
Abstract
Hybridisation in the wild between closely related species is a common mechanism of speciation in the plant kingdom and, in particular, in the grass family. Here we explore the potential for natural hybridisation in Stipa (one of the largest genera in Poaceae) between genetically distant species at their distribution edges in Mountains of Central Asia using integrative taxonomy. Our research highlights the applicability of classical morphological and genome reduction approaches in studies on wild plant species. The obtained results revealed a new nothospecies, Stipa × lazkovii, which exhibits intermediate characters to S. krylovii and S. bungeana. A high-density DArTseq assay disclosed that S. × lazkovii is an F1 hybrid, and established that the plastid and mitochondrial DNA was inherited from S. bungeana. In addition, molecular markers detected a hybridisation event between morphologically and genetically distant species S. bungeana and probably S. glareosa. Moreover, our findings demonstrated an uncertainty on the taxonomic status of S. bungeana that currently belongs to the section Leiostipa, but it is genetically closer to S. breviflora from the section Barbatae. Finally, we noticed a discrepancy between the current molecular data with the previous findings on S. capillata and S. sareptana.
Similar content being viewed by others

Introduction
Hybridisation in the wild between closely related species is a common mechanism of speciation in the plant kingdom1,2,3,4,5,6,7. Due to the prevalence of polyploidy found in angiosperms it has been estimated that around 11% of flowering plants may have arisen through hybridisation events4. In addition, speciation via hybridisation can lead to an equal ploidy number within parental and newly formed species3. In general, hybridisation is often accompanied by introgression and causes gene transfer between species via repeated backcrossing4,8,9,10,11. On the one hand it may have contributed to species diversity and speciation5,12,13, on the other, deleterious consequences of hybridisation such as decreased fitness, genetic assimilation and gene swamping may drive populations toward the brink of extinction14,15,16.
In the grass family (Poaceae) hybridisation and introgression are well studied mainly for economically important plants, such as wheats17,18, maize19,20, rice21, 22, barley23,24, oats25,26, rye27,28, sugarcanes29,30, and sorghums31,32. Nowadays new molecular markers and technologies that first came to the field of agriculture are becoming widely used in studies of wild populations with little or no previous genomic information. For instance, genotyping-by-sequencing (GBS) and GBS-like approaches that were initially developed for maize and barley33 help to detect hybridisation and introgression events in many wild plant genera34,35,36,37,38.
The genus Stipa L. belongs to the subfamily Pooideae and alongside with Bambusoideae (bamboos), and Oryzoideae (rices) form the so-called BOP clade39. The BOP species are known as the "cool season" or "pooid" grasses and all are C3 and distributed in temperate climates40. Following Tzvelev (1974), the genus Stipa includes six main sections Barbatae Junge, Leiostipa Dumort, Pseudoptilagrostis Tzvelev, Regelia Tzvelev, Stipa, and Smirnovia Tzvelev41, and comprises over 150 species native to Asia, Europe and North Africa42,43. In its strict sense, the genus is monophyletic44,45, but subdivisions within the genus are not consistently supported by available molecular data43,46. Species of the genus are dominants and/or subdominants in steppe plant communities47,48,49,50, can be used for their classification51, and in studies related to climate change52,53,54. Moreover, the species are of great economic importance mainly as pasture and fodder plants, especially in the early phases of development55, they can be used for soil remediation processes56, and as ornamental plants (e.g. S. capillata L., S. pulcherrima K. Koch, S. pennata L.).
For decades it has been hypothesised that some Stipa taxa arose via hybridisation57,58,59,60. According to our observations, Stipa hybrids reproduce vegetatively and, less frequently, sexually60. It recently was shown that hybrids in Stipa can produce fertile pollen grains and therefore are able to backcross with both parental species61. In addition, based on morphology, a hybrid origin can be attributed to ca. 30% of Stipa species where only in Middle Asia 23 of 72 species are regarded as nothospecies43. For instance, to such taxa belong S. × czerepanovii Kotukhov (= S. orientalis Trin. × S. richteriana Kar. & Kir.); S. × fallax M. Nobis & A. Nowak (S. drobovii (Tzvel.) Czer. × S. macroglossa P. A. Smirn. subsp. macroglossa); S. × gegarkunii P. A. Smirn. (= S. caucasica Schmalh. × S. pulcherrima K. Koch); S. × hissarica M. Nobis (= S. lipskyi Roshev. × S. orientalis Trin.); S. × tzveleviana Kotukhov (= S. orientalis × S. macroglossa subsp. kazachstanica); and S. × zaissanica Kotukhov (= S. orientalis × S. hohenackeriana Trin. & Rupr.)43,60,62,63.
Heretofore, all putative hybrid taxa within Stipa were described based exclusively on morphological comparison. The only exception is Stipa × heptapotamica Golosk., whose origin has been established using molecular methods61. Although its parental species Stipa richteriana Kar. & Kir and S. lessingiana Trin. & Rupr. were morphologically distant and affiliated to different sections Leiostipa and Subbarbatae Tzvelev41,58,64, genetically they are closely related65,66 and able to hybridise with each other61.
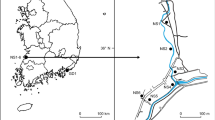
During field studies in eastern Kyrgyzstan in 2015 and 2017, interesting specimens of Stipa, combining characters not observed in the previously described taxa, were found on the south shore of Lake Issyk-Kul (Fig. 1). Due to these specimens seeming to be morphologically intermediate between two species from the same locality, we hypothesised that they can be hybrids between S. krylovii Roshev. and S. bungeana Trin. Although, traditionally both putative parental taxa were assigned to the section Leiostipa58, they are distant phylogenetically and belong to two different clades61,65. Both of them have wide distribution ranges, Stipa krylovii occurs in the Russian Far East and Southern Siberia, Mongolia, China, Northern Nepal, Southern Tajikistan, Eastern Kazakhstan, and Eastern Kyrgyzstan43,67, whereas S. bungeana is distributed in Southern Mongolia, China, and Eastern Kyrgyzstan68,69 (Fig. 1a).

Distribution map represents (a) general ranges of S. krylovii (green) and S. bungeana (red) with the dashed line indicating the hypothetical border, (b) localities of the examined specimens used for the molecular analysis. The current map is based on Google Maps.
Since hybrids between genetically distant Stipa species have not been observed previously in nature, in the current study by using integrative taxonomy based on morphology and high density genome wide genotyping-by-sequencing data, we aim to (1) obtain insight into the extent of hybridisation between S. krylovii and S. bungeana on macro- and micromorphological levels; (2) assess levels of inter-species gene flow (if present) between the examined Stipa taxa; (3) analyse the usefulness of SilicoDArT and SNPs markers for genomic studies in Stipa.
Results
Numerical analysis
The factor analysis of mixed data (FAMD) revealed six markedly differentiated groups of OTUs in accordance with the taxonomic classification of the examined taxa (Fig. 2). The first three dimensions explained 41.71%, 13.64%, and 10.14%, of the total variability, respectively. The first dimension is composed, in order of descending contribution, by the quantitative variables AL, Col1L, CL, LG, CvH (Supplementary Table S2, for character abbreviations see Table 1). The second dimension is composed, in order of descending contribution, by the quantitative variables DDL, LHTA, LigIV, WVS, LHD, SL, and the qualitative variable HTTA (Supplementary Table S2). The third dimension is composed, in order of descending contribution, by the quantitative variables HLCol2, HLCol1, WCol1, CBW, and the qualitative variable AdSVL (Supplementary Table S2). The two dimensional plot revealed the overlapping of OTUs belonging to S. breviflora and S. bungeana, whereas OTUs of S. sareptana are slightly overlapped with OTUs of S. krylovii and S. capillata (Fig. 2a). A clear dispersal of the OTUs could be seen in the three-dimensional plot, where differences between the studied species are explained by the third principal axis (Fig. 2b and in the interactive three-dimensional plot available at https://plot.ly/~eugenebayahmetov/3/). In particular, the third axis differentiates S. breviflora and S. bungeana as clear non-overlapped clouds of OTUs.

Factor analysis of mixed data performed on 22 quantitative and three qualitative characters of the six examined species of Stipa. (a) Plot of the two principal axes. (b) Plot of the three principal axes. The figure was created using the R-packages factoextra v.1.0.6 (Fig. a), https://CRAN.R-project.org/package=factoextra/, and plotly v.4.9.2 (Fig. b), https://plotly.com/r/getting-started/.
In addition, the notch plots of variables showed significant differences between means and the strong evidence of differing medians within all the taxa for CL; AL demonstrates the difference within all the taxa except the pair S. krylovii and S. sareptana; Col1L exhibits the difference within all the taxa except the pair S. bungeana and S. breviflora; LG indicates the difference within all the taxa except the pairs S. capillata and S. sareptana, as well as S. breviflora and the putative hybrid (S. bungeana × S. krylovii), here and below named as S. × lazkovii; the SL variable shows the difference within all the taxa except the pairs S. × lazkovii and S. capillata, and S. krylovii and S. sareptana (Supplementary Fig. S1).
Seven notch plots of variables show significant differences between means and the strong evidence of differing medians within S. bungeana, S. krylovii, and their putative hybrid: AL, CL, Col1L, SL, WCol1, LG, and WVS (Supplementary Fig. S1). At the same time, S. bungeana, S. krylovii and S. × lazkovii share six characters that have no significant differences between their means: CvH, CBL, CBW, DVL, HLCol1, and HLCol2. Further, S. × lazkovii and S. krylovii share seven characters with no significant differences between their means, but differ with S. bungeana: CdH, DDL, Col2L, LigC, LigIV, AW, LHTA. Finally, only two characters LHD and LHV have no significant differences between means within pairs S. × lazkovii and S. krylovii, and S. × lazkovii and S. bungeana, but have significant differences between means of S. krylovii and S. bungeana (Supplementary Fig. S1).
Micromorphology
The micromorphological examination of Stipa bungeana, S. krylovii and their putative hybrid revealed the pattern of lemma that is typical for the genus Stipa (Fig. 3)45,60,62,70,71. In all three taxa, the fundamental long cells are rectangular to more or less square in shape. The side walls of long cells are raised and undulate. Silica bodies are sparse or absent, but if present, they are reniform to ovate, whereas cork cells are absent. Hooks are frequent and oriented towards the lemma apex, whereas prickles are present mostly near the lemma apex (Fig. 3). Macrohairs are straight or bent near the base, cylindrical and/or string-like, with a bulbous base and a needle-like apex. They are organised in seven lines. The lemma apex is scabrous due to abundant hooks, prickles and short macrohairs (present especially in S. bungeana and in the hybrid), surpassed by a ring of unequal macrohairs. The pattern of lemma apex shows clearly intermediate character of S. × lazkovii between the two putative parents (Figs. 3a, 3f, 3k).

Micromorphological patterns of Stipa krylovii (a-e), S. × lazkovii (f-j) and S. bungeana (k–o): top of lemma (a, f, k), lemma abaxial surface (b-c, g-h, l-m), adaxial surface of leaf blade (d, i, n), abaxial surface of leaf blade (e, j, o). Abbreviations: h – hooks, lc – long cells; mh – macrohairs, pr – prickles; sb – silica bodies.
DArTseq analysis
A total of 137,437 SilicoDArT and 125,850 SNPs markers were obtained using a DArTseq high-density assay, of which 76,604 silicoDArT and 19,133 SNPs markers were kept after the filtering steps. The first two axes of principal coordinates analysis (PCoA) explained 77% and 91% of the total genetic divergence within the studied taxa based on the SilicoDArT and SNPs markers, respectively, whereas the third axes explained only 6.3% and 3% (Fig. 4).

Principal Coordinates Analysis plot based on genetic distances between samples. (a) Plot of the two principal axes based on SilicoDArT markers. (b) Plot of the three principal axes based on SilicoDArT markers. (c) Plot of the two principal axes based on SNPs markers. (d) Plot of the three principal axes based on SNPs markers. The figure was created using the R-packages ggplot2 v.3.3.0 (Figs a and c), https://ggplot2.tidyverse.org/, and plotly v.4.9.2 (Figs b and d), https://plotly.com/r/getting-started/.
In general, based on genetic similarities both markers revealed six markedly differentiated groups (Fig. 4). Most of the specimens are grouped together accordingly to their taxonomical classifications. However, one sample (ID0494394), which morphologically was somewhat similar to S. breviflora, is grouping together with S. bungeana OTUs and far distant to the rest of OTUs belonging to S. breviflora. All S. × lazkovii specimens have an intermediate position between S. bungeana and S. krylovii, suggesting an admixed origin. In addition, on the basis of two axes both markers are not allowed to differentiate two taxa, S. capillata and S. sareptana. On the other hand, the difference can be marked in the three-dimensional plot based on SilicoDArT markers (Figs. 4b, the interactive plot available at https://plot.ly/~eugenebayahmetov/5/), but not in SNPs markers (Fig. 4d, https://plot.ly/~eugenebayahmetov/7/).
A fastSTRUCTURE analysis of the SilicoDArT markers revealed the most likely number of clusters at K value of 5 (Fig. 5a). For the SNPs markers, the 'best' K was inferred in fastSTRUCTURE as K = 4 (Fig. 5b). Both analyses defined S. breviflora, S. bungeana, and S. krylovii as clear taxa with the exception of the specimen ID0494394 (Fig. 5) that shares 73% of markers with S. bungeana and 27% with probably S. glareosa, indicating their first backcross generation progeny (Fig. 5a). The last-mentioned taxon was not present in the analyses, however, it is common in the locality, where the specimen ID0494394 was growing. In case of SNPs markers, the specimen ID0494394 has 75% of markers with S. bungeana, 19% with S. capillata/S. sareptana, and 6% with S. krylovii, suggesting a possible hybridisation between these species followed by backcrossing with S. bungeana (Fig. 5b).

FastSTRUCTURE results based on (a) SilicoDArT markers for K = 5 and (b) SNPs markers for K = 4. The figure was created using an in-house R script in RStudio v.1.1.463, https://rstudio.com/products/rstudio/.
The fastSTRUCTURE analyses revealed F1 hybrid specimens between S. krylovii and S. bungeana due to samples of S. × lazkovii have admixture between these clusters in a range of 55% and 45% for the SilicoDArT markers (Fig. 5a), and 50/50% for the SNPs markers (Fig. 5b). The fastSTRUCTURE output for the SilicoDArT markers exhibits no difference between S. capillata and S. sareptana resulting in clustering them together (Fig. 5a), whereas the analysis of the SNPs shows an admixture between S. capillata/S. sareptana and S. krylovii in a range of 53% and 47%, respectively (Fig. 5b).
The results of the UPGMA cluster analyses revealed a clear division of samples into two major clades (Fig. 6). According to the clustering obtained with the SilicoDArT markers, the first clade is subdivided into four smaller clusters, specifically, comprising samples of: (1) S. × lazkovii; (2) S. krylovii; (3) S. sareptana; (4) S. capillata (Fig. 6a). The first two species are genetically closely related to each other and distant to S. sareptana and S. capillata that together form one sub-cluster. The second clade is composed of three clusters comprising samples of: (1) S. breviflora; (2) S. bungeana; (3) the sample ID0494394 that is genetically closer to S. bungeana than to S. breviflora. The UPGMA cluster analysis of the SNPs markers demonstrated the subdivision of samples into the same number of clusters as were obtained for the SilicoDArT markers. However, in this case, specimens of S. × lazkovii are genetically closer to S. bungeana, but not to S. krylovii.

Unweighted Pair Group Method with Arithmetic Mean cluster analyses based on Jaccard's similarity coefficients generated from (a) SilicoDArT markers and (b) SNPs markers. The figure was created using the R-package stats v.3.6.2, https://www.rdocumentation.org/packages/stats/versions/3.6.2/.
Genetic mapping onto chloroplast genomes of Stipa species and mitochondria of specimens from the Poaceae family (Supplementary Table S3) revealed 11 SilicoDArT markers assigned to chloroplast DNA and 27 loci assigned to mitochondrial DNA. The downstream neighbour-joining cluster analysis showed grouping of Stipa taxa into two main clades (Fig. 7). In the first clade three species could be defined: S. krylovii, S. capillata (bootstrap support 90%), S. sareptana (bootstrap support 84%), with an exception of the specimen ID0494394 that is grouped together with S. krylovii (Fig. 7). The second clade comprises a group of S. bungeana and S. × lazkovii (bootstrap support 79%), and the rest of S. breviflora specimens with a good bootstrap support of 87% (Fig. 7). All S. × lazkovii samples are grouping alongside with S. bungeana, and one S. bungeana specimen (ID0459867) is placed outside the main group of S. bungeana and S. × lazkovii with a bootstrap support of 79% (Fig. 7).

Neighbor-joining tree reconstructed based on the SilicoDArT markers derived from chloroplast and mitochondrial genomes. The bootstrap values > 50% obtained from 10,000 replicates are shown above the branches. The figure was created using Figtree v1.4.4, https://tree.bio.ed.ac.uk/software/figtree/.
Discussion
Although many interspecific hybrids have been described in the genus Stipa43,57,58,59, so far only a single molecular investigation was performed to verify the origin of one such species, Stipa × heptapotamica61 that appeared to be a hybrid between genetically closely related species65,66. The current study is the first report of hybridisation between two genetically distant Stipa species, S. krylovii and S. bungeana61,65, at their distribution edges in Mountains of Central Asia (Fig. 1a).
Analyses of morphological variation resulted in a clear delimitation of the studied species (Fig. 2b). Particularly, the main morphological characters (Table 1) show that species S. capillata, S. sareptana, S. krylovii, S. bungeana, S. × lazkovii representing the section Leiostipa are quite distant to S. breviflora which traditionally has been affiliated to the section Barbatae41. As expected, the hybrid specimens of S. × lazkovii were mostly characterised by intermediate morphological traits between the parental taxa S. krylovii and S. bungeana (Figs. 2 and 3, Supplementary Fig. S1). In addition, some OTUs of S. sareptana were slightly overlapped with OTUs of S. krylovii and S. capillata. However, S. sareptana and S. krylovii are easy to distinguish based on morphology of leaves (scabrous in S. sareptana and glabrous in S. krylovii) and the lemma apex (with a poorly developed ring of hairs in S. sareptana and with a well-developed ring of hairs in S. krylovii)43,58,67. As for S. sareptana and S. capillata, these taxa can be delimited by characteristics of their vegetative leaves (scabrous in S. sareptana and glabrous in S. capillata) and characters of lemma (hairs on the top in S. sareptana and glabrous in S. capillata)43,58,67.
Both PCoA and fastSTRUCTURE analyses confirmed that S. × lazkovii is the F1 hybrid of S. krylovii and S. bungeana (Figs. 4 and 5). In addition, the neighbour-joining cluster analysis identified S. bungeana as the source of maternal DNA for all hybrid specimens, suggesting unidirectional hybridisation (Fig. 7). However, due to the small sample size, we cannot exclude either an opposite combination or interspecific gene flow through introgression that could exist in nature, especially since in this area of Issyk-Kul Lake populations of both parental species are extremely large.
Although morphologically S. bungeana is considered as a member of the section Leiostipa58, molecular analyses demonstrated that it is quite distant from S. krylovii, S. capillata, and S. sareptana from the same section (Figs. 4, 6 and 7). These findings support our previous molecular results for these taxa based on a nuclear region61,65. In addition, the results of the distance based clustering algorithms UPGMA and NJ revealed that S. bungeana is closer to S. breviflora then to the rest of Leiostipa taxa from the study (Figs. 6 and 7). This result demands further investigations on S. bungeana to establish its proper taxonomic place in the genus Stipa.
Analyses of molecular markers also revealed that the genetic relationships within some studied taxa are more complex than expected. Firstly, the sample ID0494394, which morphologically was somewhat similar to S. breviflora, appeared to be an introgressive hybrid that shares 73% of markers with S. bungeana and 27%, more likely, with S. glareosa (Fig. 5a). Here, we presume that hybridisation events are happening between S. bungeana and S. glareosa, because the introgressive hybrid was found on the north shore of Lake Issyk-Kul, where only three Stipa taxa were recorded (S. bungeana, S. breviflora, and S. glareosa). However, due to S. glareosa was absent in the analyses, a new study focusing on hybridisation should be performed to verify if the gene flow is a common event within these taxa. Secondly, our research demonstrates the discordance between the results of fastStructure and PCoA analyses from one side and the UPGMA and NJ from the other. The first two represent no or almost no difference between S. capillata and S. sareptana (Figs. 4 and 5). Notwithstanding, the UPGMA and NJ dendrograms show that genetically these taxa can be delimited (Figs. 6 and 7) that supports our previous molecular investigations on these taxa61,65. However, in the current research S. capillata and S. sareptana are grouped together, whereas based on the nuclear Intergenic Spacer (IGS) the last taxon is closer to S. krylovii61,65. Due to the limited number of analysed specimens in the present and previous studies, we believe that a bigger sample size combining genetics and traditional taxonomy should be undertaken in order to better resolve the relationship between these species.
Until now the DArTseq approach has been used mostly in commercially important plant species72,73,74,75,76,77,78 and its implication in genomic studies in wild species is still limited79,80. Thus, the current study highlights the applicability of genome reduction approaches such as DArTseq in studies on natural hybridisation in wild, and specifically in a grass genus Stipa. The high density genome wide genotyping-by-sequencing resulted in a total of 137,437 silicoDArT and 125,850 SNPs markers, of which 76,604 silicoDArT and 19,133 SNPs provided robust information of the Stipa genome in the absence of the reference sequence information. Such number of markers is several 100-fold higher than was achieved in our previous study on natural hybridisation in Stipa61. In particular, by using inter simple sequence repeat markers (ISSR) we were able to detect only 105 polymorphic bands for the S. heptapotamica hybrid complex. In addition, dominant markers were used in several genomic studies in Stipa and resulted in 372 polymorphic ISSR bands for S. bungeana81, 34 polymorphic ISSR bands for S. ucrainica and S. zalesskii82, 212 polymorphic ISSR bands for S. tenacissima83, 231 polymorphic random amplified polymorphic DNA (RAPD) bands for S. krylovii84, 310 polymorphic RAPD bands for S. grandis85, and 504 polymorphic sequence-related amplified polymorphism bands for S. bungeana81. Thus, both silicoDArT and SNPs markers may better suit for genetic diversity studies in Stipa. Furthermore, the current study demonstrated the usefulness of silicoDArT markers as a tool to detect chloroplast and mitochondrial loci and thus may help to clarify the maternal inheritance of hybrid species.
Taxonomic treatment
Stipa × lazkovii M. Nobis & A. Nowak, nothosp. nov. (Fig. 3f-j, Supplementary Figs S2 and S3). TYPE: Kyrgyzstan, between Kongurlen and Kultor, 17 km SW from coast of Issyk-Kul, semidesert, N 42°5′47.07′' / E 76°39′6.22′', elev. 1940 m, wp. 930, 6 July 2017, M. Nobis, E. Klichowska, A. Wróbel, A. Nowak sn. (holotype KRA 495,093! (specimen in the middle part of the sheet); isotypes KRA 487,067!, 487,066!, 481,608!).
Diagnosis: Stipa × lazkovii differs from S. krylovii Roshev. by having shorter anthecium (7.3–8.5 mm vs. 9.0–11.5), shorter callus (1.8–2.2 vs. 2.3–3.8 mm long), shorter glumes (15–17 vs. 18–28 mm long) as well as by having long prickle-hairs below the top of the anthecium (Fig. 3). Having long prickle-hairs below the top of the anthecium Stipa × lazkovii is also similar to S. bungeana, however differs from it by longer anthecium (7.3–8.5 vs. 4.8–6.0 mm long), longer callus (over 1.8 vs. up to 1.3 mm long), longer glumes (over 15 vs. up to 15 mm long) and narrower leaves (0.5–0.6 vs. 0.6–1.0 mm wide).
Description: Plants perennial, densely tufted, with a few culms and numerous vegetative shoots; culms 35–55 cm tall, 3-noded, glabrous at and below the nodes. Leaves of vegetative shoots: sheaths glabrous, at margins ciliate; ligules truncate, up to 0.2 mm ciliate at margins; blades convolute, up to 25 cm long, 0.5–0.6(–0.7) mm in diameter, adaxial surface densely pubescent with up to 0.1 mm long hairs (prickles), adaxial surface glabrous, rarely very slightly scabrous. Cauline leaves: sheaths glabrous and with white edge, shorter than internodes; ligules 0.5–5 mm long, acute and glabrous; blades glabrous, up to 12 cm long. Panicle up to 25 cm long contracted, at base enclosed by sheath of uppermost leaf, branches erect, setulose, single or paired. Glumes subequal, 15–25 mm long, narrowly lanceolate, tapering into long hyaline apex. Anthecium 7.3–8.5 mm long and 0.7–0.9 mm wide. Callus 1.8–2.2 mm long, densely pilose on ventral and dorsal surfaces, callus base acute, cuneate, scar elliptic. Lemma pale green, on dorsal surface with abundant hooks and with 7 lines of ascending hairs, hairs up to 0.5 mm long, ventral line of hairs terminates at 1.3–1.7 mm below top of lemma and dorsal line terminates at 1.5–2.2 mm below top of lemma; top of lemma scabrous due to hooks and prickles and at apex with a ring of hairs up to 0.5 mm long. Palea equals to lemma in length. Awn 95–118 mm long, bigeniculate; lower segment of column 19–25 mm long, twisted, scabrous due to prickles and short hairs up to 0.15; upper segment of column 11.5–13 mm long, twisted, scabrous due to prickles and short hairs up to 0.2 mm in long; seta flexuous 65–80 mm long, hairs in the lower part of the seta 0.1–0.2 mm long, gradually decreasing in length towards apex. Anthers yellow, 4–5 mm long, glabrous.
Etymology: The name of the taxon honours prof. dr Georgy A. Lazkov (Academy of Sciences, Bishkek, Kyrgyzstan), the eminent botanist, taxonomists and expert of vascular plants of Middle Asian Mountains.
Other specimens studied (paratypes): Kyrgyzstan, western Tian-Shan, Kongurlen Valley, steppe grasslands near the road, 3 km E of Kongurlen settl., to the S of SW part of Issyk-Kul Lake, N 42°5′53.97′' / E 76°38′37.28′', elev. 1945 m, wp. 644, 10 July 2015, M. Nobis, A. Nowak sn. (KRA 476,871, 476,870!, 476,869!, WA!).
An identification key to central Asian species of Stipa that have scabrous awns or awns that are throughout covered by 0.1–0.3 mm long hairs is given in Supplementary S4.
Materials and methods
Plant material
Morphological examination is based on plant specimens deposited in the KRA herbarium (the acronym from Thiers86). In total, 188 fully developed Stipa samples were studied under a light microscope SMZ800 (Nikon, Japan) including 40 specimens of S. krylovii, 40 of S. bungeana, 6 of S. × lazkovii, 22 of S. breviflora, 40 of S. capillata, and 40 of S. sareptana.
For molecular analysis, we collected leaves of plants from localities where S. krylovii and S. bungeana grow together with their putative hybrid, as well as from areas where S. krylovii and S. bungeana grow separately from each other (Fig. 1b). Additionally, we included Stipa taxa that frequently occur in the area near of Issyk-Kul Lake. In total, we selected 20 specimens of S. krylovii, 20 specimens of S. bungeana, 6 specimens of S. × lazkovii, 10 specimens of S. breviflora, 2 specimens of S. capillata, and 2 specimens of S. sareptana. Only one taxon, S. glareosa, is not presented in the study due to it was not found in the locality of S. × lazkovii. Moreover, S. glareosa belongs to the section Smirnovia41 and exhibits unique characters (e.g. long and pilose awns with a single geniculation), which were not observed in any Stipa taxa in this region.
All voucher specimens used in the molecular analysis are preserved at KRA (Supplementary Table S1). The names of plants were adopted from the WCSP87.
Macromorphological analyses
For the morphometric analyses, 188 specimens were used as operational taxonomic units (OTUs)88. As a first step, the Shapiro–Wilk test was used in the R-package MVN89 to assess the normality of the distribution of each character. The non-parametric Spearman's correlation coefficient was used in the R-package MVN to examine relations between the studied characters. The 22 most informative quantitative and three qualitative morphological characters, commonly used in keys and taxonomic descriptions were chosen for the analyses (Table 1).
A Factor Analysis of Mixed Data (FAMD)90 was performed in the R-package FactoMineR91 to characterise variation within and among groups of taxa without a priori taxonomic classification and to extract the variables that best identified them. The number of principal components included in the analysis was chosen based on Scree’s test92. The R-package factoextra93 was used to visualise the first two components, whereas the R-package plotly94 was chosen to illustrate the first three.
Notch plots were created in the R-package ggplot295 to explore distributional relationships between each response variable and the studied taxa (Supplementary Fig. S1). The notched box plots display a confidence interval around the median, which is normally based on the median ± 1.57 × interquartile range/square root of n. According to this graphical method for data analysis, if the notches of the two boxes do not overlap, there is "strong evidence" (95% confidence) that their medians differ. Additionally, to reveal significant differences between means of particular characters across all examined taxa the nonparametric Kruskal–Wallis test followed by the Wilcoxon rank sum test for post hoc group comparisons were calculated. To address the multiplicity of comparison, the Bonferroni method was applied to calculate corrected p-values.
Micromorphological examination
The lemma and lamina micromorphology within Stipa × lazkovii, S. krylovii, and S. bungeana were examined using scanning electron microscopy (SEM). The dried samples were coated with a gold layer using a Quorum Q150R S coater (Quorum, UK). The SEM images were obtained by a scanning electron microscope S-4700 (Hitachi, Japan). Further, we examined the adaxial and abaxial surfaces of lamina, and five sets of diagnostic characters of lemma micromorphology: (1) long cells, (2) silica bodies, (3) hooks, (4) prickles, (5) macrohairs.
DNA extraction, amplification, and DArT sequencing
Isolation of genomic DNA was performed from dried leaf tissues using a Genomic Mini AX Plant Kit (A&A Biotechnology, Poland). Quality check, quantification and concentration adjustment for sequencing and genotyping were accomplished using a NanoDrop One (Thermo Scientific, USA) and agarose gel electrophoresis visualisation. The concentration of each sample was adjusted to 50 ng/μL. Purified DNA samples (1 μg for each sample) were sent to Diversity Arrays Technology Pty Ltd (Canberra, Australia) for sequencing and marker identification.
DArTseq represents a combination of a DArT complexity reduction methods and next generation sequencing platforms96,97,98,99,100. The technology is optimised for each organism and application in order to select the most appropriate complexity reduction method (both the size of the representation and the fraction of a genome selected for assays). Based on testing several enzyme combinations for complexity reduction Diversity Arrays Technology Pty Ltd selected the PstI-MseI method for Stipa.
DNA samples were processed in digestion/ligation reactions as described previously97, but replacing a single PstI-compatible adaptor with two different adaptors corresponding to two different Restriction Enzyme (RE) overhangs. The PstI-compatible adapter was designed to include Illumina flowcell attachment sequence, sequencing primer sequence and "staggered", varying length barcode region, similar to the sequence previously reported33. Reverse adapter contained flowcell attachment region and MseI-compatible overhang sequence. Only "mixed fragments" (PstI-MseI) were effectively amplified by PCR using an initial denaturation step of 94 °C for 1 min, followed by 30 cycles with the following temperature profile: denaturation at 94 °C for 20 s, annealing at 58 °C for 30 s and extension at 72 °C for 45 s, with an additional final extension at 72 °C for 7 min. After PCR equimolar amounts of amplification products from each sample of the 96-well microtiter plate were bulked and applied to c-Bot (Illumina, USA) bridge PCR followed by sequencing on Hiseq2500 (Illumina, USA). The sequencing (single read) was run for 77 cycles.
Sequences generated from each lane were processed using proprietary DArT analytical pipelines. In the primary pipeline, the fastq files were first processed to filter away poor quality sequences, applying more stringent selection criteria to the barcode region compared to the rest of the sequence. In that way the assignments of the sequences to specific samples carried in the "barcode split" step were very reliable. Approximately 2.5 mln sequences per barcode/sample were identified and used in marker calling.
DArTseq data analysis
DArTseq produce two types of data: (1) co-dominant single nucleotide polymorphisms (SNPs) markers, and (2) dominant SilicoDArT markers that represent the presence or absence of restriction fragments. All molecular analyses with the DArTseq data (SNPs and SilicoDArT) sets were performed after filtering steps in the R-package dartR101 with the following parameters: (1) a scoring reproducibility of 100%, (2) at least 95% loci called (the respective DNA fragment had been identified (= called) in greater than 95% of all individuals), (3) monomorphic loci were removed, (4) SNPs that shared secondaries (had more than one sequence tag represented in the dataset) were randomly filtered out to keep only one random sequence tag.
Three approaches were used to analyse genetic structure of the studied taxa: (1) Principal Coordinates Analysis (PCoA), (2) fastSTRUCTURE analysis, and (3) Unweighted Pair Group Method with Arithmetic Mean (UPGMA). The PCoA analyses based on Euclidean distance matrices were performed using R-packages dartR and visualised by using ggplot2 to show the first two components, and plotly to illustrate the first three components. Genetic structure was then investigated using the fastSTRUCTURE software, which implements the Bayesian clustering algorithm STRUCTURE, assuming Hardy–Weinberg equilibrium between alleles, in a fast and resource-efficient manner102. A number of clusters (K-values) ranging from 2 to 10 were tested using the default convergence criterion of 10−6 and priors. The most likely K-value was estimated with the best choice function implemented in fastSTRUCTURE. In case of a range of K values, the true K was determined as a value between the estimates predicted by fastSTRUCTURE and based on what made most biological sense. The output matrices for the best K-values were reordered and plotted using an in-house R script in RStudio (Version 1.1.463)103. The threshold of 0.10 < q < 0.90 was applied as the most widely utilised measure for the assessment of hybridisation104,105,106,107. Contributions from each cluster in a range between 45 and 55% were considered as F1 hybrids, while first‐ and second‐generation backcrosses with one parent were considered at values 0.25 and 0.125, respectively108. The UPGMA cluster analyses based on Jaccard's distance matrices were performed using R-packages dartR and visualised with stats109.
Finally, the SilicoDArT tags were used to determine maternal inheritance of the putative hybrid Stipa × lazkovii. The trimmed sequences of the parental species S. krylovii and S. bungeana, and the rest of studied taxa were mapped onto chloroplast genomes of Stipa species and mitochondrions of specimens from the Poaceae family (Supplementary Table S3) by using Minimap2110. The final binary data matrix was used to generate a neighbor-joining tree (NJ) derived from Jaccard's genetic distances in the fingerprint analysis with missing data software v1.31 (FAMD)111 with a set of 10,000 bootstrap replicates. The resulting tree was visualised and edited using Figtree v1.4.4112.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding authors upon request.
References
Grant, V. Plant Speciation 2nd edn. (Columbia University Press, New York, 1981).
Abbott, R. J. Plant invasions, hybridization and the evolution of new plant taxa. Trends Ecol. Evol. 7, 401–405. https://doi.org/10.1016/0169-5347(92)90020-C (1992).
Rieseberg, L. H. Hybrid origins of plant species. Annu. Rev. Ecol. Syst. 28, 359–389. https://doi.org/10.1146/annurev.ecolsys.28.1.359 (1997).
Arnold, M. L. Evolution Through Genetic Exchange (Oxford University Press, Oxford, 2006).
Mallet, J. Hybrid speciation. Nature 446, 279–283. https://doi.org/10.1038/nature05706 (2007).
Abbott, R. J. et al. Hybridization and speciation. J Evol Biol 26, 229–246. https://doi.org/10.1111/j.1420-9101.2012.02599.x (2013).
Goulet, B. E., Roda, F. & Hopkins, R. Hybridization in plants: old ideas. New Techniques. Plant Physiol. 173, 65–78. https://doi.org/10.1104/pp.16.01340 (2017).
Rieseberg, L. H. & Wendel, J. F. In Hybrid zones and the evolutionary process (ed. Harrison, R. G.) 70–109 (Oxford University Press, Oxford, 1993).
Mallet, J. Hybridization as an invasion of the genome. Trends Ecol. Evol. 20, 229–237. https://doi.org/10.1016/j.tree.2005.02.010 (2005).
Hamilton, J. A. & Miller, J. M. Adaptive introgression as a resource for management and genetic conservation in a changing climate. Conserv. Biol. 30, 33–41. https://doi.org/10.1111/cobi.12574 (2015).
Suarez-Gonzalez, A., Lexer, C. & Cronk, Q. C. B. Adaptive introgression: a plant perspective. Biol. Lett. 14, 20170688. https://doi.org/10.1098/rsbl.2017.0688 (2018).
López-Caamal, A. & Tovar-Sánchez, E. Genetic, morphological, and chemical patterns of plant hybridization. Revista Chilena Hist. Nat. 87, 1–14. https://doi.org/10.1186/s40693-014-0016-0 (2014).
Abbott, R. J., Barton, N. H. & Good, J. M. Genomics of hybridization and its evolutionary consequences. Mol. Ecol. 25, 2325–2332. https://doi.org/10.1111/mec.13685 (2016).
Lepais, O. et al. Species relative abundance and direction of introgression in oaks. Mol. Ecol. 18, 2228–2242. https://doi.org/10.1111/j.1365-294X.2009.04137.x (2009).
Gómez, J. M., González-Megías, A., Lorite, J., Abdelaziz, M. & Perfectti, F. The silent extinction: climate change and the potential hybridization-mediated extinction of endemic high-mountain plants. Biodivers. Conserv. 24, 1843–1857. https://doi.org/10.1007/s10531-015-0909-5 (2015).
Mota, M. R., Pinheiro, F., Leal, B. S. S., Wendt, T. & Palma-Silva, C. The role of hybridization and introgression in maintaining species integrity and cohesion in naturally isolated inselberg bromeliad populations. Plant Biol. 21, 122–132. https://doi.org/10.1111/plb.12909 (2019).
Parisod, C., Definod, C., Sarr, A., Arrigo, N. & Felber, F. Genome-specific introgression between wheat and its wild relative Aegilops triuncialis. J. Evol. Biol. 26, 223–228. https://doi.org/10.1111/jeb.12040 (2013).
Cheng, H. et al. Frequent intra- and inter-species introgression shapes the landscape of genetic variation in bread wheat. Genome Biol. 20, 136. https://doi.org/10.1186/s13059-019-1744-x (2019).
Hufford, M. B. et al. The genomic signature of crop-wild introgression in maize. PLoS Genet. 9, e1003477. https://doi.org/10.1371/journal.pgen.1003477 (2013).
Gonzalez-Segovia, E. et al. Characterization of introgression from the teosinte Zea mays ssp. mexicana to Mexican highland maize. PeerJ 7, e6815. https://doi.org/10.7717/peerj.6815 (2019).
Xia, H. B., Wang, W., Xia, H., Zhao, W. & Lu, B. R. Conspecific crop-weed introgression influences evolution of weedy rice (Oryza sativa f. spontanea) across a geographical range. PLoS ONE 6, e16189. https://doi.org/10.1371/journal.pone.0016189 (2011).
Civán, P. & Brown, T. A. Role of genetic introgression during the evolution of cultivated rice (Oryza sativa L.). BMC Evol. Biol. 18, 57. https://doi.org/10.1186/s12862-018-1180-7 (2018).
Molnár-Láng, M. & Linc, G. Wheat-barley hybrids and introgression lines. In Alien Introgression in Wheat: Cytogenetics, Molecular Biology, and Genomics (eds Molnár-Láng, M. et al.) 315–345 (Springer, Berlin, 2015).
Pankin, A. & von Korff, M. Co-evolution of methods and thoughts in cereal domestication studies: a tale of barley (Hordeum vulgare). Curr. Opin. Plant Biol. 36, 15–21. https://doi.org/10.1016/j.pbi.2016.12.001 (2017).
Yan, H. et al. High-density marker profiling confirms ancestral genomes of Avena species and identifies D-genome chromosomes of hexaploid oat. Theor. Appl. Genet. 129, 2133. https://doi.org/10.1007/s00122-016-2762-7 (2016).
Liu, Q., Lin, L., Zhou, X., Peterson, P. M. & Wen, J. Unraveling the evolutionary dynamics of ancient and recent polyploidization events in Avena (Poaceae). Sci. Rep. 7, 41944. https://doi.org/10.1038/srep41944 (2017).
Martis, M. M. et al. Reticulate evolution of the rye genome. Plant Cell 25, 3685–3698. https://doi.org/10.1105/tpc.113.114553 (2013).
Hagenblad, J., Oliveira, H. R., Forsberg, N. E. G. & Leino, M. W. Geographical distribution of genetic diversity in Secale landrace and wild accessions. BMC Plant Biol. 16, 23. https://doi.org/10.1186/s12870-016-0710-y (2016).
Almaraj, V. A. & Balasundaram, N. On the taxonomy of the members of 'Saccharum complex’. Genet. Resour. Crop. Evol. 53, 35–41. https://doi.org/10.1007/s10722-004-0581-1 (2006).
Pachakkil, B. et al. Cytogenetic and agronomic characterization of intergeneric hybrids between Saccharum spp. hybrid and Erianthus arundinaceus. Sci. Rep. 9, 1748. https://doi.org/10.1038/s41598-018-38316-6 (2019).
Barnaud, A. et al. A weed-crop complex in sorghum: the dynamics of genetic diversity in a traditional farming system. Am. J. Bot. 96, 1869–1879. https://doi.org/10.3732/ajb.0800284 (2009).
Ohadi, S., Hodnett, G., Rooney, W. & Bagavathiannan, M. Gene flow and its consequences in Sorghum spp. Crit. Rev. Plant Sci. 36, 367–385. https://doi.org/10.1080/07352689.2018.1446813 (2018).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6, e19379. https://doi.org/10.1371/journal.pone.0019379 (2011).
Hamlin, J. A. & Arnold, M. L. Determining population structure and hybridization for two iris species. Ecol. Evol. 4, 743–755. https://doi.org/10.1002/ece3.964 (2014).
Qi, L. et al. Genotyping-by-sequencing uncovers the introgression alien segments associated with sclerotinia basal stalk rot resistance from wild species-I. Helianthus argophyllus and H. petiolaris. Frontiers in genetics 7, 219. https://doi.org/10.3389/fgene.2016.00219 (2016).
Schilling, M. P., Gompert, Z., Li, F. W., Windham, M. D. & Wolf, P. G. Admixture, evolution, and variation in reproductive isolation in the Boechera puberula clade. BMC Evol. Biol. 18, 61. https://doi.org/10.1186/s12862-018-1173-6 (2018).
Edet, O. U., Gorafi, Y. S. A., Nasuda, S. & Tsujimoto, H. DArTseq-based analysis of genomic relationships among species of tribe Triticeae. Sci. Rep. 8, 16397. https://doi.org/10.1038/s41598-018-34811-y (2018).
Hodkinson, T. R., Perdereau, A., Klaas, M., Cormican, P. & Barth, S. Genotyping by sequencing and plastome analysis finds high genetic variability and geographical structure in Dactylis glomerata L. in Northwest Europe despite lack of ploidy variation. Agronomy 9, 342. https://doi.org/10.3390/agronomy9070342 (2019).
Saarela, J. M. et al. A 250 plastome phylogeny of the grass family (Poaceae): Topological support under different data partitions. PeerJ 6, 4299. https://doi.org/10.7717/peerj.4299 (2018).
Soreng, R. J. et al. A worldwide phylogenetic classification of the Poaceae (Gramineae) II: An update and a comparison of two 2015 classifications. J. Syst. Evol. 55, 259–290. https://doi.org/10.1111/jse.12262 (2017).
Tzvelev, N. N. Notulae de tribu Stipeae Dum. (fam. Poaceae) in URSS. Novosti Sistematiki Vyssih Rastenij 11, 4–20 (1974).
Nobis, M. Taxonomic revision of the Central Asiatic Stipa tianschanica complex (Poaceae) with particular reference to the epidermal micromorphology of the lemma. Folia Geobot. 49, 283–308. https://doi.org/10.1007/s12224-013-9164-2 (2014).
Nobis, M., Gudkova, P., Nowak, A., Sawicki, J. & Nobis, A. A revision of the genus Stipa (Poaceae) in Middle Asia, including a key to species identification, an annotated checklist and phytogeographic analysis. Ann. Mo. Bot. Gard. 105, 1–63. https://doi.org/10.3417/2019378 (2020).
Hamasha, H. R., von Hagen, K. B. & Röser, M. Stipa (Poaceae) and allies in the Old World: molecular phylogenetics realigns genus circumscription and gives evidence on the origin of American and Australian lineages. Plant Syst. Evol. 298, 351–367. https://doi.org/10.1007/s00606-011-0549-5 (2012).
Romaschenko, K. et al. Systematics and evolution of the needle grasses (Poaceae: Pooideae: Stipeae) based on analysis of multiple chloroplast loci, ITS, and lemma micromorphology. Taxon 61, 18–44. https://doi.org/10.1002/tax.611002 (2012).
Kellogg, E. A. Subfamily Pooideae. In The families and genera of vascular plants (ed. Kubitzki, K.) 199–229 (Springer, Berlin, 2015).
Yunatov, A. A. Main patterns of the vegetation cover of the Mongolian People’s Republic. Proc. Mongolian Commission 39, 233 (1950).
Dashnyam, B. Flora and vegetation of Eastern Mongolia 78–115 (Mongolian Academy of Sciences, Mongolia, 1974).
Lavrenko, E. M., Karamasheva, Z. V. & Nikulina, R. I. Eurasian steppe 143 (Nauka, Nauka, 1991).
Nowak, A., Nowak, S., Nobis, A. & Nobis, M. Vegetation of feather grass steppes in the western Pamir Alai Mountains (Tajikistan, Middle Asia). Phytocoenologia 46, 295–315. https://doi.org/10.1127/phyto/2016/0145 (2016).
Danzhalova, E. V. et al. Indicators of Pasture Digression in Steppe Ecosystems of Mongolia. Explor. into Biol. Resour. Mong. 12, 297–306 (2012).
Yang, Y. Q. et al. Transcriptome analysis reveals diversified adaptation of Stipa purpurea along a drought gradient on the Tibetan Plateau. Funct. Integr. Genomics 15, 295–307. https://doi.org/10.1007/s10142-014-0419-7 (2015).
Lv, X., He, Q. & Zhou, G. Contrasting responses of steppe Stipa ssp. to warming and precipitation variability. Ecol. Evol. 9, 9061–9075. https://doi.org/10.1002/ece3.5452 (2019).
Schubert, M., Grønvold, L., Sandve, S. R., Hvidsten, T. R. & Fjellheim, S. Evolution of cold acclimation and its role in niche transition in the temperate grass subfamily pooideae. Plant Physiol. 180, 404–419. https://doi.org/10.1104/pp.18.01448 (2019).
Maevsky, V. V. & Amerkhanov, H. H. The note of Poaceae species from former USSR flora, recommended as fodder for agricultural production. Bull. Bot. Garden Saratov State Univ. 6, 80–83 (2007).
Brunetti, G., Soler-Rovira, P., Farrag, K. & Senesi, N. Tolerance and accumulation of heavy metals by wild plant species grown in contaminated soils in Apulia region, Southern Italy. Plant Soil. 318, 285–298. https://doi.org/10.1007/s11104-008-9838-3 (2009).
Smirnov, P. A. Stiparum Armeniae minus cognitarum descriptiones. Byull. Moskovsk. Obshch. Isp. Prir. Otd. Biol. 75, 113–115 (1970).
Tzvelev, N. N. Zlaki USSR (Nauka Press, Nauka, 1976).
Kotukhov, Y. A. Synopsis of feather grass (Stipa L.) and false needlegrasses (Ptilagrostis Griseb.) the eastern of Kazakhstan (The Kazakh Altai, Zaisan valley and Prialtayskie ranges). Botanicheskie Issledovaniya Sibiri i Kazakhstana 8, 3–16 (2002).
Nobis, M. Taxonomic revision of the Stipa lipskyi group (Poaceae: Stipa section Smirnovia) in the Pamir Alai and Tian-Shan Mountains. Plant Syst. Evol. 299, 1307–1354. https://doi.org/10.1007/s00606-013-0799-5 (2013).
Nobis, M. et al. Hybridisation, introgression events and cryptic speciation in Stipa (Poaceae): a case study of the Stipa heptapotamica hybrid-complex. Perspect. Plant Ecol. Evol. Syst. 39, 125457. https://doi.org/10.1016/j.ppees.2019.05.001 (2019).
Nobis, M. & Gudkova, P. D. Taxonomic notes on feather grasses (Poaceae: Stipa) from eastern Kazakhstan with typification of seven names and one new combination. Phytotaxa 245, 31–42. https://doi.org/10.11646/phytotaxa.245.1.3 (2016).
Nobis, M. et al. Stipa ×fallax (Poaceae: Pooideae: Stipeae), a new natural hybrid from Tajikistan, and a new combination in Stipa drobovii. Phytotaxa 303, 141–154. https://doi.org/10.11646/phytotaxa.303.2.4 (2017).
Tzvelev, N. N. Some notes on the grasses (Poaceae) of the Caucasus. Botanical Zhurnal 78, 83–95 (1993).
Krawczyk, K., Nobis, M., Nowak, A., Szczecińska, M. & Sawicki, J. Phylogenetic implications of nuclear rRNA IGS variation in Stipa L. (Poaceae). Sci. Rep. 7, 11506. https://doi.org/10.1038/s41598-017-11804-x (2017).
Krawczyk, K., Nobis, M., Myszczyński, K., Klichowska, E. & Sawicki, J. Plastid superbarcodes as a tool for species discrimination in feather grasses (Poaceae: Stipa). Sci. Rep. 8, 1924. https://doi.org/10.1038/s41598-018-20399-w (2018).
Gudkova, P. D., Olonova, M. V. & Feoktisov, D. S. The comparison of ecologo-climatic niches of two species feather grass Stipa sareptana A.K. Becker and S. krylovii Roshev. (Poaceae). Ukrainian J. Ecol. 7, 263–269. https://doi.org/10.15421/2017_115 (2017).
Lu, S. L. & Wu, Z. L. On the geographical distribution of the genus Stipa L., China. Acta Phytotaxon. Sin. 34, 242–253 (1996).
Wu, Z. L. & Phillips, S. M. Stipa. In Flora of China (eds Wu, Z. Y. et al.) 196–203 (Science Press, Beijing, 2006).
Olonova, M. V., Barkworth, M. E. & Gudkova, P. D. Lemma micromorphology and the systematics of Siberian species of Stipa (Poaceae). Nordic J. Bot. 34, 319–328. https://doi.org/10.1111/njb.00881 (2016).
Barkworth, M. E. & Everett, J. Evolution in the Stipeae: identification and relationships of its monophyletic taxa. In Grass systematics and evolution (eds Soderstrom, T. R. et al.) 251–264 (Smithsonian Institution Press, Washington, 1987).
Xia, L. et al. DArT for high-throughput genotyping of cassava (Manihot esculenta) and its wild relatives. Theor. Appl. Genetics 110, 1092–1098. https://doi.org/10.1007/s00122-005-1937-4 (2005).
Akbari, M. et al. Diversity arrays technology (DArT) for high-throughput profiling of the hexaploid wheat genome. Theor. Appl. Genetics 113, 1409–1420. https://doi.org/10.1007/s00122-006-0365-4 (2006).
Mace, E. S. et al. DArT markers: diversity analyses and mapping in Sorghum bicolor. BMC Genom. 9, 26. https://doi.org/10.1186/1471-2164-9-26 (2008).
Simko, I., Eujayl, I. & van Hintum, T. J. Empirical evaluation of DArT, SNP, and SSR marker-systems for genotyping, clustering, and assigning sugar beet hybrid varieties into populations. Plant Sci. 184, 54–62. https://doi.org/10.1016/j.plantsci.2011.12.009 (2012).
Alam, M., Neal, J., O’Connor, K., Kilian, A. & Topp, B. Ultra-high-throughput DArTseq-based silicoDArT and SNP markers for genomic studies in macadamia. PLoS ONE 13, e0203465. https://doi.org/10.1371/journal.pone.0203465 (2018).
Abu Zaitoun, S. Y., Jamous, R. M., Shtaya, M. J., Eid, I. S. & Ali-Shtayeh, M. S. Characterizing Palestinian snake melon (Cucumis melo var flexuosus) germplasm diversity and structure using SNP and DArTseq markers. BMC Plant Biol. 18, 246. https://doi.org/10.1186/s12870-018-1475-2 (2018).
Bello, E. B. et al. Genetic diversity analysis of selected sugarcane (Saccharum spp. hybrids) varieties using DArT-Seq technology. Philippine J. Sci. 148, 103–114 (2019).
Rutherford, S. et al. Speciation in the presence of gene flow: population genomics of closely related and diverging Eucalyptus species. Heredity 121, 126–141. https://doi.org/10.1038/s41437-018-0073-2 (2018).
Ivanizs, L. et al. Unlocking the genetic diversity and population structure of a wild gene source of wheat, Aegilops biuncialis Vis., and its relationship with the heading time. Front Plant Sci. 10, 1531. https://doi.org/10.3389/fpls.2019.01531 (2019).
Yu, J., Jing, Z. B. & Cheng, J. M. Genetic diversity and population structure of Stipa bungeana, an endemic species in Loess Plateau of China, revealed using combined ISSR and SRAP markers. Genet. Mol. Res. 13, 1097–1108. https://doi.org/10.4238/2014.February.20.11 (2014).
Kopylov-Guskov, Y. O. & Kramina, T. E. Investigating of Stipa ucrainica и Stipa zalesskii (Poaceae) from Rostov Oblast using morphological and ISSR analyses. Bull. Moscow Soc. Nat. Biol. Ser. 119, 46–53 (2014).
Boussaid, M., Benito, C., Harche, M., Naranjo, T. & Zedek, M. Genetic variation in natural populations of Stipa tenacissima from Algeria. Biochem. Genet. 48, 857–872. https://doi.org/10.1007/s10528-010-9367-7 (2010).
Zhao, N. X., Gao, Y. B., Wang, J. L. & Ren, A. Z. Genetic diversity and population differentiation of the dominant species Stipa krylovii in the Inner Mongolia Steppe. Biochem. Genet. 44, 513–526. https://doi.org/10.1007/s10528-006-9054-x (2006).
Zhao, N. X., Gao, Y. B., Wang, J. L., Ren, A. Z. & Xu, H. RAPD diversity of Stipa grandis populations and its association with some ecological factors. Acta Ecol. Sin. 26, 1312–1319. https://doi.org/10.1016/S1872-2032(06)60023-1 (2006).
Thiers, B. Index Herbariorum: a Global Directory of Public Herbaria and Associated Staff. New York Botanical Garden’s Virtual Herbarium https://sweetgumnybg.org/science/ih (2018).
WCSP. World Checklists of Selected Plant Families https://wcsp.science.kew.org/home.do (2019).
Sokal, R. R. & Sneath, P. H. A. Principles of Numerical Taxonomy (W.H. Freeman, San Francisco, 1963).
Korkmaz, S., Goksuluk, D. & Zararsiz, G. Mvn: an r package for assessing multivariate normality. R J. 6, 151–162. https://doi.org/10.32614/rj-2014-031 (2014).
Pagès, J. Analyse Factorielle de Donnees Mixtes. Revue Statistique Appliquee 4, 93–111 (2004).
Husson, F., Josse, J., Le, S. & Mazet, J. FactoMineR: multivariate exploratory data analysis and data mining https://factominer.free.fr (2015).
Cattell, R. B. The scree test for the number of factors. Multivariate Behav Res. 1, 245–276 (1966).
Kassambara, A. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses https://rdrr.io/cran/factoextra (2015).
Sievert, C. et al. plotly: create interactive web graphics via 'plotly.js' https://rdrr.io/cran/plotly (2017).
Wickham, H. ggplot2: elegant graphics for data analysis (Springer, New York, https://ggplot2.tidyverse.org (2016).
Sansaloni, C. et al. Diversity arrays technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc. 5, 54. https://doi.org/10.1186/1753-6561-5-S7-P54 (2011).
Kilian, A. et al. Diversity arrays technology: a generic genome profiling technology on open platforms. Methods Mol. Biol. 888, 67–89. https://doi.org/10.1007/978-1-61779-870-2_5 (2012).
Courtois, B. et al. Genome-wide association mapping of root traits in a Japonica rice panel. PLoS ONE 8, e78037. https://doi.org/10.1371/journal.pone.0078037 (2013).
Cruz, V. M. V., Kilian, A. & Dierig, D. A. Development of DArT marker platforms and genetic diversity assessment of the US collection of the new oilseed crop lesquerella and related species. PLoS ONE 8, e64062. https://doi.org/10.1371/journal.pone.0064062 (2013).
Raman, H. et al. Genome-wide delineation of natural variation for pod shatter resistance in Brassica napus. PLoS ONE 9, e101673. https://doi.org/10.1371/journal.pone.0101673 (2014).
Gruber, B., Georges, A., Berry, O. & Unmack, P. dartR: importing and analysing snp and Silicodart data generated by genome-wide restriction fragment analysis https://cran.rproject.org/web/packages/dartR/index.html (2017).
Raj, A., Stephens, M. & Pritchard, J. K. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197, 573–589. https://doi.org/10.1534/genetics.114.164350 (2014).
RStudio Team. RStudio: Integrated Development for R www.rstudio.com (2016).
Burgarella, C. et al. Detection of hybrids in nature: application to oaks (Quercus suber and Q. ilex). Heredity 102, 442–452. https://doi.org/10.1038/hdy.2009.8 (2009).
Winkler, K. A., Pamminger-Lahnsteiner, B., Wanzenböck, J. & Weiss, S. Hybridization and restricted gene flow between native and introduced stocks of Alpine whitefish (Coregonus sp.) across multiple environments. Mol. Ecol. 20, 456–472. https://doi.org/10.1111/j.1365-294X.2010.04961.x (2011).
Dierking, J. et al. Anthropogenic hybridization between endangered migratory and commercially harvested stationary whitefish taxa (Coregonus spp.). Evol. Appl. 7, 1068–1083. https://doi.org/10.1111/eva.12166 (2014).
Malde, K. et al. Whole genome resequencing reveals diagnostic markers for investigating global migration and hybridization between minke whale species. BMC Genom. 18, 76. https://doi.org/10.1186/s12864-016-3416-5 (2017).
Beugin, M. P., Gayet, T., Pontier, D., Devillard, S. & Jombart, T. A fast likelihood solution to the genetic clustering problem. Methods Ecol. Evol. 9, 1006–1016. https://doi.org/10.1111/2041-210X.12968 (2018).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. https://doi.org/10.1093/bioinformatics/bty191 (2018).
Schlüter, P. M. & Harris, S. A. Analysis of multilocus fingerprinting data sets containing missing data. Mol. Ecol. Notes 6, 569–572. https://doi.org/10.1111/j.1471-8286.2006.01225.x (2006).
Rambaut, A. Figtree v1.4.4 https://tree.bio.ed.ac.uk/software/figtree (2018).
Acknowledgements
The study was supported by a RSF grant (project no. 19-74-10067); partially by a DS grant of the Jagiellonian University (DS/MND/WB/IB/1/2018) and by the National Science Centre, Poland (grant no. 2018/29/B/NZ9/00313). The authors would like to thank Dr. Ewelina Klichowska and Anna Wróbel (Jagiellonian University, Kraków) for their help in conducting the field studies. We would like to express our gratitude to Daria Kareva (Tomsk State University, Russia) for preparing the line drawing of the nothospecies. Finally, we thank two anonymous reviewers for providing valuable comments on the manuscript.
Author information
Authors and Affiliations
Contributions
E.B. and M.N. designed and supervised the study. M.N. and A.N. performed the field studies. M.N. collected the specimens for the morphological and molecular analyses and performed the micromorphological analysis on SEM. E.B. and P.D.G. performed the macromorphological analysis. E.B. performed the molecular analysis. E.B. and M.N. analysed all the data and wrote the manuscript with the other authors' helps. All authors revised the draft, provided comments, and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baiakhmetov, E., Nowak, A., Gudkova, P.D. et al. Morphological and genome-wide evidence for natural hybridisation within the genus Stipa (Poaceae). Sci Rep 10, 13803 (2020). https://doi.org/10.1038/s41598-020-70582-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-70582-1
This article is cited by
-
Insight into the phylogeny and responses of species from the genus Sergia (Campanulaceae) to the climate changes predicted for the Mountains of Central Asia (a world biodiversity hotspot)
BMC Plant Biology (2024)
-
Insights into adaptive evolution of plastomes in Stipa L. (Poaceae)
BMC Plant Biology (2022)
-
Evidence for extensive hybridisation and past introgression events in feather grasses using genome-wide SNP genotyping
BMC Plant Biology (2021)
-
The first draft genome of feather grasses using SMRT sequencing and its implications in molecular studies of Stipa
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
