the interaction with human angiotensin-converting enzyme 2. bioRxiv 2, 63 (2021).
Shahab, M. S., Imam, S. S. & Jahangir, M. A. A review on the contemporary status of mutating coronavirus and comparative literature study of current COVID-19 vaccines. Int. J. Pharm. Pharmacol. 5(1), 1–19 (2021).
Andreano, E. & Rappuoli, R. SARS-CoV-2 escaped natural immunity, raising questions about vaccines and therapies. Nat. Med. 27(5), 759–761 (2021).
Guo, S., Liu, K. & Zheng, J. The genetic variant of SARS-CoV-2: would it matter for controlling the devastating pandemic?. Int. J. Biol. Sci. 17(6), 1476 (2021).
Bar-On, Y. M. et al. The importance of time post-vaccination in determining the decrease in vaccine efficacy against SARS-CoV-2 variants of concern. medRxiv 17, 1361 (2021).
Ahmed, W. et al. First confirmed detection of SARS-CoV-2 in untreated wastewater in Australia: a proof of concept for the wastewater surveillance of COVID-19 in the community. Sci. Total Environ. 728, 138764 (2020).
Sherchan, S. P. et al. First detection of SARS-CoV-2 RNA in wastewater in North America: a study in Louisiana, USA. Sci. Total Environ. 743, 140621 (2020).
Kitajima, M. et al. SARS-CoV-2 in wastewater: state of the knowledge and research needs. Sci. Total Environ. 739, 139076 (2020).
Wurtzer, S. et al. Monitoring the propagation of SARS CoV2 variants by tracking identified mutation in wastewater using specific RT-qPCR. medRxiv 1, 716 (2021).
Morgan, A. A. & Rubenstein, E. Proline: the distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PloS One 8(1), e53785 (2013).
Laskowski, R. A. PDBsum new things. Nucleic Acids Res. 37(1(suppl_1)), D355–D359 (2009).
Jahn, K. et al. Detection of SARS-CoV-2 variants in Switzerland by genomic analysis of wastewater samples. medRxiv 9, 1055 (2021).
Pechlivanis, N. et al. Detecting SARS-CoV-2 lineages and mutational load in municipal wastewater; a use-case in the metropolitan area of Thessaloniki, Greece. medRxiv 7, 3019 (2021).
Randazzo, W. et al. Metropolitan wastewater analysis for COVID-19 epidemiological surveillance. Int. J. Hygiene Environ. Health 230, 113621 (2020).
Agrawal, S., Orschler, L. & Lackner, S. Long-term monitoring of SARS-CoV-2 RNA in wastewater of the Frankfurt metropolitan area in Southern Germany. Sci. Rep. 11(1), 1–7 (2021).
Vaughan, A. Delta to Dominate World (Elsevier, 2021).
Campbell, F. et al. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021. Eurosurveillance 26(24), 2100509 (2021).
Stern, A. et al. The unique evolutionary dynamics of the SARS-CoV-2 Delta variant. medRxiv 2, 1900 (2021).
Jassat, W. et al. A call to action: temporal trends of COVID-19 deaths in the South African Muslim community. South African Med. J. 5, 1009 (2021).
Cheng, M. H. et al. Impact of South African 501. V2 variant on SARS-CoV-2 spike infectivity and neutralization: a structure-based computational assessment. bioRxiv 9, 1008 (2021).
Liao, S.-M. et al. The multiple roles of histidine in protein interactions. Chem. Cent. J. 7(1), 1–12 (2013).
Bustin, S. A. et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments (Oxford University Press, 2009).
Johnson, R. et al. Qualitative and quantitative detection of SARS-CoV-2 RNA in untreated wastewater in Western Cape Province, South Africa. South Afr. Med. J. 111(3), 198–202 (2021).
Meng, B. et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B. 1.1. 7. Cell Rep. 35(13), 109292 (2021).
Umair, M. et al. Detection and whole-genome sequencing of SARS-CoV-2 B. 1.617. 2 and B. 1.351 variants of concern from Pakistan during the COVID-19 third wave. medRxiv 2, 743 (2021).
Bienert, S. et al. The SWISS-MODEL repository—new features and functionality. Nucleic Acids Res. 45(D1), D313–D319 (2017).
Salomon-Ferrer, R., Case, D. A. & Walker, R. C. An overview of the Amber biomolecular simulation package. Comput. Mol. Sci. 3(2), 198–210 (2013).
Schneidman-Duhovny, D. et al. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33((suppl_2)), W363–W367 (2005).
Mashiach, E. et al. FireDock: a web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 36((suppl_2)), W229–W232 (2008).
Acknowledgements
This research was funded in part by the Solidarity Fund, The Michael & Susan Dell Foundation (Grant Number: 96829) and the South Africa Medical Research Council (Grant numbers: 40118 and 50598).
Funding
This research was funded in part by The Michael & Susan Dell Foundation (Grant Number: 96829) and the South Africa Medical Research Council (Grant Number: 40118).
Author information
Authors and Affiliations
Contributions
R.J., J.S., P.R., N.M., C.K., A.V. and B.G. performed experiments, analyzed the data and wrote the manuscript. N.A. and C.W. performed all statistical analyses. R.J., S.S.-N., J.L., S.N., T.R., N.M., M.M., G.G., A.M., R.S., W.P. and C.M. applied for funding, provided scientific input and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Johnson, R., Sharma, J.R., Ramharack, P. et al. Tracking the circulating SARS-CoV-2 variant of concern in South Africa using wastewater-based epidemiology. Sci Rep 12, 1182 (2022). https://doi.org/10.1038/s41598-022-05110-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-05110-4
This article is cited by
-
Vibrio cholerae and Salmonella Typhi culture-based wastewater or non-sewered sanitation surveillance in a resource-limited region
Journal of Exposure Science & Environmental Epidemiology (2024)
-
Monitoring SARS-CoV-2 variants in wastewater of Dhaka City, Bangladesh: approach to complement public health surveillance systems
Human Genomics (2023)
-
Long-term monitoring of COVID-19 prevalence in raw and treated wastewater in Salvador, the largest capital of the Brazilian Northeast
Scientific Reports (2023)
-
Wastewater early warning system for SARS-CoV-2 outbreaks and variants in a Coruña, Spain
Environmental Science and Pollution Research (2023)
-
L'analyse des eaux usées, un moyen bon marché et efficace de suivre les tendances des mutations du COVID-19
Nature Africa (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
