
Abstract
Recent evidence suggests that the central nervous system (CNS) regulates plasma glucose levels, but the underlying mechanism is unclear. The present study investigated the role of dopaminergic function in the CNS in regulation of plasma glucose levels in mice. I.c.v. injection of neither the dopamine D1 receptor agonist SKF 38393 nor the antagonist SCH 23390 influenced plasma glucose levels. In contrast, i.c.v. injection of both the dopamine D2 receptor agonist quinpirole and the antagonist l-sulpiride increased plasma glucose levels. Hyperglycemia induced by quinpirole and l-sulpiride was absent in dopamine D2 receptor knockout mice. I.c.v. injection of quinpirole and l-sulpiride each increased mRNA levels of hepatic glucose-6-phosphatase and phosphoenolpyruvate carboxykinase, which are the key enzymes for hepatic gluconeogenesis. Systemic injection of the β2 adrenoceptor antagonist ICI 118,551 inhibited hyperglycemia induced by l-sulpiride, but not by quinpirole. In contrast, hyperglycemia induced by quinpirole, but not by l-sulpiride, was inhibited by hepatic vagotomy. These results suggest that stimulation of central dopamine D2 receptors increases plasma glucose level by increasing hepatic glucose production through parasympathetic nerves, whereas inhibition of central dopamine D2 receptors increases plasma glucose level by increasing hepatic glucose production through sympathetic nerves.
Similar content being viewed by others
Introduction
Previous reports have indicated that plasma glucose levels are regulated by the central nervous system (CNS). For example, antipsychotic drugs such as clozapine and olanzapine are known to increase body weight and disrupt glucose metabolism1,2. We have previously reported that olanzapine injected intracerebroventricularly increases plasma glucose levels3,4 and that this hyperglycemia may involve dopamine D2 receptors, α1-adrenoceptors and histamine H1 receptors5. These results indicate that central dopamine, noradrenaline and histamine neurons play important roles in regulation of plasma glucose levels. However, the mechanisms by which central neurons regulate plasma glucose levels are unclear.
Plasma glucose levels are regulated by the balance between glucose production and glucose utilization in the whole body. It is well known that the pancreas and liver are important in regulation of plasma glucose levels. The pancreas secretes insulin and glucagon: insulin decreases plasma glucose levels by increasing glucose uptake into organs and inhibiting hepatic glucose production, whereas glucagon increases plasma glucose levels by increasing hepatic glucose production. The liver increases plasma glucose levels by glycogenolysis and gluconeogenesis and decreases plasma glucose levels by glycogenesis and glycolysis6. In addition, the functions of the pancreas and the liver are known to be regulated by autonomic nerves. Stimulation of sympathetic nerves increases glucagon secretion and inhibits insulin secretion from the pancreas7,8, and increases gluconeogenesis in the liver through β2 adrenoceptors9,10. Thus, sympathetic nerves positively regulate plasma glucose levels. In contrast, stimulation of parasympathetic nerves increases insulin secretion from the pancreas8 and decreases gluconeogenesis and increases glycogenesis in the liver9,11. Thus, parasympathetic nerves play an inhibitory role in regulation of plasma glucose levels.
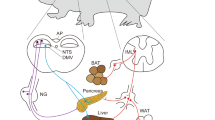
The hypothalamus is known as the center of energy homeostasis. It senses peripheral metabolic signals, including hormones and nutrients, and regulates glucose metabolism12,13. Moreover, it is reported that the hypothalamus regulates the activity of autonomic nerves. For example, sympathetic and parasympathetic nerves projecting to the pancreas are reported to be regulated by the paraventricular nucleus of hypothalamus (PVN) and lateral hypothalamus (LH), respectively14. In addition, previous reports have shown that the ventromedial hypothalamus (VMH) regulates the activity of hepatic sympathetic nerves, the LH regulates the activity of hepatic vagal nerves, and the PVN regulates both sympathetic and parasympathetic nerves9,15. Thus, the hypothalamus is thought to regulate glucose metabolism through both sympathetic and parasympathetic nerves.
We have recently reported that dopamine D1 and D2 receptors in the hypothalamus regulate feeding behavior in mice16, suggesting that central dopamine D1 and D2 receptors also regulate plasma glucose levels. However, the roles of dopamine receptor subtypes in regulation of plasma glucose levels are not consistent. For instance, it has been reported that a low dose of dopamine decreases plasma glucose levels but a high dose of dopamine increases plasma glucose levels17. Moreover, some reports have shown that stimulation of dopamine D2 receptors increases plasma glucose levels18,19, whereas other reports have shown that stimulation of dopamine D2 receptors decreases plasma glucose levels20,21. One possible reason for these discrepancies is that dopamine receptors regulate many functions related to regulation of plasma glucose levels. In fact, it is reported that dopamine D2/D3 receptors in the pancreas regulate insulin secretion22,23. In addition, dopamine D2 receptors are known to regulate the secretion of hormones such as glucocorticoids and prolactin, which regulate plasma glucose levels24,25. Thus, it is likely that dopamine receptors in both the CNS and the peripheral nervous system regulate plasma glucose levels through different mechanisms.
The present study focuses on dopaminergic functions in the CNS and investigated the role of central dopamine D1 and D2 receptors in regulation of plasma glucose levels. In addition, we investigated whether autonomic nerves and hepatic glucose metabolism are involved in regulation of plasma glucose levels by central dopaminergic function.
Results
Effect of dopamine receptor agonists and antagonists on plasma glucose levels
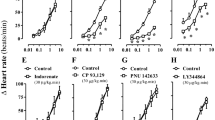
We first examined whether central dopamine D1 receptors regulate plasma glucose levels in mice using i.c.v. injections of a selective dopamine D1 receptor agonist and a selective dopamine D1 receptor antagonist. Injection of neither the dopamine D1 receptor agonist SKF 38393 (3 and 10 μg, i.c.v.; Fig. 1a) nor antagonist SCH 23390 (1 and 3 μg, i.c.v.; Fig. 1b) influenced plasma glucose levels. These results indicate that central dopamine D1 receptors are not involved in regulation of plasma glucose levels.

Effects of dopamine D1 and D2 receptor agonists and antagonists on plasma glucose levels in mice. (a) Effect of SKF 38393 (3 and 10 μg, i.c.v.) on plasma glucose levels. (b) Effect of SCH 23390 (1 and 3 μg, i.c.v.) on plasma glucose levels. (c) Effect of quinpirole (3 and 10 μg, i.c.v) on plasma glucose levels. (d) Effect of l-sulpiride (3 and 10 μg, i.c.v.) on plasma glucose levels. (e) Effect of l-sulpiride on hyperglycemia induced by quinpirole. l-Sulpiride (2 μg, i.c.v.) was co-administered with quinpirole (10 μg, i.c.v.). Each point represents the mean ± S.E.M. of 7–16 mice. **P < 0.01, ***P < 0.001 vs. vehicle group.#P < 0.05, ##P < 0.01 vs. quinpirole group.
We then examined whether central dopamine D2 receptors regulate plasma glucose levels using i.c.v. injections of a selective dopamine D2 receptor agonist and a selective dopamine D2 receptor antagonist. Injection of the dopamine D2 receptor agonist quinpirole (3 and 10 μg, i.c.v.) significantly increased plasma glucose levels (treatment: F(2,180) = 4.38, P = 0.020; treatment × time: F(10,180) = 6.61, P < 0.001; Fig. 1c). Injection of the dopamine D2 receptor antagonist l-sulpiride (3 and 10 μg, i.c.v.) also significantly increased plasma glucose levels (treatment: F(2,220) = 3.27, P = 0.047; treatment × time: F(10,220) = 3.82, P < 0.001; Fig. 1d). The hyperglycemia induced by quinpirole (10 μg, i.c.v.) was inhibited by a low dose of l-sulpiride (2 μg, i.c.v.) that alone did not significantly affect plasma glucose levels (treatment: F(3,220) = 16.07, P < 0.001, treatment × time: F(15,220) = 3.02, P < 0.001; Fig. 1e). These results indicate that both the D2 receptor agonist and the D2 receptor antagonist increase plasma glucose levels.
Effect of dopamine D2 receptor agonist and antagonist on plasma glucose levels in dopamine D2 receptor knockout mice
To confirm the involvement of dopamine D2 receptors in these processes, we repeated the studies using i.c.v. injections of the selective dopamine D2 receptor agonist and antagonist in mice with dopamine D2 receptor knockout. Hyperglycemia induced by quinpirole (10 μg, i.c.v.) and by l-sulpiride (10 μg, i.c.v.) was not observed in dopamine D2 receptor knockout mice (Fig. 2; quinpirole, treatment: F(3,115) = 24.64, P < 0.001, treatment × time: F(15,115) = 4.91, P < 0.001; l-sulpiride, treatment: F(3,125) = 6.29, P = 0.003, treatment × time: F(15,125) = 3.00, P < 0.001). These results confirm that the above actions of both the D2 receptor agonist and the D2 receptor antagonist to increase plasma glucose levels are mediated via their respective actions at dopamine D2 receptors.

Effects of the dopamine D2 receptor agonist and antagonist on plasma glucose levels in dopamine D2 receptor knockout mice (KO). Effect of quinpirole (10 μg, i.c.v.; a) and l-sulpiride (10 μg, i.c.v.; b) on plasma glucose levels in wild-type mice (WT) and KO mice. Each point represents the mean ± S.E.M. of 6–8 mice. *P < 0.05, **P < 0.01, ***P < 0.001 vs. WT-vehicle group. ##P < 0.01, ###P < 0.001 vs. WT-quinpirole or WT-l-sulpiride group.
Effect of glucocorticoid receptor antagonist on hyperglycemia induced by dopamine D2 receptor agonist and antagonist
Since dopamine D2 receptors are known to regulate glucocorticoid secretion, we investigated the role of glucocorticoids in these processes by repeating the studies using i.c.v. injections of the selective dopamine D2 receptor agonist and antagonist following pretreatment with a glucocorticoid receptor antagonist. The glucocorticoid receptor antagonist RU 486 (15 mg/kg, i.p.) did not influence hyperglycemia induced by either quinpirole (10 μg, i.c.v.; Fig. 3a) or l-sulpiride (10 μg, i.c.v.; Fig. 3b). These results indicate that stimulation or inhibition of dopamine D2 receptors do not increase plasma glucose levels through regulation of glucocorticoid secretion.

Effect of the glucocorticoid receptor antagonist on hyperglycemia induced by the dopamine D2 receptor agonist and antagonist. RU 486 (15 mg/kg, i.p.) was administered 30 min before injection of quinpirole (10 μg, i.c.v.; a) or l-sulpiride (10 μg, i.c.v.; b). Each point represents the mean ± S.E.M. of 7–9 mice. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle group. +P < 0.05, +++P < 0.001 vs. RU 486-vehicle group.
Changes in plasma glucagon and insulin levels after injection of dopamine D2 receptor agonist and antagonist
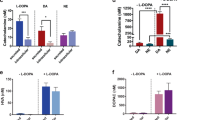
Since glucagon and insulin are known to regulate plasma glucose levels, we investigated their roles in these processes by measuring plasma glucagon and insulin levels following i.c.v. injections of the selective dopamine D2 receptor agonist and antagonist. Injection of neither quinpirole (10 μg, i.c.v.) nor l-sulpiride (10 μg, i.c.v.) significantly influenced plasma glucagon and insulin levels (Fig. 4a–d). These results indicate that hyperglycemia induced by stimulation or inhibition of dopamine D2 receptors does not involve glucagon and insulin secretion.

Effects of the dopamine D2 receptor agonist and antagonist on secretion of hormones and hepatic glucose production. (a–d) Effects of quinpirole (10 μg, i.c.v.; a, c) and l-sulpiride (10 μg, i.c.v.; b, d) on plasma glucagon (a, b) and insulin (c, d) levels. Blood samples were collected 10 min after drug injections. (e, f) Effect of quinpirole (10 μg, i.c.v.; e) and l-sulpiride (10 μg, i.c.v.; f) on hepatic glycogen levels. Liver samples were collected 30 min after drug injections. (g–j) Effect of quinpirole (10 μg, i.c.v.; g, i) and l-sulpiride (10 μg, i.c.v.; h, j) on hepatic mRNA levels of glucose-6-phosphatase (G6Pase; g, h) and phosphoenolpyruvate carboxykinase (PEPCK; i, j). Liver samples were collected 30 min after drug injections. Each point represents the mean ± S.E.M. of 6–14 mice. *P < 0.05, **P < 0.01 vs. vehicle group.
Changes in hepatic glucose production after injection of dopamine D2 receptor agonist and antagonist
Since hepatic glucose production increases plasma glucose levels, we next investigated whether stimulation or inhibition of central dopamine D2 receptors increased hepatic glucose production by glycogenolysis and gluconeogenesis. Hepatic glycogen levels were decreased by quinpirole (10 μg, i.c.v.; Fig. 4e) but not by l-sulpiride (10 μg, i.c.v.; Fig. 4f). These results indicate that stimulation of central dopamine D2 receptors increases glycogenolysis. In addition, mRNA levels of hepatic glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK), which are the key enzymes in gluconeogenesis, were increased by quinpirole (10 μg, i.c.v.; Fig. 4g,i) and l-sulpiride (10 μg, i.c.v.; Fig. 4h,j). Since G6Pase and PEPCK are rate-limiting enzymes in gluconeogenesis, these results indicate that both stimulation and inhibition of central dopamine D2 receptors increase gluconeogenesis. Taken together, these results indicate that stimulation or inhibition of central dopamine D2 receptors increases hyperglycemia through hepatic glucose production.
Effect of adrenergic receptor antagonist on hyperglycemia induced by dopamine D2 receptor agonist and antagonist
Since hepatic glucose production is regulated by sympathetic nerves through β2 adrenoceptors10, we investigated whether stimulation or inhibition of central dopamine D2 receptors increases plasma glucose levels through sympathetic nerves using a β2 adrenoceptor antagonist. The β2 adrenoceptor antagonist ICI 118,551 (2 mg/kg, i.p.) did not influence hyperglycemia induced by quinpirole (10 μg, i.c.v.; Fig. 5a) but significantly inhibited hyperglycemia induced by l-sulpiride (10 μg, i.c.v.; treatment: F(3,150) = 6.21, P = 0.002, treatment × time: F(15,150) = 6.80, P < 0.001; Fig. 5b). These results indicate that inhibition of central dopamine D2 receptors increases plasma glucose levels through stimulation of sympathetic nerves.

Role of sympathetic and parasympathetic nerves in hyperglycemia induced by the dopamine D2 receptor agonist and antagonist. (a, b) Effect of the β2 adrenoceptor antagonist ICI 118,551 on hyperglycemia induced by quinpirole or l-sulpiride. ICI 118,551 (2 mg/kg, i.p.) was administered 30 min before injection of quinpirole (10 μg, i.c.v.; a) or l-sulpiride (10 μg, i.c.v.; b). Each point represents the mean ± S.E.M. of 8–15 mice. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle-vehicle group. +P < 0.05, +++P < 0.001 vs. ICI 118,551-vehicle group. ###P < 0.001 vs. vehicle-l-sulpiride group. (c, d) Effect of hepatic vagotomy on hyperglycemia induced by quinpirole (10 μg, i.c.v.; c) and l-sulpiride (10 μg, i.c.v.; d). Hepatic vagotomy was conducted at least 1 week before drug injections. Each point represents the mean ± S.E.M. of 7–12 mice. *P < 0.05, **P < 0.01, ***P < 0.001 vs. sham-vehicle group. +P < 0.05, ++P < 0.01, +++P < 0.001 vs. vagotomy-vehicle group. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. sham-quinpirole group.
Effects of hepatic vagotomy on hyperglycemia induced by dopamine D2 receptor agonist and antagonist
To investigate the involvement of parasympathetic nerves, we examined the effects of hepatic vagotomy on the ability of stimulation or inhibition of central dopamine D2 receptors to increase plasma glucose levels. Hepatic vagotomy significantly inhibited hyperglycemia induced by quinpirole (10 μg, i.c.v.; treatment: F(3,135) = 15.53, P < 0.001, treatment × time: F(15,135) = 8.34, P < 0.001; Fig. 5c). In contrast, hepatic vagotomy did not influence hyperglycemia induced by l-sulpiride (10 μg, i.c.v.; Fig. 5d). Since stimulation of parasympathetic nerves decreases plasma glucose levels, these results indicate that stimulation of central dopamine D2 receptors increases plasma glucose levels through inhibition of parasympathetic nerves.
Discussion
The present study investigated the role of central dopaminergic function in regulation of plasma glucose levels.
Our results showed that i.c.v. injection of quinpirole dose-dependently increased plasma glucose levels. Surprisingly, i.c.v. injection of l-sulpiride also increased plasma glucose levels. Hyperglycemia induced by quinpirole was antagonized by a low dose of l-sulpiride. In addition, hyperglycemia induced both by quinpirole and by l-sulpiride was absent in dopamine D2 receptor knockout mice. Thus, these results indicate that both stimulation and inhibition of dopamine D2 receptors in the CNS increase plasma glucose levels. Some reports have shown that dopamine D2 receptors positively regulate plasma glucose levels, but other reports have shown that dopamine D2 receptors negatively regulate plasma glucose levels. For example, a previous report has shown that systemic injection of a low dose and a high dose of dopamine respectively increases and decreases plasma glucose levels17. In addition, there are reports that stimulation of dopamine D2 receptors increases plasma glucose levels18,19, whereas other reports suggest that stimulation of dopamine D2 receptors inhibits hyperglycemia induced by food intake or in diabetes20,21. Taken together, it is likely that dopamine D2 receptors regulate plasma glucose levels through more than one mechanism.
It is reported that systemic injection of dopamine D2 receptor agonists such as quinpirole and bromocriptine increase plasma glucose levels18,19. Moreover, hyperglycemia induced by systemic injection of quinpirole was inhibited by the centrally acting dopamine D2 receptor antagonist haloperidol, but not by the peripheral acting dopamine D2 receptor antagonist domperidone18. In addition, our results show that i.c.v. injection of quinpirole increases plasma glucose levels. Thus, dopamine D2 receptors in the CNS play an important role in regulation of plasma glucose levels, while dopamine D2 receptors in the pancreas are reported to regulate insulin secretion22,23.
Previous reports have pointed out that dopamine D2 receptor agonists such as bromocriptine can have a therapeutic effect in type 2 diabetes mellitus by improving glycemic control and glucose tolerance26. In preclinical research, there are contradictory results regarding the effect of bromocriptine. Some reports have shown that bromocriptine improves insulin-resistance and glucose intolerance20, whereas other reports have shown that bromocriptine induces hyperglycemia19. Since chronic treatment with dopamine D2 receptor agonists improves insulin-resistance and glucose intolerance20,21,26, it is likely that acute treatment with dopamine D2 receptor agonists increases plasma glucose levels, while chronic treatment with dopamine D2 receptor agonists decreases plasma glucose levels.
It is well known that dopamine D2 receptors inhibit hormone secretion. Among them, corticosterone is known to increase plasma glucose levels. Thus, it is possible that inhibition of dopamine D2 receptors increases plasma glucose levels through corticosterone secretion. However, our results showed that the glucocorticoid receptor antagonist RU 486 at an effective dose27 did not influence hyperglycemia induced by either quinpirole or l-sulpiride, while the β2 adrenoceptor antagonist ICI 118,551 completely blocked hyperglycemia induced by l-sulpiride. Thus, it appears that that corticosterone is not involved in regulation of glucose homeostasis by central dopamine D2 receptors.
Plasma glucose levels are regulated by hepatic glucose production6. Since glycogenolysis and gluconeogenesis increase plasma glucose levels, it is possible that central dopamine D2 receptors regulate plasma glucose levels through glycogenolysis and gluconeogenesis in the liver. The i.c.v. injection of quinpirole, but not l-sulpiride, reduced glycogen levels in the liver, suggesting that stimulation of central dopamine D2 receptors increases glycogenolysis. In addition, injection both of quinpirole and of l-sulpiride increased mRNA levels of G6Pase and PEPCK, which are key enzymes in gluconeogenesis. Since mRNA levels of G6Pase and PEPCK are thought to correlate with gluconeogenesis10, it is suggested that both stimulation and inhibition of central dopamine D2 receptors increase gluconeogenesis. Taken together, these results suggest that stimulation and inhibition of central dopamine D2 receptors increase plasma glucose levels by increasing hepatic glucose production.
It has been reported that hepatic glucose production is increased by stimulation of sympathetic nerves9,10. Thus, it is possible that stimulation and inhibition of dopamine D2 receptors increase plasma glucose levels through sympathetic nerves. Since stimulation of sympathetic nerves increases hepatic glucose production through β2 adrenoceptors10, we examined whether the β2 adrenoceptor antagonist ICI 118,551 influences hyperglycemia induced by quinpirole and l-sulpiride. The results showed that ICI 118,551 inhibited the hyperglycemia induced by l-sulpiride but did not influence the hyperglycemia induced by quinpirole. Since β adrenoceptors regulate glucagon and insulin secretion28,29, it is possible that ICI 118,551 affects glucagon and/or insulin secretion. However, the present study showed that injection of ICI 118,551 alone had no effect on plasma glucose levels, suggesting that ICI 118,551 did not affect glucagon and insulin secretion. Thus, it is unlikely that ICI 118,551 inhibits hyperglycemia induced by l-sulpiride through glucagon and insulin secretion. These findings indicate that inhibition of central dopamine D2 receptors increases plasma glucose levels through stimulation of sympathetic nerves.
In contrast, since stimulation of parasympathetic nerves has been reported to decrease hepatic glucose production9,11, it is possible that stimulation and inhibition of central dopamine D2 receptors increase plasma glucose level by inhibiting parasympathetic nerves. Therefore, we performed vagotomy to the liver. The results showed that hyperglycemia induced by quinpirole, but not l-sulpiride, was inhibited by vagotomy. These findings indicate that stimulation of central D2 receptors increases plasma glucose levels by inhibiting parasympathetic nerves.
Previous reports have suggested that activities of autonomic nerves are regulated by the hypothalamus. For instance, the VMH and the PVN regulate sympathetic nerves, whereas the LH and the PVN regulate parasympathetic nerves14,30. More specifically, stimulation of the LH increases activity of parasympathetic nerves to the liver, stimulation of the VMH facilitates sympathetic nerves to the liver, and the PVN integrates information from other areas of the hypothalamus and regulates both nerves9,15. Since dopamine D2 receptors are reported to exist in the hypothalamus31, it is possible that stimulation of dopamine D2 receptors in the LH and/or PVN increases plasma glucose levels through inhibition of parasympathetic nerves, whereas inhibition of dopamine D2 receptors in the VMH and/or PVN increases plasma glucose levels through sympathetic nerves. Future studies should further investigate both these processes and the signaling mechanisms associated with the receptor-mediated effects reported.
In conclusion, the present studies indicate that dopamine D2, but not D1, receptors in the CNS regulate plasma glucose levels. In addition, our results suggest that there are at least two mechanisms related to regulation of plasma glucose levels by central dopamine D2 receptors: one through sympathetic nerves and the other through parasympathetic nerves. It remains possible that dopamine D2 receptors in different nuclei of the hypothalamus also regulate these processes.
Materials and methods
Animals
Male ICR mice (age 6 weeks; body weight 27–39 g; Tokyo Animal Laboratories, Tokyo, Japan), male C57BL/6 N mice (wild type; age 11–23 weeks; body weight 23–34 g) and dopamine D2 receptor knockout mice (age 11–23 weeks; body weight 18–28 g) were used (see below). The mice were kept under a 12-h light/dark cycle (light on at 08:00) at a constant room temperature (24 ± 1 °C) and with free access to food and water. This study was carried out in accordance with the guidelines for the care and use of laboratory animals of Hoshi University and Niigata University, which are accredited by the Ministry of Education, Culture, Sports, Science, and Technology of Japan. The protocols were approved by the Animal Experimentation Committee of Hoshi University. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Generation of knockout mice
To produce dopamine D2 receptor (Drd2) protein-deficient mice, we generated knock-in mice by inserting a gene encoding the iCre protein into the initiation methionine of the gene. A total of 10.85 kbp of the gene region, containing 5.15 kbp upstream and 5.70 kbp downstream of the initiation codon in exon 2 of the Drd2 gene, was subcloned into pDT-MC#332 taken from the BAC clone RP-24-71N14. A targeting vector was constructed by inserting the iCre-IRES-Flag-EGFP-pgk-gb2neo-polyA fragment33 into the translational initiation site of the Drd2 gene in frame (Fig. 6a). The vector linearized was transfected into RENKA ES cells derived from the C57BL/6 N mouse strain34 and recombinant clones were identified by Southern blot hybridization analysis (Fig. 6b). The recombinant ES cells were injected into eight-cell stage embryos of the CD-1 mouse strain to obtain chimeric mice. The chimeric mice were mated with C57BL/6 N mice to establish the Drd2-iCre mice (Drd2 knockout mice) on a pure C57BL/6 N genetic background. PCR-based genotyping was performed on genomic DNA extracted from mouse ear using KOD FX neo (Toyobo life science, Osaka, Japan). The following primers were used: Drd2 (forward: 5′-CTC AGC TCT GCT AGC TCT TG-3′; wild-type reverse: 5′-GCA GCA TGG CAT AGT AGT TG-3′) and Cre (5′-CAG GAA GGC CAG GTT CCT G-3′). Expected PCR products were 257 bp for the wild-type allele and 650 bp for the knockout allele (Fig. 6c). Since insertion of the gene encoding iCre into Drd2 was expected to inhibit Drd2 expression, Drd2-iCre homozygous mice can be regarded as Drd2 knockout. To confirm loss of Drd2 expression we conducted RT-PCR, which showed that Drd2 mRNA was negligible in knockout mice (Fig. 6d). Drd2 knockout mice showed the normal Mendelian ratio of offspring (1:2:1 based on 82 animals) after breeding of Drd2-iCre+/- mice (Fig. 6e).

Generation of the Drd2-Cre mouse line. (a) Schema of Drd2 cDNA, genomic DNA, targeting vector and the targeted genome. Numbered blue boxes in cDNA are transmembrane domains, and black boxes indicate coding exons. The vector was constructed to insert an improved Cre recombinase gene (iCre), followed by an internal ribosome entry site (IRES) and a FLAG-tagged EGFP, into the translational initiation site of the Drd2 gene. Red arrows show primer positions for PCR genotyping. White boxes indicate probes for Southern blot analysis. Two frt sequences (semicircles) are attached to remove the neomycin resistance gene (Neo). B, BamH1; E, EcoT22I; and K, KpnI. (b) Southern blot analysis for genomic DNAs from wild-type (+ / +) and targeted (+ /iCre) ES cells. The first panel from left shows EcoT22I-digested genomic DNA hybridized with 3′ probe, and the other three panels show KpnI-digested DNA hybridized with Cre, BamHI digested DNA, Neo and 5′ probes, respectively. Full-length images are available in Supplementary Fig. 1. (c) Genomic PCR genotyping of Drd2+/iCre using Drd2 forward, reverse and Cre primers. Full-length images are available in Supplementary Fig. 2. (d) Expression of Drd2 mRNA in the hypothalamus of wild-type (WT) mice and Drd2 knockout (KO) mice. Each point represents the mean ± S.E.M. of 6–9 mice. ***P < 0.001 vs. WT group. (e) Observed genotypes of offspring of heterozygous intermatings, in comparison with expected genotypes calculated according to the total number of mice born (82 mice) and the expected Mendelian 1:2:1 ratio.
Drugs
The drugs used were: the dopamine D1 receptor agonist SKF 38393 hydrochloride (Sigma-Aldrich, St. Louis, MO, USA)16; the dopamine D1 receptor antagonist R( +)-SCH 23390 hydrochloride (Sigma-Aldrich)16; the dopamine D2 receptor agonist (-)-quinpirole hydrochloride (Sigma-Aldrich)16; the dopamine D2 receptor antagonist l-sulpiride (Sigma-Aldrich)5,16; the glucocorticoid receptor antagonist RU 486 (Sigma-Aldrich); the β2 adrenoceptor antagonist ICI 118,551 hydrochloride (Sigma-Aldrich). l-Sulpiride was dissolved in a minimum volume of 0.1 N HCl, neutralized with 0.1 N NaOH to pH 6–7 and adjusted to final volume with saline5,16; RU 486 was dissolved in a vehicle containing 90% saline, 5% dimethylsulfoxide (Wako Pure Chemical Industries, Osaka, Japan) and 5% cremophor EL (Sigma-Aldrich); other drugs were dissolved in saline3,4,5,16.
Drugs were injected in a volume of 10 ml/kg body weight for intraperitoneal (i.p.) injection and in a volume of 4 μl for intracerebroventricular (i.c.v.) injection3,4,5. I.c.v. injection was performed as described previously3,4,5,35; the injection site was 0.0 mm anterior, 1.0 mm lateral and 3.0 mm deep from bregma and only data from mice with correctly located injections were included in the analyses3,4,5. Doses of drugs were selected according to previous reports5,27,36,37 and optimized in preliminary experiments.
Measurement of plasma glucose levels
Plasma glucose levels were measured according to previous reports3,4,5. Blood samples were obtained from the tail vein. Immediately after the initial blood sample was collected, drugs were injected. RU 486 and ICI 118,551 were administered 30 min before i.c.v. injections of quinpirole or l-sulpiride. The intervals between treatments were based on preliminary experiments. Blood samples were collected 15, 30, 60, 90, 120 and 180 min after i.c.v. injections. Plasma glucose levels were determined using glucose CII-test Wako (Wako Pure Chemical Industries).
Measurement of plasma glucagon and insulin levels
Ten min after injection of quinpirole or l-sulpiride, blood samples were collected and aprotinin (500 KIU/ml) was added. Plasma glucagon and insulin levels were determined using Glucagon EIA Kit (YK090; Yanaihara, Shizuoka, Japan; RRID: AB_2857344) and LEBIS Mouse Insulin ELISA Kit (U-type; Fujifilm Wako Shibayagi, Gunma, Japan; RRID: AB_2857341), respectively.
Measurement of hepatic glycogen levels
The liver was collected 30 min after injection of quinpirole or l-sulpiride, then immediately frozen in liquid nitrogen and kept at -80 °C until use. Hepatic glycogen levels were determined using Glycogen Colorimetric/Fluorometric Assay Kit (K646-100; BioVision, Milpitas, CA, USA).
Reverse transcription polymerase chain reaction (RT-PCR)
Hepatic mRNA levels were measured as described previously3,16,38,39,40: the liver was collected 30 min after drug injections, then immediately frozen in liquid nitrogen and kept at -80 °C until use; total RNA was isolated from the liver using RNAiso Plus (Takara Bio, Shiga, Japan) and reverse-transcribed to cDNA using PrimeScript RT reagent Kit (Takara Bio); the primers were mixed with buffer, dNTP mix, DNA polymerase and ultrapure water, and cDNA was added to the mixture for each reaction3,16,38,39,40. The primers were: G6Pase (forward: 5′-CGA CTC GCT ATC TCC AAG TGA-3′, reverse: 5′-GTT GAA CCA GTC TCC GAC CA-3′); PEPCK (forward: 5′-CAT ATG CTG ATC CTG GGC ATA AC-3′, reverse: 5′-CAA ACT TCA TCC AGG CAA TGT C-3′); β-actin (forward: 5′-CAT CCG TAA AGA CCT CTA TGC CAA C-3′, reverse: 5′-ATG GAG CCA CCG ATC CAC A-3′)3. PCR was conducted on a thermal cycler (TP650; Takara Bio) as follows: 94 °C for 4 min 30 s, followed by 33 cycles of denaturing at 94 °C for 30 s, annealing at 58 °C for 1 min, and extension at 72 °C for 1 min3,16,38,39,40. The PCR products were analyzed by electrophoresis on 1.7% agarose gels; the gel was stained with ethidium bromide, photographed with UV transillumination and intensity of the band analyzed by computer-assisted densitometry using ImageJ image-analysis software; the value for each enzyme was normalized by the respective value for β-actin, intensities of the bands obtained from drug-treated mice were compared with those of vehicle-treated mice, and the percentage of control was quantified for each sample3,16,38,39,40.
Operation of hepatic vagotomy
Hepatic vagotomy was conducted as describe previously41. Mice were anesthetized with sodium pentobarbital (40 mg/kg, i.p.) and isoflurane (1 v/v% inhalation). The midline of the abdomen was incised, and the diaphragm was cut to reveal the esophageal-hepatic connections. Since this region contains a neurovascular bundle, including the hepatic branch of the vagus nerve, this branch was selectively cut. The diaphragm was sutured and then the abdomen was sutured. Mice were allowed to recover from surgery for at least 1 week.
Statistical analysis
Results are presented as means ± S.E.M. Two-way analysis of variance (ANOVA) for repeated measures, followed by post hoc Bonferroni tests, were used to compare groups as appropriate. Student’s t-test was used to compare two groups as appropriate. Differences were considered statistically significant when P < 0.05.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Sernyak, M. J., Gulanski, B. & Rosenheck, R. Undiagnosed hyperglycemia in patients treated with atypical antipsychotics. J. Clin. Psychiatry. 66, 1463–1467 (2005).
Llorente, M. D. & Urrutia, V. Diabetes, psychiatric disorder, and the metabolic effects of antipsychotic medications. Clin. Diabetes 24, 18–24 (2006).
Ikegami, M. et al. Olanzapine increases hepatic glucose production through the activation of hypothalamic AMPK. Diabetes Obes. Metab. 15, 1128–1135 (2013).
Ikegami, M. et al. Olanzapine induces glucose intolerance through the activation of AMPK in the mouse hypothalamus. Eur. J. Pharmacol. 718, 376–382 (2013).
Ikegami, M. et al. Olanzapine-induced hyperglycemia: Possible involvement of histaminergic, dopaminergic and adrenergic functions in the central nervous system. Neuroendocrinology 98, 224–232 (2013).
Nordlie, R. C., Foster, J. D. & Lange, A. J. Regulation of glucose production by the liver. Annu. Rev. Nutr. 19, 379–406 (1999).
Iversen, J. Adrenergic receptors and the secretion of glucagon and insulin from the isolated, perfused canine pancreas. J. Clin. Invest. 52, 2102–2116 (1973).
Ahrén, B. Autonomic regulation of islet hormone secretion - Implications for health and disease. Diabetologia 43, 393–410 (2007).
Uyama, N., Geerts, A. & Reynaert, H. Neural connections between the hypothalamus and the liver. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 280, 808–820 (2004).
Erraji-Benchekroun, L. et al. Overexpression of β2-adrenergic receptors in mouse liver alters the expression of gluconeogenic and glycolytic enzymes. Am. J. Physiol. Endocrinol. Metab. 288, E715–E722 (2005).
Niijima, A. Blood glucose levels modulate efferent activity in the vagal supply to the rat liver. J. Physiol. 364, 105–112 (1985).
Roh, E., Song, K. & Kim, M. S. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp. Mol. Med. 48, e216. https://doi.org/10.1038/emm.2016.4 (2016).
Ruud, J., Steculorum, S. M. & Brüning, J. C. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat. Commun. 8, 15259. https://doi.org/10.1038/ncomms15259 (2017).
Buijs, R. M., Chun, S. J., Niijma, A., Romijn, H. J. & Nagai, K. Parasympathetic and sympathetic control of the pancreas: a role for the suprachiasmatic nucleus and other hypothalamic centers that are involved in the regulation of food intake. J. Comp. Neurol. 431, 405–423 (2001).
Yoshimatsu, H., Niijima, A., Oomura, Y. & Katafuchi, T. Lateral and ventromedial hypothalamic influences on hepatic autonomic nerve activity in the rat. Brain Res. Bull. 21, 239–244 (1988).
Yonemochi, N. et al. Dopaminergic mechanisms in the lateral hypothalamus regulate feeding behavior in association with neuropeptides. Biochem. Biophy. Res Commun. 519, 547–552 (2019).
Keck, F. S., Foldenauer, Z., Zeller, G., Wolf, C. F. & Pfeiffer, E. F. Comparative effects of dopamine and dobutamine on glucoregulation in a rat model. Can. J. Physiol. Pharmacol. 69, 1178–1183 (1991).
Saller, C. F. & Kreamer, L. D. Glucose concentrations in brain and blood: regulation by dopamine receptor subtypes. Brain Res. 546, 235–240 (1991).
Durant, S., Coulaud, J. & Homo-Delarche, F. Bromocriptine-induced hyperglycemia in nonobese diabetic mice: Kinetics and mechanisms of action. Rev. Diabet. Stud. 4, 185–194 (2007).
Luo, S., Liang, Y. & Cincotta, A. H. Intracerebroventricular administration of bromocriptine ameliorates the insulin-resistant/glucose-intolerant state in hamsters. Neuroendecrinology. 69, 160–166 (1999).
Gibson, C. D., Karmally, W., McMahon, D. J., Wardlaw, S. L. & Korner, J. Randomized pilot study of cabergoline, a dopamine receptor agonist: effects on body weight and glucose tolerance in obese adults. Diabetes Obes. Metab. 14, 335–340 (2012).
Rubί, B. et al. Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion. J. Biol. Chem. 280, 36824–36832 (2005).
Ustione, A. & Pitson, D. W. Dopamine synthesis and D3 receptor activation in pancreatic β-cells regulates insulin secretion and intracellular [Ca2+] oscillations. Mol. Endocrinol. 26, 1928–1940 (2012).
Parks, S., Kang, S., Lee, H. W. & Ko, B. S. Central prolactin modulates insulin sensitivity and insulin secretion in diabetic rats. Neuroendocrinology 95, 332–343 (2012).
Perez Millan, M. I. et al. Selective disruption of dopamine D2 receptors in pituitary lactotropes increases body weight and adiposity in female mice. Endocrinology 155, 829–839 (2014).
Pijl, H. et al. Bromocriptine: a novel approach to the treatment of type 2 diabetes. Diabetes Care 23, 1154–1161 (2000).
Cippitelli, A. et al. Binge-like ethanol consumption increases corticosterone levels and neurodegeneration whereas occupancy of type II glucocorticoid receptors with mifepristone is neuroprotective. Addict. Biol. 19, 27–36 (2012).
Lacey, R. J., Berrow, N. S., Scarpello, J. H. B. & Morgan, N. G. Selective stimulation of glucagon secretion by β2-adrenoceptors in isolated islets of Langerhans of the rat. Br. J. Pharmacol. 103, 1824–1828 (1991).
Haffner, C. A. & Kendall, M. J. Metabolic effects of β2-agonists. J. Clin. Pharm. Ther. 17, 155–164 (1992).
Vander Tuig, J. G., Knehans, A. W. & Romsos, D. R. Reduced sympathetic nervous system activity in rats with ventromedial hypothalamus lesions. Life Sci. 30, 913–920. https://doi.org/10.1016/0024-3205(82)90619-1 (1982).
Fetissov, S. O., Meguid, M. M., Sato, T. & Zhang, L. H. Expression of dopaminergic receptors in the hypothalamus of lean and obese Zucker rats and food intake. Am. J. Regul. Integr. Comp. Physiol. 283, R905–R910 (2002).
Miyazaki, T. et al. Cav2.1 in cerebellar Purkinje cells regulates competitive excitatory synaptic wiring, cell survival, and cerebellar biochemical compartmentalization. J. Neurosci. 32, 1311–1328 (2012).
Nakayama, H. et al. Microglia permit climbing fiber elimination by promoting GABAergic inhibition in the developing cerebellum. Nat. Commun. 9, 2830. https://doi.org/10.1038/s41467-018-05100-z (2018).
Mishina, M. & Sakimura, K. Conditional gene targeting on the pure C57BL/6 genetic background. Neurosci. Res. 58, 105–112 (2007).
Haley, M. J. & McCormick, W. G. Pharmacological effects produced by intracerebral injections of drug in the conscious mice. Br. J. Pharmacol. 12, 12–15 (1957).
Yoshimura, N., Kuno, S., Chancellor, M. B., de Groat, W. C. & Seki, S. Dopaminergic mechanisms underlying bladder hyperactivity in rats with a unilateral 6-hydroxydopamine (6-OHDA) lesion of the nigrostriatal pathway. Brit. J. Pharmacol. 139, 1425–1432 (2003).
Vranjkovic, O., Hang, S., Baker, D. A. & Mantsch, J. R. β-Adrenergic receptor mediation of stress-induced reinstatement of extinguished cocaine-induced conditioned place preference in mice: roles for β1 and β2 adrenergic receptors. J. Pharmacol. Exp. Ther. 342, 541–551 (2012).
Ikeda, H. et al. Inhibition of opioid systems in the hypothalamus as well as the mesolimbic area suppresses feeding behavior of mice. Neuroscience 311, 9–21 (2015).
Ardianto, C. et al. Opioid systems in the lateral hypothalamus regulate feeding behavior through orexin and GABA neurons. Neuroscience 320, 183–193 (2016).
Yonemochi, N., Ardianto, C., Ueda, D., Kamei, J. & Ikeda, H. GABAergic function in the lateral hypothalamus regulates feeding behavior: possible mediation via orexin. Neuropsychopharmacol. Rep. 39, 289–296 (2019).
Bernal-Mizrachi, C. et al. An afferent vagal nerve pathway links hepatic PPARα activation to glucocorticoid-induced insulin resistance and hypertension. Cell Metab. 5, 91–102 (2007).
Acknowledgements
This work was partly supported by JSPS KAKENHI Grant Number JP17K07065 (HI) and by a Grant-in-Aid for Scientific Research on Innovative Areas (Comprehensive Brain Science Network) from the Ministry of Education, Science, Sports and Culture of Japan. We are grateful to Megumi Iwasaki, Ayumi Matsumaru, Mami Ohsuga, Haruna Miyake, Sae Kouno, Hitomi Yanagawa, Yuka Iwasaki, Midori Matumoto and Mamika Hamada for their excellent technical assistance.
Author information
Authors and Affiliations
Contributions
H.I. designed the experiments. R.M. and N.Y. performed the experiments. M.A., M.K., R.N. and K.S. established the dopamine D2 receptor knockout mice. H.I., R.M. and N.Y. analyzed data. H.I. wrote the manuscript. J.L.W. advised on the studies. H.I., R.M., N.Y., M.A., R.N, K.S., J.L.W. and J.K. critically read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ikeda, H., Yonemochi, N., Mikami, R. et al. Central dopamine D2 receptors regulate plasma glucose levels in mice through autonomic nerves. Sci Rep 10, 22347 (2020). https://doi.org/10.1038/s41598-020-79292-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-79292-0
This article is cited by
-
Prefrontal cortical dopamine deficit may cause impaired glucose metabolism in schizophrenia
Translational Psychiatry (2024)
-
Therapeutic potential of dopamine agonists in the treatment of type 2 diabetes mellitus
Environmental Science and Pollution Research (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
