
Abstract
Measures of microbial growth, used as indicators of cellular stress, are sometimes quantified at a single time-point. In reality, these measurements are compound representations of length of lag, exponential growth-rate, and other factors. Here, we investigate whether length of lag phase can act as a proxy for stress, using a number of model systems (Aspergillus penicillioides; Bacillus subtilis; Escherichia coli; Eurotium amstelodami, E. echinulatum, E. halophilicum, and E. repens; Mrakia frigida; Saccharomyces cerevisiae; Xerochrysium xerophilum; Xeromyces bisporus) exposed to mechanistically distinct types of cellular stress including low water activity, other solute-induced stresses, and dehydration-rehydration cycles. Lag phase was neither proportional to germination rate for X. bisporus (FRR3443) in glycerol-supplemented media (r2 = 0.012), nor to exponential growth-rates for other microbes. In some cases, growth-rates varied greatly with stressor concentration even when lag remained constant. By contrast, there were strong correlations for B. subtilis in media supplemented with polyethylene-glycol 6000 or 600 (r2 = 0.925 and 0.961), and for other microbial species. We also analysed data from independent studies of food-spoilage fungi under glycerol stress (Aspergillus aculeatinus and A. sclerotiicarbonarius); mesophilic/psychrotolerant bacteria under diverse, solute-induced stresses (Brochothrix thermosphacta, Enterococcus faecalis, Pseudomonas fluorescens, Salmonella typhimurium, Staphylococcus aureus); and fungal enzymes under acid-stress (Terfezia claveryi lipoxygenase and Agaricus bisporus tyrosinase). These datasets also exhibited diversity, with some strong- and moderate correlations between length of lag and exponential growth-rates; and sometimes none. In conclusion, lag phase is not a reliable measure of stress because length of lag and growth-rate inhibition are sometimes highly correlated, and sometimes not at all.
Similar content being viewed by others

Introduction
Chemical reactions and thermodynamic and biological processes often experience a lag period prior to reaching their maximum rate. This phenomenon that can be observed at various levels; thermodynamic processes (e.g. thermal lag), chemical reactions, biochemical activities1, cellular physiology2, microbial growth kinetics, and ecosystem functions3. The term lag, used in English since the early 14th century, has been applied to biological processes since at least the 1680s4. In the context of microbial growth kinetics, lag, once known as ‘latency’, was studied since the 1800s, including work of Louis Pasteur5. Despite this long pedigree of research into this phenomenon, however, some aspects of the biology and application of the lag phase are not yet resolved.
Accurate assessments of microbial growth must account for the various growth processes, including: mycelial extension, cell division within planktonic populations, increases in the mass of individual cells, accumulation of compatible solutes or other endogenous reserves, cell elongation or growth, sporulation, germination, biofilm development, other forms of differentiation (e.g., primordium- and fruiting-body formation), and invasion and colonization of new habitats. Regardless of this, a phase of zero- or near-zero growth commonly precedes exponential growth. The duration of this ‘lag’ phase is by definition slightly less than that of the period preceding exponential growth (see below). During early studies, lag phase was quantified by different means6, but is usually now determined according to the method of Lodge and Hinshelwood (1943)7.
Microbial cells are active during all or most of the lag phase, and the physiological and molecular changes during this period have been well-characterized8,9,10. Lag-phase metabolism may include the activation of signalling pathways11 and transcriptional changes leading to the upregulation of protein assembly, nucleotide metabolism, lipopolysaccharide biosynthesis, respiration, and other processes which are needed for differentiation and multiplication12. Collectively, such activities lead to cell division, and can facilitate exponential growth.
In relation to both laboratory-based experiments and ecological studies carried out in situ, lag phase may impact the outcomes of studies of stress tolerance, biomass yield, habitability, vigour, competitive ability, and other parameters. Cellular stress occurs in all types of microbial system; furthermore, microbes can never be entirely stress-free13. There is an element of autonomy in relation to essential processes that occur prior to germination proper, the processes of breakdown of compatible solutes and RNA degradation14. This said, cells may also experience environmental challenges that necessitate more-extensive periods of adjustment before growth can commence and so lengthen the lag phase. These can include various (mechanistically distinct) stresses. By way of example, a minimal lag phase for germination of conidia may span several hours, whereas stressors and/or temperature may lengthen this to days, months, or even longer15,16,17. The same stress parameter(s) may also constrain rates of cell division if and when exponential growth commences18,19. In other words, cellular stress may sometimes prolong lag-phase and then also cause low rates of exponential growth for a given microbial population19,20.
Single time-point determinations of optical density, cell number, dry weight and fresh biomass are therefore used to quantify stress for diverse microbial systems21,22,23,24,25,26,27. The same methodology is also used during studies of stress in plant species28,29,30,31. However, measurements obtained this way actually represent multiple parameters such as lag phase, size of inoculum, the proportion of viable cells, heterogeneity of growth rates within the population, and the rates of subsequent (exponential) growth. Lag phase is often assumed to correlate with cellular stress32,33,34,35,36,37 and has even been used as a direct measure of product-induced inhibition of growth38,39,40. Historically, cellular stress has most-commonly been defined according to empirically determined constraints on exponential growth rate25,41,42,43,44,45,46. During exponential growth under biophysical or physicochemical constraints, growth rate acts as a direct measure of stress on the system13. The validity of using single time-point determinations of biomass, colony formation or germination to quantify stress, however, depends on whether lag phase is proportional to the rate of exponential growth. Thus far, there is a paucity of information on whether lag phase is a meaningful as a proxy for stress for different types of growth phenotype (e.g. planktonic cell division versus radial growth of filamentous colonies), different phylogenetic taxa, and/or mechanistically distinct types of cellular stress.
The current study was carried out to test whether length of the microbial lag phase is a reliable measure of cellular stress. All microbes are aqueous systems that function in essentially aqueous environments, so the primary type of stress that can impact all microbial life is solute-induced stress. Solutes cause stress in microbial cells via various mechanisms: e.g. water activity reduction, osmotically (turgor-induced changes), and/or chaotropicity47. Hyper-osmotic stress dehydrates cells, and chaotropicity can cause lipid peroxidation which, in turn, induces oxidative damage42. Solute-induced and water-related stresses, arguably the most-potent that shape ecosystem and biosphere function, were therefore used as the basis for the study system. We used a range of microbial systems (and enzymes from two microbes) as models from different domains of life, that encompassed different processes (growth of hyphal cultures and planktonic cultures, germination of propagules), and ecophysiological distinct groups (e.g. mesophiles, xerophiles) (Table S1). The models were: spore germination of fungal xerophiles (12 strains) at extreme water-activities (0.741 to 0.585), Saccharomyces cerevisiae subjected to dehydration-rehydration stress, planktonic growth of Bacillus subtilis and Escherichia coli, and colony formation of the psychrophilic yeast M. frigida under mechanistically diverse solute-induced stresses. The fungal xerophiles A. penicillioides, Eurotium amstelodami, Eurotium echinulatum, Eurotium halophilicum, Eurotium repens, Xerochrysium xerophilum and Xeromyces bisporus represent the extreme fringe of the microbial biosphere in terms of their ability to function at low water-activity48,49; S. cerevisiae is important for bioethanol production and in food and other applications; the soil bacterium B. subtillis is an important research model and factory of choice for various white biotechnologies; and E. coli is a pathogen and important research model. We also analysed datasets from independent studies of microbial and enzyme systems: mycelial extension of mesophilic fungi (Aspergillus aculeatinus and Aspergillus sclerotiicarbonarius under glycerol stress), planktonic growth of mesophilic and psychrotolerant bacteria (Enterococcus faecalis, Salmonella typhimurium and Staphylococcus aureus, and Brochothrix thermosphacta and Pseudomonas fluorescens, respectively, under glycerol-, NaCl- and sucrose-induced stresses); and catalytic activity of fungal lipoxygenase and tyrosinase under acid stress. These were selected on the basis that Aspergillus aculeatinus, A. sclerotiicarbonarius, B. thermosphacta, E. faecalis, P. fluorescens, S. typhimurium and S. aureus are food-spoilage microbes; and the enzyme systems which are pertinent to food quality and/or development of human diseases. Each of the microbes used in the experiments, and those used for meta-analyses, can be exposed to dehydration-rehydration and/or extreme, solute-induced stresses in their natural habitats and/or anthropogenic systems. The specific aims of the current study were to: (i) determine windows of tolerance to solute- and water-induced stresses; (ii) determine whether there is a relationship between length of lag phase and exponential growth rates under these stresses for diverse model microbes; and (iii) consider the implications for studies of microbial stress biology.
Results and discussion
Windows of stress tolerance
Whereas some types of extremophilic Bacteria and Archaea are more stress-tolerant than their eukaryote counterparts, fungi are generally more xerophilic than prokaryotes, with the exception of extreme bacterial and archaeal halophiles17,18,50. The biotic windows for growth and germination of model microbes used for experiments in the current study are shown in Table S2. Xerophile germination occurred at the lowest water-activity (0.637) on malt extract yeast extract phosphate agar (MYPiA) supplemented with glycerol +sucrose, for X. bisporus strains (Fig. S1; Table S3). The most-xerophilic fungus known, however, is A. penicillioides strain JH06THJ which germinated down to 0.640 water activity in the current study (Fig. S1; Table S3), but at water activities down to 0.585 under other environmental conditions17. Water-activity reduction is the primary mechanism by which glycerol induces cellular stress at relatively moderate concentrations47, but chaotropicity becomes the limiting parameter when glycerol is present at concentrations of > 5 M51,52.
The highest glycerol concentration at which B. subtilis was capable of growth (i.e. 2.71 M; Fig. S2a) corresponded to a water activity of 0.941 (Table S4). The window for growth of B. subtilis on glycerol-supplemented media (∼1 to 0.941) is consistent with the limits of glycerol tolerance established for B. subtilis and closely related strains in earlier studies18,53,54. Bacillus subtilis cultures were also grown under stresses induced by ionic osmolytes (ammonium sulphate, NaCl); non-ionic osmolytes (betaine, polyethylene glycol 600, glucose, proline, sucrose); chaotropic, osmotically-active stressors (guanidine hydrochloride, MgCl2); a chaotropic non-osmotic stressor (urea); and matric forces (polyethylene glycol 6000) (Figs. 1 and S2, data for NaCl- and urea-supplemented media were from Williams, 2010)55. For S. cerevisiae strains 14 and 77, the majority of cells survived throughout the dehydration period, irrespective of the subsequent rehydration treatment (Figs. S3 and S4). For M. frigida, growth occured at 1.7 °C in media supplemented with sucrose, glucose, glycerol, NaCl, or MgCl2 and combined solute-treatments: glycerol + sucrose + KCl + NaCl and glycerol + sucrose + KCl + NaCl but there was considerable variation between replicates (Figs. 2 and S5; Table S5).

Length of lag phase versus maximum rate of growth for Bacillus subtilis 168 in Belitzki minimal medium broth, supplemented with a range of stressors. Media were supplemented with: (a) betaine at 2.56 (♦), 2.13 (■), 1.71 (▲), 1.28 (×), 0.85 (−) and 0.43 M (●), and without betaine (control; +); (b) guanidine hydrochloride at 150 (♦), 125 (■), 100 (▲), 75 (×), 50 (−) and 25 nM (●), and without guanidine hydrochloride (control; +); (c) polyethylene glycol 6000 at 50 (♦), 42 (■), 33 (▲), 25 (×), 17 (−) and 8 nM (●) and without polyethylene glycol 6000 (control; +) and (d) polyethylene glycol 600 at 400 (♦), 333 (■), 267 (▲), 200 (×), 133 (−) and 67 nM (●), and without polyethylene glycol 600 (control; +). Data were obtained from Fig. S7 (see Experimental procedures), and trend lines showing a strong correlation between data are indicated by a solid black line (c,d), while lines fitted which indicated inconsistency between lag and growth rates (i.e. no correlation was observed) are shown as a dashed grey line (a,b).

Growth curves for Mrakia frigida (DSM 70883) on malt-extract, yeast-extract phosphate agar (MYPiA) at 1.7 °C, supplemented with a range of stressors: (a) glycerol (1.1 M); (b) glucose (1.7 M); (c) sucrose (0.7 M); and (d) combined solute-treatment 1 (10% glycerol, 10% sucrose, 1% KCl plus 5% NaCl [w/v]). Data were obtained using Spot Test Assays (see Experimental procedures). For each treatment, there were six replicates; these are shown in red, blue, orange, green, purple, and turquoise on each plot.
Escherichia coli BL21 (syn. DE3) was exposed to a range of stressors including ethanol, butanol and urea at concentrations of up to 850 mM, 120 mM, and 837 mM respectively, and the bacterium was able to grow at all concentrations tested, regardless of the stressor. Extrapolations indicate theoretical growth windows of 0 to 931 and 0 to >853 mM ethanol, and urea, respectively (Fig. S6). These upper-concentration limits are in the same concentration range as those established for E. coli strain BL21 (syn. DE3) in the presence of ethanol56 or butanol57 and E. coli strain 31 in the presence of urea58.
During the current study, we exposed each of the model microbes to a range of stresses/stressors. This revealed considerable variation in the relationship (or lack of relationship) between lag and growth rates for each individual microbe, depending on stressor. It also revealed that the same stressors do not necessarily give the same result for different microbes. For instance, r2 values for aspergilli and Bacillus subtilis on glycerol-supplemented media ranged from 0.020 to 0.972 (Figs. 3 and S2a). Whereas there have been earlier studies in which an individual microbial strain has been exposed to a number of mechanistically distinct solute-induced stresses59,60,61,62, tolerance to six or more individual solute stressors has only been studied for a relatively small number of microbial strains. For fungi, these belong to the following species: the extreme xerophile Aspergillus wentii47; mesophilic ascomycetes Aspergillus oryzae63; Saccharomyces cerevisiae32,64; fission yeasts Schizosaccharomyces japonicas, Schizosaccharomyces malidevorans, Schizosaccharomyces octosporus, Schizosaccharo-myces pombe, and Schizosaccharomyces slooffiae65; and the entomopathogens Beauveria bassiana, Metarhizium anisopliae, and Paecilomyces farinosus66,67,68. For bacteria: Bacillus subtilis (current study); the solvent-tolerant Pseudomonas putida32,69,70,71; the environmentally tenacious Mycobacterium parascrofulaceum and Mycobacterium smegmatis72; and psychrophiles: Glaciecola punicea, Psychrobacter glacincola, Psychrobacter urativorans, and Sporosarcina psychrophilia73; the xerotolerant and acid-tolerant bacterium Gluconacetobacter diazotrophicus74 and the halotolerant Ochrobactrum sp.75. The bacteria that were used in other studies are generally robust, stress-tolerant species, yet B. subtilis (168) exhibited comparable solute-tolerances to many of them. For instance, Glaciecola punicea, Psychrobacter glacincola, Psychrobacter urativorans, and the mycobacteria could grow up to ≥1000 mM MgCl2, and the limit for B. subtilis is >1710 mM (Fig. S2d). Similarly, the most-NaCl tolerant of the bacteria studied previously (M. parascrofulaceum and M. smegmatis) which can grow up to ≥1710 mM NaCl, was only able to tolerate 50% more NaCl than B. subtilis 168 (upper limit = 1140 mM55). Pseudomonas putida and the mycobacteria (M. parascrofulaceum and M. smegmatis) grow up to 2120 and 4792 mM glycerol, respectively, and the tolerance limit for B. subtilis is >2710 mM (Fig. S2a), and all these bacteria have a comparable upper limit for urea; i.e. 950, 832, and 755 mM, respectively32,55,72. The water-activity values at which the xerophiles under study can germinate are considerably lower than the values at which mycelial growth or spore germination ceases (i.e. in the range 0.880–0.920) for non-xerophilic species such as Beauveria bassiana, Metarhizium anisopliae, Paecilomyces farinosus, S. cerevisiae and A. oryzae63,76,77,78.

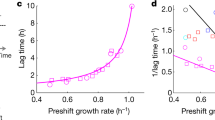
Length of lag phase versus maximum rate of mycelial extension for three Aspergillus strains on Robusta coffee-meal extract agar (CMEA) supplemented with glycerol: (a) Aspergillus sclerotiicarbonarius CBS 121853 at 30 °C; (b) Aspergillus aculeatinus CBS 121872 at 35 °C; (c) A. aculeatinus CBS 121872 at 30 °C; (d) and A. aculeatinus CBS 121875 at 30 °C. Media were adjusted, using glycerol (23, 9.2, 2.7 g l−1), to 0.900 (♦), 0.950 (▲) and 0.980 (■) water activity, respectively (primary data were taken from Akbar and Magan80, see Experimental procedures). Control media (no glycerol added) had a water activity of 0.990 (+). In the original experiment, three replicates of each water-activity treatment were incubated at 25, 30 and 35 °C for 9 days (Akbar and Magan80). Trend lines showing a strong correlation between data are indicated by a solid black line (c,d), while dashed grey lines indicate inconsistency between lag and growth rates (i.e. no correlation was observed) (a,b).
Lag phase can sometimes vary independently of maximum rates of germination/growth
There was no correlation between lag phase and maximum rates of germination/growth for some of the fungal germination- and bacterial growth datasets. In general, length of lag phase increased progressively at lower water-activities for X. bisporus FRR 3443 (Fig. S1b), but maximum rates of germination did not decline progressively regardless of type of stressor/stressor combination (Figs. 4b and S1b; maximum gradients of the linear trend lines are shown in Table S6). Similarly, there was inconsistency between length of lag and the maximum growth rates for the B. subtilis 168 cultures in guanidine hydrochloride- and betaine-supplemented media (Figs. 2a,b and S7a,b). For these xerophile strains and B. subtilis cultures, plots of lag-phase versus germination/growth rates were scattered, and/or the line of best fit was approximately horizontal. The r2 value for plots of B. subtilis growth in betaine- and guanidine hydrochloride-supplemented media were 0.232 and 0.335, respectively (Fig. 1a,b), and for X. bisporus (FRR 3443) and A. penicillioides JH06THH, r2 values were only 0.012 and 0.103, respectively (Fig. 4a,b; Table S6). Other xerophiles for which the lag phase and germination rates did not correlate were X. bisporus FRR 0025 and E. echinulatum FRR 5040 (r2 = 0.208 and 0.298, respectively; Fig. S8g,d). The psychrophilic yeast M. frigida DSM 70883 also revealed a disconnect between lag-phase and growth rates for 10% w/v glycerol- and 30% w/v glucose-supplemented media (r2 = 0.144 and 0.415, respectively; Fig. S9c,d). For diverse microbes, attempts to fit trend lines to a number of plots (for diverse microbes) yielded lines that are close to horizontal (Figs. 3a, 4b, 5b, 6b and 7a). For instance, the gradients for trend lines of A. sclerotiicarbonarius in glycerol-supplemented media over a range of temperature (Fig. 3a) and water activity (Fig. 7a) were −0.395 and 0.0907, respectively (Table S6). Similarly, for S. cerevisiae that had been dehydrated and rehydrated rapidly in sterile distilled water or with a 1 M xylitol solution at 23 °C, the gradient of the trend line for lag and maximum growth rates was −1.31 (Fig. 5b; Table S6). A horizontal trend line shows that length of lag phase does not change even though maximum rate of germination varies. This inconsistency indicates that lag phase cannot be used to predict germination rates.

Length of lag phase versus maximum rate of germination for four strains of extremophilic fungi: (a) Aspergillus penicillioides JH06THH; (b) X. bisporus FRR 3443; (c) X. bisporus FRR 1522; and (d) Xerochrysium xerophilum FRR 0530. Length of the pre-germination phase and maximum rate of germination were determined on malt-extract yeast-extract phosphate agar (MYPiA) supplemented with diverse stressor(s) and incubated at 30 °C for up to 50 days. Media were supplemented with: glycerol (red dots), at 7.0 and 7.1 M, with water-activity values of 0.707 and 0.664, respectively; glycerol (5.5 M) + NaCl at 0.5, 1.0, 1.5, 1.6 and 1.7 M (green dots), with water-activity values of 0.765, 0.741, 0.709, 0.692 and 0.668, respectively; glycerol (5.5 M) + sucrose at 0.25, 0.50, 0.65 and 0.80 M (blue dots), with water-activity values of 0.734, 0.699, 0.674 and 0.637, respectively; glycerol (5.5 M) + glucose at 0.8 and 1.0 M + fructose at 0.80 and 1.0 M (yellow dots), with water activities of 0.694 and 0.649, respectively; and glycerol (5.5 M) + NaCl (0.5 M) + sucrose at 0.3 and 0.5 M (black dots), with water-activity values of 0.701 and 0.685, respectively. Data were obtained from Fig. S1 (see Experimental procedures), and trend lines showing a correlation between data (whether strong or moderate, see Experimental procedures) are indicated by a solid black line (c,d) while lines fitted which indicated inconsistency between lag and germination rates (i.e. no correlation was observed) are shown as a dashed grey line (a,b).

Length of lag phase versus maximum rate of growth at 23 °C: strain 14 in salt-supplemented nutrient broth (SSNB) (a) and yeast extract peptone dextrose broth (YPDB) (b); and strain 77 in SSNB (c) and YPDB (d). Cells suspended in water (not subjectied to any dehydration treatment) (pink squares), and cells suspended in 1 M xylitol (no dehydration treatment) (orange squares), were used as controls. For dehydrated cells, rehydration treatments were either rapid, using water (grey squares); rapid, using 1 M xylitol (brown dots); gradual, using water vapour then rapid in water (purple squares); or gradual rehydration with water vapor, then rapid in 1 M xylitol (turquoise squares). For strong correlations, trend lines are indicated by a solid black line (d), while inconsistency between lag and growth rates (i.e. no correlation was observed) is indicated by a dashed grey line (a–c).

Length of lag phase versus maximum rate of growth for Saccharomyces cerevisiae at 30 °C: strain 14 in salt-supplemented nutrient broth (SSNB) (a) and yeast extract peptone dextrose broth (YPDB) (b); and strain 77 in SSNB (c) and YPDB (d). Cells suspended in water (not subjected to any dehydration treatment) (pink squares), and cells suspended in 1 M xylitol (no dehydration treatment) (orange squares), were used as controls. For dehydrated cells, rehydration treatments were either rapid, using water (grey squares); rapid, using 1 M xylitol (brown squares); gradual, using water vapour then rapid in water (purple squares); or gradual rehydration with water vapor, then rapid in 1 M xylitol (turquoise squares) or for xylitol dehydrated cells; gradual, using water vapour then rapid in water (×); and gradual, using water vapour then rapid in 1 M xylitol (▲). Trend lines showing a strong correlation between data are indicated by a solid black line (d), while inconsistency between lag and growth rates (i.e. no correlation was observed) is indicated by a dashed grey line (a–c).

Length of lag phase versus maximum rate of mycelial extension for three Aspergillus strains on Robusta coffee-meal extract agar (CMEA) supplemented with glycerol: (a) Aspergillus sclerotiicarbonarius CBS 121853 at a water activity (aw) of 0.950; (b) Aspergillus aculeatinus CBS 121872 at a water activity of 0.990; (c) A. aculeatinus CBS 121872 at a water activity of 0.980; (d) and A. aculeatinus CBS 121875 at a water activity of 0.980 (primary data were taken from Akbar and Magan80, see Experimental procedures). Three replicates of each treatment were incubated at 20 (+), 25 (■), 30 (▲), 35 (♦), and 37 °C (●) for 9 days. Trend lines showing a strong correlation are indicated by a solid black line (c,d), while dashed grey lines indicate inconsistency between lag and maximum rate of mycelial extension (i.e. no correlation was observed) (a,b).
In some of the cases where there was no correlation between length of lag phase and subsequent rates of germination/growth, there is nevertheless evidence that cultures experienced considerable stress during the lag phase. For instance, the culture conditions under which lag phase and growth rates were determined for B. subtilis and A. penicilliodes were more stressful than those of the preculture conditions (because the former included the presence of solute stressors in the medium) (Table S2). Accordingly, growth rates were reduced at high concentrations of stressors (Figs. 1a,b, 4a,b, S1a,b, S7a,b and S8d,g). Aspergillus. aculeatinus was subcultured at optimal temperature, however the temperatures under which lag phase and growth rates were determined were higher than optimal, adding to the considerable stress during the lag phase. The disconnects between the lag phase and growth rate that are apparent in Figs. 1a,b, 3a, 4a,b, 5a–c, 6a–c, 7a,b, 8a,b, S1b, S3a,b, S7a,b, S8d,g and S9c,d are consistent with the findings of some other studies. For instance, Ding et al.79, characterized lag-phase adaptation in mutants of S. cerevisiae which over-expressed genes implicated in acid-stress tolerance. Over-expression of genes which code for a protein thought to increase vacuolar proton-pumping ATPase activity, PEP3, also shortened the lag phase. However, over-expression of PEP3 decreased length of lag phase but did not alter the rate of subsequent growth. In a study of two Aspergillus species, food-spoilage strains were cultivated over a range of water-activity values, in glycerol-supplemented nutrient media, and incubated over a range of temperatures80. We analysed the relationship between lag phase and maximum rate of mycelial extension of these fungi and found that, in some cases there was inconsistency between these parameters; for A. sclerotiicarbonarius CBS 12183 at 0.950 water activity and A. aculeatinus CBS 121872 at 0.990 water activity, r2 = 0.0002 and 0.393 respectively (Fig. 7a,b). For A. aculeatinus CBS 121872, there was a decrease of growth rate at high temperatures (a phenomenon often seen at supra-optimal water activity49,66) which contributed to the scatter. Furthermore, when plots were constructed for the same fungal strains in relation to each individual temperature, data for A. sclerotiicarbonarius CBS 121853 at 30 °C, and A. aculeatinus CBS 121872 at 35 °C were also scattered (Fig. 3a,b respectively).

Length of lag phase versus maximum rate of growth Mrakia frigida (DSM 70883) on malt-extract, yeast-extract phosphate agar (MYPiA) at 1.7 °C, supplemented with a range of stressors: (a) glycerol (1.1 M); (b) glucose (1.7 M); (c) sucrose (0.7 M); and (d) combined solute-treatment 1 (10% glycerol, 10% sucrose, 1% KCl plus 5% NaCl [w/v]). For each treatment, there were six replicates; these are shown in red, blue, orange, green, purple, and turquoise on each plot. Data were obtained from Fig. 2; see Experimental procedures. Trend lines showing a strong correlation between data are solid and black (c,d), while dashed grey lines indicate inconsistency between lag and growth rates (i.e. no correlation was observed) (a,b).
A study of a food-borne, pathogenic bacterium, Chronobacter, revealed that lag phase increased after acid stress, but lag was not proportional to subsequent growth rate26. Similarly, in relation to enzyme systems, studies of T. claveryi lipoxygenase activity during exposure 13- and 9-hydroperoxide derivatives (HPOD) of linoleic acid (i.e., 13-HPOD and 9-HPOD respectively) indicated that lag phase was not proportional to tolerance of oxidative stress. Data for rates of catalytic activity during steady state, when plotted against lag phase, were scattered, with r2 values of 0.105 for 9-HPOD and 0.043 for 13-HPOD (Fig. 9a,b). For a microbial population that has been unable to grow due to unfavourable environmental conditions, cells will sense changes in the intra- and/or extracellular milieu that facilitate growth, thus triggering the lag phase. Whereas there is a substantial literature about the end of lag and transition to exponential growth rate, there is little information about how (in terms of cellular biology) we define the start of lag. According to Penfold4, “By bacterial lag, we understand, the interval between the inoculation of a bacterial culture and the time of commencement of its maximum rate of growth.” It may be more appropriate to define lag from the time when genetic and metabolic adaptations begin. When microbial cells are placed into a fresh culture medium, changes in gene expression have been detected within 4 minutes, for 1119 genes12. The question remains, both in conceptual and practical terms, how to determine that lag has begun? Do we assume this occurred at time 081,82 or at some point between 0 and 4 mins? Regardless of this, and as detailed by Pirt83, “the requisite conditions for growth of biomass in culture are:
-
a viable inoculum,
-
an energy source,
-
nutrients to provide the essential materials from which biomass is synthesized (it may be that metabolites leaking from the cell can at as nutrients to facilitate entry into lag proper),
-
absence of inhibitors which prevent growth, [and]
-
suitable physicochemical conditions.”

Length of lag phase versus steady-state rate for T. claveryi lipoxygenase activity exposed to two hydroperoxy octadecadienoic acids; (a) 9-HPOD and (b) 13-HPOD; and A. bisporus tyrosinase exposed to a carboxylic acid; (c) anisic acid. For the hydroperoxy octadecadienoic acids, media composition consisted of 180 µM linoleic acid, and 0.01 unit of Terfezia claveryi LOX in sodium borate buffer at pH 10.0 and media were supplemented with: (a) 9-HPOD at 16.28 (×), 14.99 (●), 7.54 (♦), 5.23 (▲), and 2.66 mM (■) and without 9-HPOD (control; +); (b) 13-HPOD at 19.54 (×), 0.81 (●), 0.32 (♦), 0.24 (▲), and 0.16 mM (■) and without 13-HPOD (control; +). The reaction medum (3 ml) for the carboxylic acid consisted of 0.5 mM of tyrosine in 50 mM sodium phosphate buffer (pH 6.8), mushroom tyrosinase 33.3 µg/ml and media were supplemented with (c) anisic acid at 3 (●), 2 (■), 1 (▲), and 0.4 (♦) mM, and without anisic acid (control; +). Trend lines showing a strong correlation between data are solid and black (c,d), while dashed grey lines indicate inconsistency between lag and steady-state rate (i.e. no correlation was observed) (a,b).
Each of these conditions is characterized by complexity and so represents a black box when considering implications for microbial cell biology. However, it is well-established that many phenotypic, physiological and molecular changes occur during lag phase that are not stress responses, and the environmental changes that can induce lag phase do not necessarily induce stress. Genes or enzymes that are generally expressed or required to keep cellular systems functional are said to be involved in housekeeping84,85; these can include genes/proteins involved in DNA replication/repair, glycolysis, and construction of the cytoskeleton. Levels of mRNA for housekeeping genes are not always constant because housekeeping activities can vary in intensity depending on environmental and other factors. Furthermore, some housekeeping proteins are stable, so the corresponding genes are expressed at low levels. This said, there are housekeeping activities that are imperative to the lag phase of microbes. Accordingly, concentrations of DNA repair proteins (e.g. endonucleases) and DNA precursers increase during lag, thus facilitating the repair of any damaged DNA prior to chromosome replication8,12. The (picomolar) concentrations of metabolic intermediates in central carbon metabolism typically increase in the cytosol during the lag phase. Some of these facilitate increased glycolytic flux helping to fuel increased protein synthesis8,12. In turn, increases in cell-available energy, and protein synthesis, enable increased lipopolysaccharide production needed for cell-membrane repair and synthesis, and cellular division12,86.
Growth rates can sometimes vary with minimal variations in lag phase
Under some stress regimes, microbial growth rates varied considerably whereas lag phase was relatively consistent. For example, B. subtilis exhibited growth rates between 0.0068 and 0.651 OD units h−1 when grown in liquid media supplemented with ammonium sulphate (over a range of concentrations; Figs. 1c and S7c). In each case, lag phase was relatively short (between 1.5 and 7.5 hours), so whereas there was a 96-fold difference in growth rates, the lag phases of these cultures only showed a five-fold variation (Figs. 1c and S7c). The same phenomenon was seen for S. cerevisiae strains that had been exposed to different dehydration-rehydration treatments and then inoculated into a liquid medium. For instance, S. cerevisiae 14 grown at 23 °C YPDB (Figs. 5b and S3b) exhibited growth rates that ranged from 0.14 to 0.27. The maximum rate was 100% greater than the minimum, whereas lag phases varied from only 14 to 16 hours. For mycelial extension of A. sclerotiicarbonarius when grown over a range of water-activity values (at 30 °C; Fig. 3a), growth rates varied by 11.5-fold whereas lag phase varied by only two-fold. As discussed above (in Lag phase can sometimes vary independently of maximum rates of germination/growth), it might be expected that the length of lag changes when environmental conditions and/or the degree of stress changes. Indeed, it seems counterintuitive that very different levels of stress – as evidenced by a wide variation in growth rates – can occur with a small or negligible change in lag phase when the inoculum came from a single source (Figs. 1c, 3a, 5b, 6b and S7c). These data imply that, despite the considerable difference in the intensity of stress, there was a minimal or very rapid change required to adapt to the conditions imposed. This phenomenon was observed in E. coli, and was mostpronounced under urea-induced stress where the variation in growth rates was an order of magnitude greater than variation in lag. Whereas maximum growth rate decreased by 8.4-fold as urea concentration increased (between 600 to 900 mM), the corresponding extension of lag phase was only 1.7-fold (Fig. S10c). In some of the cases where growth rates varied considerably but lag phase was relatively consistent, cultures may have experienced considerable stress during the lag phase (Figs. 1c, 3a and S7c), but not in others (5b; 6b; S3b and S4b). For instance, the culture conditions for determinations of lag phase and growth rates for A. sclerotiicarbonarius were more stressful than preculture conditions; due to solute stressors and supra-optimal temperatures (Table S2).
Lag phase can sometimes correlate with the maximum rates of germination or growth
Generally, the microbes under study are more tolerant to solute-induced stresses than other filamentous fungi (for the xerophiles), yeasts (for S. cerevisiae) and mesophilic bacteria (especially B. subtilis)16,17,18,48. In relation to the aim of our study, however, the question remains whether lag phase is ever indicative of stress.
We found strong correlations between length of lag and subsequent rates of exponential growth under some conditions; for B. subtilis, germination for some fungal xerophiles, and for colony formation of M. frigida (Figs. 1c,d, 4c,d, 8c,d and S2b–f; Table S6). In the cases where strong correlations were observed (r2 > 0.810), the gradient (m) varied between −3.878 and −4567 (Table S6). For instance, the fungal xerophile X. bisporus (FRR 1522) was progressively more stressed on media with lower water-activity; lengths of lag phase were longer and gradients of exponential-phase growth curves less steep on media with high concentrations of glycerol (Fig. S1c). The length of lag phase was strongly correlated with subsequent rates of germination for X. bisporus (FRR 1522) (r2 = 0.916; Figs. 4c and S1c; Table S6). In addition, there was a number of other plots of lag versus germination rate where r2 ranged from 0.639 to 0.714, indicating moderately good correlations (Figs. 4 and S8). These included X. xerophilium FRR 0530, X. bisporous FRR 2347, E. repens JH06JPD and E. amstelodami FRR 2792 (r2 = 0.643, 0.639, 0.666 and 0.714; Figs. 4d and S8c,f,h; Table S6). Whereas lag phase of S. cerevisiae 14 correlated weakly with exponential growth rate (Figs. 5a,b and 6a,b), lag phase and growth rate showed a moderately good correlation for S. cerevisiae 77 on YPDB at 23 °C and on SSNB at 30 °C (r2 = 0.678 and 0.773, respectively ; Figs. 5d and 6d). The lag phase of B. subtilis strongly correlated with exponential growth rate in media supplemented with ammonium sulphate, polyethylene glycol 600, proline, MgCl2, sucrose and polyethylene glycol 6000 (r2 = 0.880, 0.961, 0.941, 0.901, 0.876 and 0.973, respectively; Figs. 1c,d and S11b,d–f). In addition, there was a moderately good correlation for the B. subtilis in media supplemented with glycerol; r2 = 0.593 (Fig. S2a). The lag phase of M. frigida was strongly correlated with subsequent rates of growth on media supplemented with sucrose (0.7 M), sucrose (1.5 M), glycerol (2.7 M), NaCl (2.5 M), and a combination of solutes (i.e., 0.55 M glycerol, 0.3 M sucrose, 0.1 M KCl plus 2.5 M NaCl) (r2 = 0.999, 0.956, 0.947, 0.842 and 0.999 respectively; Figs. 8c,d and S9a,d,e). In addition, there were moderately good correlations for M. frigida on media supplemented with MgCl2 (1 M), glycerol (4.3 M), glucose (2.8 M), and a combination of solutes (1.1 M glycerol, 0.3 M sucrose, 0.1 M KCl plus 2.5 M NaCl) (r2 = 0.771, 0.703, 0.687 and 0.743 respectively; Fig. S9b,c,f,g).
Correlations between lag and growth rates can also be found in the datasets we analysed from other researchers. For example, Li and Torres87 carried out a study of Gram-negative and Gram-positive bacteria (of the phyla Proteobacteria and Firmicutes) in liquid media supplemented with the mechanistically diverse solute-stressors NaCl, glycerol, and sucrose47. Analyses of length of lag versus exponential growth rate from 15 cultures/treatments87 revealed correlations between these two paremeters (13 strong correlations and two moderate correlations), regardless of bacterium or stressor (Fig. S12). For instance, the Gram-negative bacteria Pseudomonas fluorescens and Salmonella typhimurium, exhibited strong correlations between lag phase and exponential growth rate in media supplemented with NaCl, glycerol, and sucrose (r2 = 0.893; 0.921; 0.875, respectively for P. fluorescens [Fig. S12a,f,k] and 0.895; 0.963 and 0.892, respectively for S. typhimurium [Fig. S12d,I,n]). There were also strong correlations for the Gram-positive Brochothrix thermosphacta when media was supplemented with NaCl and glycerol (r2 = 0.940 and 0.915, respectively; Fig. S12b,g), and moderately a good correlation for media supplemented with sucrose (r2 = 0.723; Fig. S12l). For the Gram-positive bacterium Enterococcus faecalis, lag phase and exponential growth rate correlated strongly in media supplemented with NaCl (r2 = 0.842; Fig. S12c), and correlated moderately for media supplemented with glycerol or sucrose (r2 = 0.607 and 0.762, respectively; Fig. S12h,m). For S. aureus, the lag phase correlated strongly with exponential growth rate in media supplemented with NaCl, glycerol, and sucrose (r2 = 0.833, 0.913 and 0.857, respectively; Fig. S12e,j,o).
Where there were strong correlations between lag phase and maximum rates of subsequent germination/growth, there is evidence that cultures experienced considerable stress during the lag phase. For instance, determinations of length of lag and growth rates for Pseudomonas fluorescens and Salmonella typhimurium were carried out under more-stressful conditions (due to the solutes present) than those of the precultures (no stressors present) (Fig. S12a,d,f,I,k,n; Table S2). That lag phase can sometimes indicate the level of stress is also consistent with the data reported by others. For instance, studies of oxidative stress in S. cerevisiae BY4741 grown in batch culture found that length of lag phase is in approximate agreement with rates of exponential growth 120. In the Aspergillus study by Akbar and Magan80, there was a strong correlation between length of lag phase and maximum growth rate for some strains, including A. aculeatinus CBS 121872 at 0.980 water activity (r2 = 0.904) and A. aculeatinus CBS 121875 at 0.980 water activity (r2 = 0.950) (Fig. 7c,d). Furthermore, when growth curves of these strains were obtained for cultures on glycerol-supplemented media (over a range of water activities), there were also strong correlations between lag phase and maximum growth rates, depending on strain and temperature (Fig. 7c,d). Similarly, functional analyses of mutant strains have shown that lag phase is proportional to glycolaldehyde-induced inhibition, in S. cerevisiae BY474343, and osmotic stress, in Salmonella enterica88. A correlation between lag phase and stressor concentration was also observed in Bacillus strain TCL when exposed to oxidative stress induced by chromium36. A study of Listeria innocua exposed to stresses caused by UV-C and oxidative damage found that lag increased 13-fold whereas the growth rate was reduced by only 45%; nevertheless, the former still correlated with the latter89.
Many changes and adaptations which must take place during the microbial lag phase (whether for vegetative growth, germination, or other processes) relate directly to cellular stress and response and adaptation to stress. For instance, desiccated spores that imbibe water (a process that can be, in part at least, driven by ATP90,91) experience rehydration-related and dilution stresses (which may include hypo-osmotic shock); or new environmental conditions may necessitate the production of protein-stabilisation proteins, compatible solutes, or systems to remove reactive oxygen species92,93,94. It is therefore no surprise that lag can correlate with subsequent rates of growth or (presumably) with other indicators of cellular stress. Conversely, length of lag may not be determined by intensity of stress and, accordingly, lag may correlate weakly with growth rate, or not at all (see above).
The activities of housekeeping genes or protiens may increase (or decrease) under stress and vice versa. For instance, cellular changes in response to mild- to moderate levels of stress typically include increases in protein synthesis and upregulation of energy generation62. These, in turn, may require increased flux through glycolysis and/or upregulation of other so-called housekeeping functions. Stress-specific responses can also be upregulated during lag phase; these may relate to rehydration-induced stresses (when propagules imbibe water for instance) or other stressful events/stress parameters.
Other stress-induced changes that occur during lag phase include: protein-stabilisation proteins which mitigate against the chaotropicity of ethanol in E. coli94; increased expression of proline- and glycine-betaine transporters in Bacillus cereus under NaCl (osmotic) stress91; trehalose accumulation in response to a heat ramp from 28 to 41 °C in Saccharomyces cervisiae93; and acid-shock protein production in Salmonella typhimurium at low pH95.
It would therefore be expected that length of lag can sometimes act as a useful indicator of stress, and that at other times it may do so only weakly or not at all. Whereas both the lag- and stationary phases of batch cultures are characterized by very slow (or even zero) growth rates, it could be argued that the lag phase differs from the both the stationary- and late exponential phases in this way. Exponential growth can be seen as an expression of completely functional cell division processes, though these rarely, if ever, attain their theoretical optima. A variety of things can impede or prevent cell division; oxidative stress, lack of a key nutrient, cell division inhibitors, environmental stress signals, etc. So, observations of a lack of correlation between lag phase/exponential growth rate ratios and microbial responses to stress might be expected. When viewed this way, microbial lag phases seem analogous to cell senescence of animal and plant cells…cell senescence is generally viewed to occur with blockage of eukaryotic cell cycle check points [S, G2, M, G1] while metabolism continues. Markers for cell scenescence are known - e.g. p16, p21, p53, etc. - and nearly all are inhibitors of specific cell cycle check point targets [e.g. kinase inhibitors or activators] or upstream effectors of these inhibitors. We beleive that analogous processes are going on during microbial lag phases. Consistent with this, although there were strong correlations between lag phase and rates of exponential growth/germination in some cases and no correlation for others, the r2 values for Figs. 1–7, S2, S8–S10 and S12 were more or less proportional to the steepness of gradients (see also Table S6). There was a total of 16 datasets exhibiting a moderately good correlation (see above), and two plots which indicate a weak correlation. The latter were A. penicillioides JH06THJ on glycerol+NaCl−; glycerol+sucrose− and glycerol+NaCl+ sucrose-supplemented media and E. coli BL21 (syn. DE3) on Luria-Bertani broth supplemented with ethanol (r2 = 0.512 and 0.544 respectively; Figs. S8b and S10a; Table S6).
Concluding remarks
Definitions and studies of lag phase have almost exclusively related to microbes in batch cultures. Lag, however, occurs in virtually all types of microbial habitat within the biosphere, including times when populations come out of a prolonged stationary phase. A lag phase may also occur as a deviation from exponential growth. Furthermore, lag takes many forms and is characterised by complexity in relation to heterogeneity within populations of individual microbes, and in the context of evolutionary biology. For instance, a cell’s environmental and life history can determine rates (and other aspects) of adaption including length of lag96. Many studies also show phenotypic and behavioural heterogenity within populations of individual strains97,98,99,100. For instance, a study of S. cerevisiae cells from a glucose-containing (sole carbon source) medium were placed into a maltose-containing medium revealed profound differences in lag phase times. Indeed, up to 90% of cells never left the lag phase during the experimental period101, which may indicate genetic and/or epigenetic variation within the population; either of which can provide the basis for evolutionary changes99. Further examples can be found in the recent review by Bertrand86. In some cases, microbial cells can remain dormant for an indefinite period (independently of environmental conditions) even though they are viable102,103. This can sometimes be mistaken for lag. So, whereas much is known about lag phase, some aspects of lag also remain unpredictable and enigmatic. It should also be noted that the lag phase can involve the up-regulation as well as down-regulation of stress responses104. In relation to lag as a measure of vitality or stress tolerance, we believe that measures of lag (or single-point determinations of biomass) represent a precarious means to assess cellular stress.
In the current study, we analysed phylogenetically and ecophysiologically diverse microbes under various types of solute- and water-induced stress. Given that life is based on water, these stresses are omnipresent and potent. Indeed, they act as a paradigm for evolution and adaptation, the environmental challenges that life is continutously exposed to, and the functionality and survival microbial cells and their communities13,105. The study revealed that lag phase can be proportional to subsequent rates of growth/germination and so, in these cases at least, might be considered an indicator of cellular stress. However, we also found that length of lag is frequently unrelated to rates of growth/germination. For each type of microbe and each type of stress, length of lag phase can both correlate with, and be independent of, stress. Lag phase can vary independently of exponential growth rate and, in other cases, growth rates can vary when lag phase is constant. Regardless of the microbial system or microbial process, therefore, length of lag phase can correlate with and can be inconsistent with growth rate. Both these trends were observed whether stress treatments are differed quantitatively (Figs. 1 and S2) or qualitatively (Figs. 5 and 6); for fungi as well as bacteria (Figs. 1, 3–7, S2, S8–S10 and S12); in relation to diverse environmental parameters and cellular-stress treatments (water activity, chaotropicity, temperature, dehydration-rehydration); whether cultures were planktonic (Figs. 4, 5 and 7) or germinating or growing on a solid surface (Figs. 3, 4 and 7); and even for enzyme systems (Fig. 9). When the same B. subtilis strain is subjected to solute-induced stresses, lag phase can be strongly proportional to, independent of, or weakly correlated with the level of stress; depending on the solute stressor, but regardless of whether the latter is ionic or non-ionic (Figs. S7 and S11). Similarly, when A. aculeatinus CBS 121872 is cultured at different water activities, the lag phase may be inconsistent with or strongly correlated with exponential growth rate, depending on water activity (Fig. 7). Furthermore, when the same strain was incubated at a range of temperatures, both scatter plots and strong correlations were obtained (Fig. 3). It may frequently be difficult, if not impossible, to predict which cultures and/or stressors/stress regimes and/or stress parameters/mechanisms will be produce a lag phase that is primarily determined by stress, and which will not. In conclusion, therefore, single-point determinations of growth, should not be assumed to be indicative of microbial stress. In other words, lag phase is not a reliable measure of microbial stress. Whereas exponential growth rate is not ideal to define or quantify cellular stress13, it may actually be the best proxy we currently have.
The findings of this study give rise to a number of unanswered questions in addition to those discussed above:
-
1.
Apart from cellular growth, a lag phase can occur at various levels of complexity within biology; the macromolecular system106,107, the organism108,109, the ecosystem110,111, and evolutionary biology112,113. Furthermore, the biophysical events and chemical reactions which can drive biological processes may have their own lag phases; e.g. in relation to phase shifts in oscillatory processes (such cytoskeletal force generation and circadian clocks) or various types of friction (e.g. because of diffusion-rate limits) that might delay signal and response114. We analyzed datasets for studies of two enzyme systems, and once again found that length of lag phase in some cases is inconsistent with, and in other cases correlates with steady-state rate (Fig. 9a,b) and oxidation rate (Fig. 9c) (Fig. 9)115,116. For [9-HPOD] and [13-HPOD] (metabolites produced under stress) (from Terfezia claveryi Chatin), there was no apparent correlation (r2 = 0.105, 0.043; Fig. 9a,b). By contrast, for mushroom tyrosine (EC 1.14.18.1) there was a strong correlation between length of lag phase and oxidation rate (Fig. 9c) (r2 = 0.956; Fig. 9c). However, more work is needed to determine whether findings such as those reported here occur across all the levels of complexity, even though the underlying mechanisms in each case may differ.
-
2.
Studies of halophile communities (and other microbes found in extreme environments) suggest that lag phase in natural ecosystems can be prolonged. In such communities, it is possible that cells may, under some circumstances, experience lag phase for an indeterminate period. There is currently a paucity of data to demonstrate prolonged lag phases which progress at an infinitesimally slow rate. However, we hypothesize that extended lag phases are commonplace for the majority of the microbes in Earth’s biosphere and that, like for in-vitro cultures, lag phase is a measure of adaptation rather than stress per se for their in-situ populations.
-
3.
The findings also raise questions about the meaning of ‘stress’ in the context of cellular systems, as well as the way we identify or measure it13. Batches of cells of a given microbe with different phenotypes (in different physiological conditions) likely respond differently from each other during lag phase. Also, the combination of conditions in which they existed and the new conditions that have triggered lag, will determine which changes occur in the cell. It may be, therefore, that molecules sometimes considered biomarkers, are reliable indicators of lag because they depend on cellular history, past environmental conditions, and new environmental conditions. Further work, including nuanced experimentation, is required to disentangle these issues in relation to potential biomarkers. The microbial lag phase can be easily identified and determined from a basic growth curve, and yet complexities arising from this simple parameter continue to generate scientific miscalculations in studies of microbiology.
Experimental procedures
Bacillus subtilis and Escherichia coli strains, nutrient medium, and culture conditions
A culture of Bacillus subtilis 168 (also known as DSM 402 or ATCC 23857) was obtained from M. Hecker of the University of Greifswald (Germany), and maintained on Belitzki minimal medium (15 mM [NH4]2SO4, 8 mM MgSO4:7H20, 27 mM KCl, 7 mM Na3-citrate:2H2O, 50 mM tris-HCl, 0.6 mM KH2PO4, 2 mM CaCl2:2H2O, 1 µM FeSO4:7H2O, 10 µM MnSO4:4H20, 11.1 mM glucose, 62.4 µM tryptophan and 4.5 mM glutamic acid) at 37 °C. The pH of the medium was buffered pre-sterilisation to 7.5 using KOH, HCl and NaOH. The pH was measured post-sterilisation using a Mettler Toldeo Seven Easy, pH probe (Mettler Toldeo; Zurich, Switzerland).
A culture of Escherichia coli BL21 (syn. DE3) was obtained from New England Biolabs. (Hitchin, UK) and maintained in Luria–Bertani (LB) broth (per litre: 10 g yeast extract and 5 g peptone [peptic digest from meat; Fluka Analytical]; pH 6.1) at 25 °C. The pH was measured post-sterilisation using Pehanon pH test strips (Macherey-Nagel; Düren, Germany).
Culture of B. subtilis and E. coli under solute-induced stresses
Growth rates for B. subtilis 168 were determined in Belitzki minimal medium (control) and modified Belitzki minimal media, which had been supplemented with a range of concentrations of the following compounds: betaine, guanidine hydrochloride, ammonium sulphate, polyethylene glycol 600, glycerol, proline, glucose, MgCl2, sucrose, and polyethylene glycol 6000 (Table S4). All media were sterilised, using 500-ml aliquots in 1-litre glass Schott Duran bottles which were immersed in a water bath for 60 minutes at 100 °C, following which they were allowed to cool to 37 °C. For each medium, water activity was then determined, as described below, and values are given in Table S4. Aliquots (50 ml) of each medium were put into 250-ml conical flasks, which were then sealed with non-absorbent cotton wool. An exponential phase B. subtilis culture, grown in Belitzki minimal medium, was used to inoculate each medium to give a starting optical density500 nm of ∼0.05. Cultures were incubated at 37 °C with shaking at 180 revolutions per minute (rpm). Optical densities500 nm were determined every 40 minutes (or as required), and all treatments were carried out in triplicate.
Growth rates for E. coli BL21 (syn. DE3) were determined using cultures grown in LB broth (control) and LB broth supplemented with ethanol, butanol and urea over a range of concentrations (Table S4). Media were autoclaved, cooled to 23 °C and then ethanol or butanol was added using a Gilson Pipetman micropipette with the pipette tip inserted beneath the surface of the broth. Urea was weighed into a sterile vessel before being added to the media and swirled to dissolve. For each medium, pH was determined using Pehanon pH test strips and found to be between 5.9 and 6.1. Aliquots of each medium (50 ml) were placed into 100-ml Supelco serum bottles (Sigma-Aldrich, Dorset, UK) and sealed with aluminium foil. An E. coli culture grown to exponential phase in LB broth was used to inoculate each medium at a starting optical density (OD)560 nm of 0.01–0.02. Cultures were placed on a shaking incubator (120 rpm) at 20 °C and OD560 nm was determined every 1–4 hours.
Determinations of lag phase, and exponential growth rate, for B. subtilis and E. coli
Mean optical density values were plotted against incubation time for each stressor type (Figs. S7 and S11). Lag phase was determined as described above7, and exponential growth rates were calculated according to Pirt83. The relationship between lag phase and exponential growth rate was characterized, for each medium type, as described below.
Extremophilic fungal strains, nutrient media, and culture conditions
Eurotium repens JH06JPD were isolated by Williams and Hallsworth51 and are available from the corresponding author of the current article. The fungal xerophiles Eurotium amstelodami FRR 2792, Eurotium echinulatum FRR 5040, Eurotium halophilicum FRR 2471, Xerochrysium xerophilum FRR 0530, and Xeromyces bisporus FRR 0025, FRR 1522, FRR 2347, and FRR 3443 were obtained from CSIRO Food and Nutritional Sciences Culture Collection (North Ryde, NSW, Australia). Please note that E. echinulatum, E. halophilicum, and E. repens have recently been renamed as Aspergillus brunneus, Aspergillus halophilicus and Aspergillus pseudoglaucus, respectively117. Cultures were maintained on malt-extract yeast-extract phosphate agar (MYPiA; 1% w/v malt-extract (Oxoid; Hampshire, UK), 1% w/v yeast extract (Oxoid; Hampshire, UK), 1.5% w/v agar (Acros; Loughborough, UK), and 0.1% w/v anhydrous K2HPO4)supplemented with glycerol (5.5 M; 0.821 water activity) at 30 °C, as for previous studies16,17,49.
Assays of fungal germination at low water-activity
For each xerophile strain, ability to germinate was assessed at biologically hostile water activities (0.765–0.570) previously, using a range of 36 culture media designed to recreate physicochemical stresses experienced by microbes in natural and anthropogenic systems16. They were based on MYPiA, but supplemented with glycerol + NaCl; glycerol + sucrose; glycerol + glucose + fructose; glycerol only; glycerol + NaCl + sucrose; and glycerol + NaCl + KCl + sucrose (Table S3). Generally, media were sterilized by autoclaving at 121 °C at 1 atm. For those supplemented with glucose + fructose, however, 500 ml aliquots of medium were instead placed into a 1-litre glass Schott Duran bottle (Sigma-Aldrich, Dorset, UK), and immersed in a water bath at 80 °C for 30 minutes. Regardless of the sterilization method, media were allowed to cool before pouring into Petri plates. Once media had solidified, water activity was determined as described below.
MYPiA + glycerol (5.5 M) medium was inoculated using 2-mm-diameter plugs of agar taken from the periphery of exponential phase cultures growing on medium of the same composition, and plates were placed in a sealed bag of low-density polyethylene to maintain a constant relative humidity (thus maintaining water activity), while allowing gaseous exchange. For strains of A. penicillioides, E. amstelodami, E. echinulatum and E. repens, plates were incubated for 10–14 days at 30 °C; for strains of X. bisporus, X. xerophilum and E. halophilicum, plates were incubated for 21–28 days at 30 °C. Spores were then harvested by covering Petri plates with sterile solutions of 5.5 M glycerol (15 ml) and aerial spores were dislodged by gently brushing with a sterile glass rod. The spore suspensions were then passed through sterile glass-wool twice to remove hyphal fragments as described in earlier studies73,76. Each suspension was adjusted, by adding sterile 5.5 M glycerol solution, to a final spore concentration of 1 × 106 spores ml−1.
Spore suspension (150 µl) was used to inoculate each germination-assay medium by pipetting onto the agar surface and then spreading using a sterilize glass spreader. Plates of each type of medium were incubated in sealed bag of low-density polyethylene to maintain a constant relative humidity (in the dark; 30 °C). Periodically, a 4-mm agar disc was removed and used to immediately quantify percentage germination, via counts of 200 spores, using a light microscope. Upon removal of the disc, plates were resealed and placed back in the incubator. Germination was recorded for spores with a germ tube that was longer than the spore diameter, for isolated spores only (those located in clumps were not included in the assessment), as described previously76,118. These examinations were carried out at least daily, throughout a 50-d period, and all measurements were carried out in triplicate. These data were analysed as described below.
Determinations of lag phase before, and rates of, fungal germination
Percentage germination was plotted against incubation time by Stevenson et al.16 (Fig. S1). The length of the lag phase was determined by extrapolating trend lines to a point of 0% spore germination for each media type, as described previously16. The plots of percentage germination versus time (Fig. S1) were also used to determine maximum rate of progress of germination; i.e. during the exponential phase (Fig. 4). The relationship between length of lag phase and germination rate was characterized, for each xerophile species, as described below.
Saccharomyces cerevisiae strains, nutrient medium, and culture conditions
Saccharomyces cerevisiae strains (14 and 77) were obtained from the Yeast Strains Collection of Laboratory of Cell Biology, Institute of Microbiology and Biotechnology, University of Latvia. Each strain was maintained on yeast extract, peptone, dextrose agar (YPDA: 1% yeast extract, 2% peptone and 2% glucose and 2% w/v agar l−1) at 4 °C (Table S7). Each was cultivated in both a nutrient-rich, complex medium (yeast extract peptone dextrose broth; YPDB: 1% yeast extract, 2% peptone and 2% w/v glucose) and a salt-supplemented nutrient broth (SSNB: 0.7 g MgSO4, 0.5 g NaCl, 3.3 g (NH4)2SO4, 1 g KH2PO4, 0.13 g K2HPO4, 20 g glucose, and 3 g yeast extract l−1, adjusted to pH 5.5 by addition of KOH prior to autoclaving) at 23 or 30 °C (see below).
Saccharomyces cerevisiae dehydration-rehydration experiments
For each strain, cells were inoculated into YPDB (50 ml in a 250-ml conical flask) and incubated in an orbital shaking incubator (180 rpm, 30 °C) for 24 h, by which time they were in the stationary growth phase (Table S7). A 0.5-ml aliquot was then removed from each flask and used to inoculate fresh YPDB or SSNB (100 ml in a 500-ml conical flask) and incubated (180 rpm, 30 °C) for 48 h. Cells were harvested by centrifugation at 2000 rpm (Eppendorf 5810 R, Hamburg, Germany) for 10 min. The supernatant was then discarded and the cell pellet was blotted with filter paper to remove any residual liquid. Following this, the pellet was pressed through a metal sieve (pore size = 1 mm), and then divided into two fractions. One was dried to a constant weight at 105 °C (±0.1 mg), using a fan-free oven. The other was placed on a piece of filter paper in a vented Petri plate and allowed to dehydrate by placing at 30 °C in the fan-free oven, until the water content of cells was reduced to 8–10% w/w according to the difference between the weights obtained at 105 °C and the initial weight. At this water content, the S. cerevisiae cells are known to enter a state of anhydrobiosis during which time a high level of viability is retained119,120,121.
The water content of these cells was reduced to 8–10% w/w (as described above). For determinations of cell viability after rapid rehydration, sterile distilled water or a 1 M xylitol solution (10 ml) was added to 50 mg of dried cells and shaken gently for 10 min at 20 °C122. Survival rates of dehydrated cultures were then determined by fluorescence microscopy. Five microlitres of 1 mg ml−1 stock of fluorochrome primuline (Sigma Aldrich) was added to 5 µl yeast suspension and placed on a microscope slide, under a cover slip, for assessment123,124. The percentage viable cells were determined by subtracting the number which appeared by eye to be bright under fluorescence (these were dead) from the total number that was observed under phase contrast. For each treatment there were two replicate samples and for each sample, a minimum of 600 cells were assessed. In some experiments gradual rehydration of dry cells in the vapour of water was performed. This was carried out by placing cells (50 mg) on parchment paper in a 3-l glass chamber (its volume 3 l) containing 200 ml of distilled water in the base (giving an equilibrium relative humidity of 100%) at 37 °C for 2 hours122. After this procedure, cells were subjected to a rapid rehydration, by suspending them into either distilled water or a 1 M xylitol solution for 10 mins.
Determinations of lag phase, and rates of exponential growth, for Saccharomyces cerevisiae post-rehydration
After the rapid rehydration of cells, each suspension was standardized to OD600 = 0.5 by adding water and 10 µl was taken from each. These aliquots were used to inoculate YPDB or SSNB (190 µl) in a 96-well microplate at 30 °C for 24 h. Optical density600 nm was then monitored in an Infinite M200Pro Microplate Reader (Tecan, Switzerland). Mean values of optical density were plotted against incubation time for each treatment as well as the control. Lag phase was determined according to Lodge and Hinshelwood7 (data not shown), and exponential growth rates were calculated according to Pirt83. The relationship between lag phase and exponential growth rate was characterized as described below.
Mrakia frigida strains, nutrient medium, and culture conditions
Mrakia frigida strain DSM 70883 was obtained from DSMZ (Brunswick; Germany) and was cultured on MYPiA (0.997 water activity) at 1.7 °C73. Spot Test Assays were carried out at 1.7 °C: assessments of colony density (from 0 to 5 arbitrary units) were made at frequent intervals over a period of 210 days as described previously52,73,125,126,127,128,129. All Petri plates containing the same medium were sealed in a bag of low-density polyethylene to maintain a constant relative humidity (thus maintaining water activity), while allowing gaseous exchange. Experiments were carried out in triplicate, and plotted values are the means of independent treatments.
Comparison with extant data for food-spoilage fungi and bacteria and enzyme systems
In order to determine whether the findings of the current study were applicable to mesophilic spoilage fungi and/or for enzyme systems, extant datasets were located, analyzed, and then compared with the model systems used in the current study. For the mesophilic fungi Aspergillus aculeatinus and Aspergillus sclerotiicarbonarius, that are known to cause food-spoilage, growth data80 were used to construct length of lag phase-versus-maximum growth-rate plots, as for B. subtilis (see above). As described by Akbar and Magan80, media was prepared by boiling 300 g of ground green coffee beans in 1 litre of distilled water for 30 min, then filtering through a double layer of muslin. All media contained 6% [w/v] concentrated coffee extract, agar (2% w/v) and glycerol; the latter was used to adjust water activity. Glycerol concentrations were 23, 9.7 or 2.7% w/v, and the water activites of these media were 0.900, 0.950 and 0.980, respectively. For the food-spoilage microbes, the mesophilic bacteria E. faecalis, S. aureus and S. typhimurium and the psychrotolerant bacteria B. thermosphacta and P. fluorescens, length of lag- and growth-rate data87 were used to construct length of lag phase-versus-maximum growth-rate plots, as described above for B. subtilis.
For in-vitro activity of lipoxygenase (extracted from the ascomycete Terfezia claveryi), the assay medium contained linoleic acid solution (10 µl from a stock solution of 18 mM, in ethanol), sodium borate buffer (0.1 M, pH 10), lipoxygenase (20 µl in 0.1 M sodium borate buffer at pH 10.0), and the hydroperoxy octadecadienoic acids (HPODs) 9-HPOD and 13-HPOD over a range of concentrations (0 to 19.3 µM); made up to a final volume of 1 ml. The length of the lag phase was quantified by Pérez-Gilabert et al.116 by measuring activity spectrophotometrically (234 nm) for a period of time to construct a reaction progress curve, constructing a straight line through the maximal rate section of this curve, and then determining the value at which this straight line intercepts the time axis. The steady-state rate was calculated according to the maximal rate of the reaction progress curve116. These data were used in the current study to construct plots values of length of lag phase versus steady-state rate of catalytic activity (Fig. 9a,b).
For in-vitro activity of tyrosinase (extracted from the basodiomycete Agaricus bisporus), the assay medium (3 ml) contained sodium phosphate buffer (50 mM, pH 6.8), anisic acid over a range of concentrations (0 to 4 mM), tyrosinase (0.1 ml from a 1.0 mg ml−1 stock solution in 0.1 M phosphate buffer, pH 6.8), and tyrosine (2 mM)115. Kubo et al.115 state that “0.1 ml of the sample solution and 0.1 ml of the aqueous solution of the mushroom tyrosinase (0.2 mg ml−1 in 0.1 M phosphate buffer, pH 6.8) were added in this order to the mixture. This solution was immediately monitored [at 30 °C]. The enzyme activity was monitored by dopachrome formation at 475 nm”, so absorbance (475 nm) was monitored continuously using a spectrophotometer115. The length of lag phase was also determined by Kubo et al.115. Oxidation rates were calculated in the current study using the absorbance measurements to detect dopachrome that is produced via the enzyme-catalyzed oxidation of the substrate115. Plots of length of lag phase versus oxidation rates were constructed (Fig. 9c).
Characterization of relationship between microbial lag phases and stress
For microbial systems, maximum rates of growth or germination have traditionally been used as a reliable indicator for, and measure of, cellular stress (see above). For growth of Bacillus, S. cerevisiae, M. frigida, E. coli or mesophilic fungi, germination of fungal xerophiles, mesophilic bacteria, psychrotolerant bacteria and catalytic activity of fungal enzymes, length of lag phase was plotted against maximum rates (Figs. 1, 3–7, 9, S2, S8–S10 and S12). Using Microsoft Excel, 2016 MSO, Microsoft Corporation, California, USA, trend lines which provided the closest fit and highest regression coefficient (r2) value were added. Plots were fitted with linear, exponential, logarithmic, power- and polynomial (orders 2, 3, 4 and 5) trend lines and, in each case, the curve which best-described the data and gave the highest r2 value selected as described in earlier studies16. Values for r2 of >0.810 were considered to represent a strong correlation, those in the range 0.580 to 0.773 a moderately good correlation, those in the range 0.512 to 0.544 suggested a weak correlation, and those of <0.500 indicated inconsistency between lag phase and rate of growth/germination.
Quantitation of water activity
For all culture media, water-activity values were determined empirically using a Novasina Humidat IC-II water-activity machine fitted with an alcohol-resistant humidity sensor and eVALC alcohol filter (Novasina, Pfäffikon, Switzerland). The instrument was calibrated between each measurement using saturated salt solutions of known water activity130, and measurements were taken at the same temperature at which cultures were to be incubated. The water activity of each medium type was determined three times, and variation was within ±0.001. A number of additional precautions were taken to ensure accuracy of readings, as described previously18,131.
References
Das, B. & Gangopadhyay, G. Stochastic theory of interfacial enzyme kinetics: A kinetic Monte Carlo study. Chem. Phys. 393, 58–64 (2012).
Boekhoven, J., Hendriksen, W. E., Koper, G. J., Eelkema, R. & van Esch, J. H. Transient assembly of active materials fueled by a chemical reaction. Science. 349, 1075–1079 (2015).
Toth, L. T., Kuffner, I. B., Stathakopoulos, A. & Shinn, E. A. A 3,000-year lag between the geological and ecological shutdown of Florida’s coral reefs. Global. Change. Biol. 24, 5471–5483 (2018).
Plot, R. & Bobart, J. A discourse concerning the effects of the great frost, on trees and other plants anno 1683. drawn from the answers to fame Queries sent into divers Countries by Dr. Rob. Plot S. R. S. and from several observations made at Oxford, by the skilful Botanist Mr. Jacob Bobart. Phil. Trans. R. Soc. 14 (1684).
Pasteur, L. Memoire sur les corpuscles organises qui existent dan l ‘atmosphere. Examen de la doctrine des generations spontanees. Annales des Sciences Naturelles. Z Biol Animale 16, 5–98 (1861).
Penfold, W. J. On the nature of bacterial lag. J. Hyg. (Lond). 14, 215–241 (1914).
Lodge, R. M. & Hinshelwood, C. N. Physicochemical aspects of bacterial growth. Part IX. The lag phase of Bact. lactis aerogenes. J Chem Soc: 213–219 (1943).
Watanabe, S. et al. Intensive DNA replication and metabolism during the lag phase in cyanobacteria. PLOS. ONE. 10, e0136800, https://doi.org/10.1371/journal.pone.0136800 (2015).
Vanacloig-Pedros, E., Bets-Plasencia, C., Pascual-Ahuir, A. & Proft, M. Coordinated gene regulation in the initial phase of salt stress adaptation. J. Biol. Chem. 290, 10163–10175 (2015).
Voyles, J. et al. Diversity in growth patterns among strains of the lethal fungal pathogen Batrachochytrium dendrobatidis across extended thermal optima. Oecologia. 184, 363–373 (2017).
Osherov, N. & May, G. Conidial germination in Aspergillus nidulans requires RAS signaling and protein synthesis. Genetics. 155, 647–656 (2000).
Rolfe, M. D. et al. Lag phase is a distinct growth phase that prepares bacteria for exponential growth and involves transient metal accumulation. J. Bacteriol. 194, 686–701 (2012).
Hallsworth, J. E. Stress-free microbes lack vitality. Fungal. Biol. 122, 379–385 (2018).
Dijksterhuis, J. Fungal spores: Highly variable and stress-resistant vehicles for distribution. Food. Microbiol. 81, 2–11 (2019).
Dagnas, S., Gougouli, M., Onno, B., Koutsoumanis, K. P. & Membré, J.-P. Quantifying the effect of water activity and storage temperature on single spore lag times of three moulds isolated from spoiled bakery products. Int. J. Food. Microbiol. 240, 75–84 (2017).
Stevenson, A. et al. Glycerol enhances fungal germination at the water- activity limit for life. Environ. Microbiol. 19, 947–967 (2017).
Stevenson, A. et al. Aspergillus penicillioides differentiation and cell division at 0.585 water activity. Environ. Microbiol. 19, 687–697 (2017).
Stevenson, A. et al. Is there a common water-activity limit for the three domains of life? ISME. J. 9, 1333–1351 (2015).
Calderini, E. et al. Comparative proteomics of oxidative stress response of Lactobacillus acidophilus NCFM reveals effects on DNA repair and cysteine de novo synthesis. Proteomics. 17, 1600178 (2017).
Altintas, A. et al. High-resolution kinetics and modeling of hydrogen peroxide degradation in live cells. Free. Radic. Biol. Med. 101, 143–153 (2016).
Mellefont, L. A., McMeekin, T. A. & Ross, T. Viable count estimates of lag time responses for Salmonella typhimurium M48 subjected to abrupt osmotic shifts. Int. J. Food. Microbiol. 105, 399–410 (2005).
Zhou, K., George, S. M., Metris, A., Li, P. L. & Baranyi, J. Lag phase of Salmonella enterica under osmotic stress conditions. Appl. Environ. Microbiol. 77, 1758–1762 (2011).
Lima, S., Guo, M. S., Chaba, R., Gross, C. A. & Sauer, R. T. Dual molecular signals mediate the bacterial response to outer-membrane stress. Science. 340, 837–841 (2013).
Kar, J. R., Hallsworth, J. E. & Singhal, R. Fermentative production of glycine betaine and trehalose from acid whey using Actinopolyspora halophila (MTCC 263). Environ. Technol. Innov. 3, 68–76 (2015).
Dijkstra, A. R. et al. Strain-dependent transcriptome signatures for robustness in Lactococcus lactis. PLOS ONE 11, e0167944 (2016).
Margot, H., Zwietering, M. H., Joosten, H. & Stephan, R. Determination of single cell lag times of Chronobacter spp. strains exposed to different stress conditions: Impact on detection. Int. J. Food. Microbiol. 236, 161–166 (2016).
Ferro, P., Vaz-Moreira, I. & Manaia, C. M. Association between gentamicin resistance and stress tolerance in water isolates of Ralstonia pickettii and R. mannitolilytica. Folia Microbiol. 64, 63–72 (2019).
Callaway, R. et al. Positive interactions among alpine plants increase with stress. Nature. 417, 844–848 (2002).
Fazal, H., Abbasi, B. H., Ahmad, N., Ali, M. & Ali, S. Sucrose induced osmotic stress and photoperiod regimes enhanced the biomass and production of antioxidant secondary metabolites in shake-flask suspension cultures of Prunella vulgaris L. Plant Cell. Tiss. Org’ 124, 573–581 (2016).
Hosseini, F., Mosaddeghi, M. R., Dexter, A. R. & Sepehri, M. Effect of endophytic fungus Piriformospora indica and PEG-induced water stress on maximum root growth pressure and elongation rate of maize. Plant. Soil. 435, 423–436 (2019).
Kollárová, K., Kusá, Z., Vatehová-Vivodová, Z. & Lišková, D. The response of maize protoplasts to cadmium stress mitigated by silicon. Ecotoxicol. Environ. Saf. 170, 488–494 (2019).
Cray, J. A. et al. The biology of habitat dominance; can microbes behave as weeds? Microbiol. Biotechnol. 6, 453–492 (2013).
Azevedo, R. F. F., Souza, R. K. F., Braga, G. U. L. & Rangel, D. E. N. Responsiveness of entomopathogenic fungi to menadione-induced oxidative stress. Fungal. Biol. 118, 990–995 (2014).
Singh, S., Fatima, Z. & Hameed, S. Insights into the mode of action of anticandidal herbal monoterpenoid geraniol reveal disruption of multiple MDR mechanisms and virulence attributes in Candida albicans. Arch. Microbiol. 198, 459–472 (2016).
Jabeen, B. et al. Ectopic expression of plant RNA chaperone offering multiple stress tolerance in E. coli. Mol. Biotechnol. 59, 66–72 (2017).
Banerjee, S., Misra, A., Chaudhury, S. & Dam, B. A Bacillus strain TCL isolated from Jharia coalmine with remarkable stress responses, chromium reduction capability and bioremediation potential. J. Hazard. Mater. 367, 215–223 (2019).
Araújo, A. S., et al. Osmotolerance as a determinant of microbial ecology: A study of phylogenetically diverse fungi. Fungal. Biol doi.org/10.1016/j.funbio.2019.09.001 (in press).
Liu, Z. L. et al. Adaptive response of yeasts to furfural and 5-hydroxymethylfurfural and new chemical evidence for HMF conversion to 2,5-bis-hydroxymethylfuran. J. Ind. Microbiol. Biotechnol. 31, 345–352 (2004).
Liu, Z. L. Genomic adaptation of ethanologenic yeast to biomass conversion inhibitors. Appl. Microbiol. Biotechnol. 73, 27–36 (2006).
Ma, M. & Liu, Z. L. Comparative transcriptome profiling analyses during the lag phase uncover YAP1, PDR1, PDR3, RPN4, and HSF1 as key regulatory genes in genomic adaptation to the lignocellulose derived inhibitor HMF for Saccharomyces cerevisiae. BMC Genomics. 11, 660 (2010).
Dutrochet, H. L’Agent Immédiat Du Mouvement Vital Dévoilé Dans La Nature Et Dans Son Mode d’action, Chez Les Végétaux Et Les Animaux. (Baillière, J.B, 1826).
Hallsworth, J. E., Heim, S. & Timmis, K. N. Chaotropic solutes cause water stress in Pseudomonas putida. Environ. Microbiol. 5, 1270–1280 (2003).
Jayakody, L. N. et al. SUMO expression shortens the lag phase of Saccharomyces cerevisiae yeast growth caused by complex interactive effects of major mixed fermentation inhibitors found in hot-compressed water-treated lignocellulosic hydrolysate. Appl. Microbiol. Biotechnol. 99, 501–515 (2015).
Jin, J. H. et al. Investigation of growth phase-dependent acid tolerance in Bifidobacteria longum BBMN68. Curr. Microbiol. 73, 660–667 (2016).
Tremonte, P. et al. Homology-based modeling of universal stress protein from Listeria innocua up-regulated under acid stress conditions. Front. Microbiol. 7, 2–17 (2016).
Miladi, H., Elabed, H., Ben Slama, R., Rhim, A. & Bakhrouf, A. Molecular analysis of the role of osmolyte transporters opuCA and betL in Listeria monocytogenes after cold and freezing stress. Arch. Microbiol. 199, 259–265 (2017).
de Lima Alves, F. et al. Concomitant osmotic and chaotropicity-induced stresses in Aspergillus wentii: compatible solutes determine the biotic window. Curr. Genet. 61, 457–477 (2015).
Stevenson, A. et al. Multiplication of microbes below 0.690 water activity: implications for terrestrial and extraterrestrial life. Environ. Microbiol. 2, 257–277 (2015).
Stevenson, A., Hamill, P. G., Dijksterhuis, J. & Hallsworth, J. E. Water-, pH- and temperature relations of germination for the extreme xerophiles Xeromyces bisporus (FRR 0025), Aspergillus penicillioides (JH06THJ) and Eurotium halophilicum (FRR 2471). Microbiol. Biotechnol. 10, 330–340 (2017).
Lee, C. J. D. et al. NaCl-saturated brines are thermodynamically moderate, rather than extreme, microbial habitats. FEMS. Microbiol. Rev. 42, 672–693 (2018).
Williams, J. P. & Hallsworth, J. E. Limits of life in hostile environments; no limits to biosphere function? Environ. Microbiol. 11, 3292–3308 (2009).
Cray, J. A., Russell, J. T., Timson, D. J., Singhal, R. S. & Hallsworth, J. E. A universal measure of chaotropicity and kosmotropicity. Environ. Microbiol. 15, 287–296 (2013).
Marshall, B. J., Ohye, D. F. & Christian, J. H. B. Tolerance of bacteria to high concentrations of NaCl and glycerol in the growth medium. Appl. Microbiol. 21, 363–364 (1971).
Cray, J. A. et al. Biocontrol agents promote growth of potato pathogens, depending on environmental conditions. Microbiol. Biotechnol. 9, 330–354 (2016).
Williams, J. P. Exploring the Impacts of Environmentally Relevant Solute Activities on the Limits of Cellular Function. PhD Thesis. Belfast, Northern Ireland, UK: Queen’s University Belfast. (2010).
Lee, S. J. et al. Increased ethanol resistance in ethanolic Escherichia coli by insertion of heat-shock genes BEM1 and SOD2 from Saccharomyces cerevisiae. Biotechnol. Bioproc. E. 15, 770–776 (2010).
Bui, L. M. et al. Improved n-butanol tolerance in Escherichia coli by controlling membrane related functions. J. Biotech. 204, 33–44 (2015).
Chambers, S. & Kunin, C. M. The osmoprotective properties of urine for bacteria: the protective effect of betaine and human urine against low pH and high concentrations of electrolytes, sugars, and urea. J. Infect. Dis. 152, 1308–1316 (1985).
Ishida, A., Kawatake, Y. & Ono, N. Osmotic-stress conditioning for induction of acquired osmotolerance in Escherichia coli. J. Gen. Appl. Microbiol. 40, 35–42 (1994).
Abadias, M., Teixido, N., Usall, J., Vinas, I. & Magan, N. Solute stresses affect growth patterns, endogenous water potentials and accumulation of sugars and sugar alcohols in cells of the biocontrol yeast Candida sake. J. Appl Microbiol. 89, 1009–1017 (2000).
Ramirez, M. L., Chulze, S. N. & Magan, N. Impact of osmotic and matric water stress on germination, growth, mycelial water potentials and endogenous accumulation of sugars and sugar alcohols in Fusarium graminearum. Mycologia. 96, 470–478 (2004).
Sartori, M., Nesci, A. & Etcheverry, M. Impact of osmotic/matric stress and heat shock on environmental tolerance induction of bacterial biocontrol agents against Fusarium verticillioides. Res. Microbiol. 161, 681–686 (2010).
Hallsworth, J. E. Ethanol-induced water stress in yeast. J. Ferment. Bioeng. 85, 125–137 (1998).
Kets, E. P., Galinski, E. A., de Wit, M., de Bont, J. A. & Heipieper, H. J. Mannitol, a novel bacterial compatible solute in Pseudomonas putida S12. J. Bacteriol. 178, 6665–6670 (1996).
Ganthala, B. P., Marshall, J. H. & May, J. W. Xerotolerance in fission yeasts and the role of glycerol as compatible solute. Arch. Microbiol. 162, 108–113 (1994).
Hallsworth, J. E. & Magan, N. Effects of KCl concentration on accumulation of acyclic sugar alcohols and trehalose in conidia of three entomopathogenic fungi. Lett. Appl. Microbiol. 18, 8–11 (1994).
Hallsworth, J. E. & Magan, N. Effect of carbohydrate type and concentration on polyols and trehalose in conidia of three entomopathogenic fungi. Microbiology. 140, 2705–2713 (1994).
Hallsworth, J. E. & Magan, N. Water and temperature relations of growth of the entomogenous fungi Beauveria bassiana, Metarhizium anisopliae and Paecilomyces farinosus. J. Invertebr. Pathol. 74, 261–266 (1999).
Holden, P. A., Hunt, J. R. & Firestone, M. K. Toluene diffusion and reaction in unsaturated Pseudomonas putida biofilms. Biotechnol. Bioeng. 56, 656–670 (1997).
Halverson, L. J. & Firestone, M. K. Differential effects of permeating and nonpermeating solutes on the fatty acid composition of Pseudomonas putida. Appl. Environ. Microbiol. 66, 2414–2421 (2000).
Svenningsen, N. B. et al. Pseudomonas putida mt-2 tolerates reactive oxygen species generated during matric stress by inducing a major oxidative defense response. BMC Microbiol. 15, 202 (2015).
Santos, R., de Carvalho, C. C. R., Stevenson, A., Grant, I. R. & Hallsworth, J. E. Extraordinary solute-stress tolerance contributes to the environmental tenacity of mycobacteria. Environ. Microbial. Rep. 7, 746–764 (2015).
Chin, J. P. et al. Solutes determine the temperature windows for microbial survival and growth. Proc. Natl. Acad. Sci. USA 107, 7835–7840 (2010).
De Oliveira, M. V. V. et al. Differential effects of salinity and osmotic stress on the plant growth-promoting bacterium Gluconacetobacter diazotrophicus PAL5. Arch. Microbiol. 198, 287–294 (2016).
Príncipe, A., Jofré, E., Alvarez, F. & Mori, G. Role of a serine-type D-alanyl-D-alanine carboxypeptidase on the survival of Ochrobactrum sp. 11a under ionic and hyperosmotic stress. FEMS. Microbiol. Lett. 295, 261–273 (2009).
Hallsworth, J. E. & Magan, N. Manipulation of intracellular glycerol and erythritol enhances germination of conidia at low water availability. Microbiology. 141, 1109–1115 (1995).
Hallsworth, J. E., Nomura, Y. & Iwahara, M. Ethanol- induced water stress and fungal growth. J. Ferment. Bioeng. 86, 451–456 (1998).
Cray, J. A. et al. Chaotropicity: a key factor in product tolerance of biofuel-producing microorganisms. Curr. Opin. Biotechnol. 33, 228–259 (2015).
Ding, J. et al. PEP3 overexpression shortens lag phase but does not alter growth rate in Saccharomyces cerevisiae exposed to acetic acid stress. Appl. Microbiol. Biotechnol. 99, 8667–8680 (2015).
Akbar, A. A. & Magan, N. The impact of water and temperature interactions on lag phase, growth and potential ochratoxin A production by two new species, Aspergillus aculeatinus and A. sclerotiicarbonarius, on a green coffee-based medium. Int. J. Food. Microbiol. 188, 116–121 (2014).
Buchanan, R. L. & Cygnarowicz, M. L. A mathematical approach toward defining and calculating the duration of the lag phase. Food. Microbiol. 7, 237–240 (1990).
Bréand, S., Fardel, G., Flandrois, J. P., Rosso, L. & Tomassone, R. A model describing the relationship between lag time and mild temperature increase duration. Int. J. Food. Microbiol. 38, 157–167 (1997).
Pirt, S. J. Principles of Microbe and Cell Cultivation. (Blackwell, 1975).
Pancholi, V. & Chhatwal, G. S. Housekeeping enzymes as virulence factors for pathogens. Int. J. Med. Microbiol. 293, 391–401 (2003).
De Ferrari, L. & Aitken, S. Mining housekeeping genes with a Naive Bayes classifier. BMC Genomics 7, 1–14 (2006).
Bertrand, R. L. Lag phase is a dynamic, organized, adaptive, and evolvable period that prepares bacteria for cell division. J. Bacteriol. 201, e00697–18 (2019).
Li, K. Y. & Torres, J. A. Water activity relationships for selected mesophiles and psychrotrophs at refrigeration temperature. J. Food. Protect. 56, 612–615 (1993).
Shiroda, M., Pratt, Z. L., Dopfer, D., Wong, A. C. L. & Kaspar, C. W. RpoS impacts the lag phase of Salmonella enterica during osmotic stress. FEMS Microbiol. Lett. 357, 195–200 (2014).
Aguirre, J. S. et al. Characterization of damage on Listeria innocua surviving to pulsed light: Effect on growth, DNA and proteome. Int. J. Food. Microbiol. 284, 63–72 (2018).
Liu, S. L., Wu, W. J. & Yu, P. T. Effect of sonic stimulation on Bacillus endospore germination. FEMS. Microbiol. Lett. 363, fnv217 (2016).
Turgeman, T. et al. The role of aquaporins in pH dependent germination of Rhizopus delemar spores. PLOS ONE. 11, e0150543 (2016).
Den Besten, H. M., Mols, M., Moezelaar, R., Zwietering, M. H. & Abee, T. Phenotypic and transcriptomic analyses of mildly and severely salt-stressed Bacillus cereus ATCC 14579 cells. Appl. Environ. Microbiol. 75, 4111–4119 (2009).
Mensonides, F. I. C. et al. A new regulatory principle for in vivo biochemistry: Pleiotropic low affinity regulation by the adenine nucleotides – Illustrated for the glycolytic enzymes of Saccharomyces cerevisiae. FEBS Lett. 587, 2860–2867 (2013).
Soufi, B., Krug, K., Harst, A. & Macek, B. Characterization of the E. coli proteome and its modifications during growth and ethanol stress. Front. Microbiol. 6, 1–11 (2015).
Eriksson, S., Luchinni, S., Thompson, A., Rhen, M. & Hinton, J. C. D. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol. Microbiol. 47, 103–118 (2003).
Perez-Samper, G. et al. The crabtree effect shapes the Saccharomyces cerevisiae lag phase during the switch between different carbon sources. mBio 9, e01331–18 (2018).
Chitarra, G. S., Dijksterhuis, J., Breeuwer, P., Rombouts, F. M. & Abee, T. Differentiation inside multicelled macroconidia of Fusarium culmorum during early germination. Fungal. Genet. Biol. 42, 694–703 (2005).
Touzain, F. et al. Small variable segments constitute a major type of diversity of bacterial genomes at the species level. Genome. Biol. 11, R45 (2010).
van Leeuwen, M. R. et al. The effect of natamycin on the transcriptome of conidia of Aspergillus niger. Stud. Mycol. 74, 71–85 (2013).
Vermeersch, L. et al. On the duration of the microbial lag phase. Curr. Genet. 65, 1–7 (2019).
Cerulus, B. et al. Transition between fermentation and respiration determines history-dependent behavior in fluctuating carbon sources. eLife 7, e39234 (2018).
Feofilova, E. P., Ivashechkin, A. A., Alekhinc, A. I. & Sergeevaa, Y. E. Fungal spores: dormancy, germination, chemical composition, and role in biotechnology (review). Appl. Biochem. Microbiol. 48, 1–11 (2012).
Peddireddy, V., Doddam, S. N. & Ahmed, N. Mycobacterial dormancy systems and host responses in tuberculosis. Front. Immunol. 8, 1–19 (2017).
Pin, C. et al. Network analysis of the transcriptional pattern of young and old cells of Escherichia coli during lag phase. BMC. Syst. Biol 3, 108 (2009).
Ball, P. Water as an active constituent in cell biology. Chem. Revs. 108, 74–108 (2008).
Ali, A. J. & Lohman, T. M. Kinetic measurement of the step size of DNA unwinding by Escherichia coli UvrD helicase. Science 275, 377–380 (1997).
Rashno, F. et al. Very rapid amyloid fibril formation by a bacterial lipase in the absence of a detectable lag phase. BBA-Proteins Proteom. 1865, 652–663 (2017).
Gatenby, R. A. & Frieden, B. R. Information dynamics in carcinogenesis and tumor growth. Mutat. Res. 568, 259–273 (2004).
Pascoe, P. L., Parry, H. E. & Hawkins, A. J. S. Observations on the measurement and interpretation of clearance rate variations in suspension-feeding bivalve shellfish Aquat. Biol. 6, 181–190 (2009).
Marsico, T. D. et al. Underutilized resources for studying the evolution of invasive species during their introduction, establishment, and lag phases. Evol. Appl. 3, 203–219 (2010).
Smith, B. T. et al. The role of historical and contemporary processes on phylogeographic structure and genetic diversity in the Northern Cardinal, Cardinalis cardinalis. BMC Evol. Biol. 11, 136 (2011).
Clements, D. R. & Ditommaso, A. Climate change and weed adaptation: can evolution of invasive plants lead to greater range expansion than forecasted? Weed. Res. 51, 227–240 (2011).
Zhou, A. F. et al. Rapid selective sweep of pre-existing polymorphisms and slow fixation of new mutations in experimental evolution of Desulfovibrio vulgaris. ISME. J. 9, 2360–2372 (2015).
Kruse, K. & Jülicher, F. Oscillations in cell biology. Curr. Opin. Cell. Biol. 17, 20–26 (2005).
Kubo, I., Chena, Q. X., Niheia, K. I., Calderón, J. S. & Céspedes, C. L. Tyrosinase inhibition kinetics of anisic acid. Z. Naturforsch. 58, 713–718 (2003).
Pérez-Gilabert, M., Sánchez-Felipe, I., Morte, A. N. & García-Carmona, F. Kinetic properties of lipoxygenase from desert truffle (Terfezia claveryi) Chatin ascocarps: effect of inhibitors and activators. J. Agric. Food. Chem. 53, 6140–6145 (2005).
Hubka, V., Kolarik, M., Kubátová, A. & Peterson, S. W. Taxonomic revision of the genus Eurotium and transfer of species to Aspergillus. Mycologia 105, 912–937 (2013).
Hallsworth, J. E. et al. Compatible solutes protect against chaotrope (ethanol)-induced, nonosmotic water stress. Appl. Environ. Microbiol. 69, 7032–7034 (2003).
Beker, M. J. & Rapoport, A. I. Conservation of yeasts by dehydration. Adv. Biochem. Engin. Biotechnol. 35, 127–171 (1987).
Rapoport, A. Anhydrobiosis and dehydration of yeasts. In: Biotechnology of Yeasts and Filamentous Fungi (ed. Sibirny A.). (Springer, 2017).
Rapoport, A., Golovina, E. A., Gervais, P., Dupont, S. & Beney, L. Anhydrobiosis: Inside yeast cells. Biotechnol. Adv. 37, 51–67 (2019).
Rapoport, A. I., Khroustalyova, G. M., Camanis, G. J. & Beker, M. J. Yeast anhydrobiosis: permeability of the plasma membrane. Microbiology 64, 229–232 (1995).
Rapoport, A. I. & Meissel, M. N. Survival rates of yeast organisms after dehydration as determined by fluorescence microscopy. Microbiology 54, 53–55 (1985).
Rapoport, A., Rusakova, A., Khroustalyova, G. & Walker, G. Thermotolerance in Saccharomyces cerevisiae is linked to resistance to anhydrobiosis. Process. Biochem. 49, 1889–1892 (2014).
Albertyn, J., Hohmann, S., Thevelein, J. M. & Prior, B. GPD1, which encodes glycerol-3-phosphate dehydrogenase, is essential for growth under osmotic stress in Saccharomyces cerevisiae, and its expression is regulated by the high osmolarity glycerol response pathway. Mol. Cell. Biol. 14, 4135–4144 (1994).
Toh, T. H. et al. Implications of FPS1 deletion and membrane ergosterol content for glycerol efflux from Saccharomyces cerevisiae. FEMS Yeast. Res. 1, 205–211 (2001).
Tejero-Sariñena, S., Barlow, J., Costabile, A., Gibson, G. R. & Rowland, I. In vitro evaluation of the antimicrobial activity of a range of probiotics against pathogens: Evidence for the effects of organic acids. Anaerobe 8, 530–538 (2012).
Stevenson, A. & Hallsworth, J. E. Water and temperature relations of soil Actinobacteria. Environ. Microbiol. Rep. 6, 744–755 (2014).
Issa, R. et al. Antibiofilm potential of purified environmental bacteriophage preparations against early stage Pseudomonas aeruginosa biofilms. J. Appl. Microbiol. 126, 1657–1667 (2019).
Winston, P. W. & Bates, P. S. Saturated salt solutions for the control of humidity in biological research. Ecology 41, 232–237 (1960).
Hallsworth, J. E. & Nomura, Y. A simple method to determine the water activity of ethanol-containing samples. Biotechnol. Bioeng. 62, 242–245 (1999).
Acknowledgements
M. Hecker (University of Greifswald) kindly provided Bacillus subtilis [168]. Useful discussion was provided by Kathleen C. Benison (West Virginia University, USA); Gilberto U. L. Braga (University of São Paulo, Brazil); Jan Disksterhuis (Westerdijk Fungal Biodiversity Institute, The Netherlands), Elis C. A. Eleutherio (Federal University of Rio de Janeiro, Brazil); Kostya Panov (Queen’s University Belfast, Northern Ireland), David J. Timson (The University of Brighton, UK). Funding was received from the Biotechnology and Biological Sciences Research Council (BBSRC, United Kingdom) project BBF00347. Doctoral fellowships were provided by the Department of Education and Learning (Northern Ireland) who supported J. P. Williams and from the Department of Agriculture, Environment and Rural Affairs (Northern Ireland) who supported A. Stevenson, P. G. Hamill and P. E. McMullan.
Author information
Authors and Affiliations
Contributions
The study was conceived by J.E.H. and experimental work and data analyse were carried out by P.G.H., A.S., P.E.M., J.P.W. and G.K.; meta-analyses of data were carried out by P.G.H., A.D.R.L. and J.E.H.; the manuscript was written by J.E.H., P.G.H., A.R., P.E.M., K.D.F., G.K., J.P.Q., J.Y.T., K.E.S. and S.S; the final manuscript was reviewed by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hamill, P.G., Stevenson, A., McMullan, P.E. et al. Microbial lag phase can be indicative of, or independent from, cellular stress. Sci Rep 10, 5948 (2020). https://doi.org/10.1038/s41598-020-62552-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62552-4
This article is cited by
-
Nematicidal lipopeptides from Bacillus paralicheniformis and Bacillus subtilis: A comparative study
Applied Microbiology and Biotechnology (2023)
-
Sugarcane bagasse pretreated by different technologies used as support and carbon source in solid-state fermentation by Aureobasidium pullulans LB83 to produce bioemulsifier
Biomass Conversion and Biorefinery (2023)
-
A sustainable approach to enhance heavy hydrocarbons removal in landfarming treatment
Biodegradation (2023)
-
Examining the impact of zinc on horizontal gene transfer in Enterobacterales
Scientific Reports (2022)
-
Optimizing the Culture Medium of Lasiodiplodia sp. to Improve the Yield of Ethyl Acetate Extract as an Antimicrobial Source
Current Microbiology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
