
Abstract
Retinitis pigmentosa (RP) is described as a bilateral disease with inter-eye symmetry that presents on short-wavelength fundus autofluorescence (SW-AF) imaging with hyperautofluorescent (hyperAF) rings with an ellipsoid shape and regular borders. Nevertheless, both asymmetry and irregular ring morphologies are also observed. In this retrospective study of 168 RP patients, we characterize the degree of inter-eye asymmetry and frequency of irregular hyperAF ring morphologies according to mode of inheritance and disease-causing gene by using SW-AF imaging and spectral-domain optical coherence tomography (SD-OCT) scans. We observed that from 336 eyes, 290 (86%) presented with regular hyperAF rings and 46 (14%) presented with irregular shapes. From the 168 patients, 23 (14%) presented with asymmetric disease, with 16 (70%) of these patients also presenting with irregular ring shapes. Patients with autosomal dominant RP (adRP) had the highest proportion of irregular ring shapes (21%) and disease asymmetry (23%) in comparison to other modes of inheritance. Furthermore, both RP1 and RHO-adRP had the highest proportions of both disease asymmetry and irregular ring morphology. Our results suggest that in patients presenting with either irregular ring shapes or asymmetric disease, emphasis should be placed in targeted gene sequencing of genes known to cause adRP, such as RHO and RP1.
Similar content being viewed by others
Introduction
Retinitis pigmentosa (RP) is a group of heterogenous rod-cone retinal dystrophies caused at a cellular level by the degeneration of photoreceptors1,2. In most cases, the genetic defect is exclusive to the rods, whose degeneration leads to secondary cone death2,3. Patients clinically present with night blindness, constricted visual fields, and an eventual decrease in central vision that ultimately leads to blindness in the late stages. Other clinical findings on examination of the posterior pole include intraretinal pigment migration associated with retinal pigment epithelium (RPE) thinning, attenuated blood vessels, and pallor of the optic nerve4. The prevalence of RP is estimated to be 1 in 4,000, and the disease can be inherited in an autosomal dominant (30–40%), autosomal recessive (50–60%), or X-linked (5–15%) manner1.
RP is often described as a bilateral disease that presents and progresses symmetrically between both eyes. Exceptions are rare, but unilateral RP may account for 5% of the total population of RP patients5. Other pathologies that have been reported to cause unilateral pigmentary retinopathy include infection, inflammation, and trauma6,7,8,9. Nevertheless, few studies exist that analyze the degree of disease asymmetry among RP patients. An early study by Biro et al. suggested that a feature of RP is the symmetrical development of pigmentation, while a study by Massof et al. reported a high degree of symmetry as measured by visual fields10,11. More recent studies by Sujirakul et al. and Fakin et al. have also reported a high degree of symmetry, although asymmetry does occur12,13. Nevertheless, these studies did not explore the role that mode of inheritance and specific disease-causing genes play in affecting disease asymmetry in RP.
In this study, we explore disease asymmetry between fellow eyes in a cohort of RP patients by measuring the dimensions of the hyperautofluorescent (hyperAF) ring on short-wavelength fundus autofluorescence (SW-AF) imaging and the width of the ellipsoid zone (EZ) line on spectral-domain optical coherence tomography (SD-OCT) scans, parameters that are often used to track disease progression in RP14,15. In addition, we explore the degree of asymmetry and its relationship to mode of inheritance, patient genotype, and shape of the hyperAF ring.
Methods
Patients and clinical examination
The study procedures were defined and informed patient consent was obtained as outlined by the protocol #AAAR0284 approved by the Institutional Review Board at Columbia University Medical Center. The study is adherent to the tenets of the Declaration of Helsinki. The data presented in this study, including images and genetic testing results, is not identifiable to individual patients. A retrospective review of patients with a clinical diagnosis of RP by an inherited retinal disease specialist (SHT) at the Department of Ophthalmology at Columbia University was conducted. The clinical diagnosis was based on presenting symptoms, family history, fundus examination, and subsequently supported by clinical imaging, full-field electroretinography (ffERG), and/or genetic testing. Ophthalmic examinations included a slit-lamp and dilated funduscopic examination, best-corrected visual acuity (BCVA), short-wavelength fundus autofluorescence (SW-AF, 488 nm excitation), and spectral domain optical coherence tomography (SD-OCT). The inclusion criteria for this study were the diagnosis of RP and genetic characterization of the disease, while the exclusion criteria precluded monocular patients or those without SD-OCT or SW-AF imaging.
Imaging across all modalities was conducted after pupil dilation (>7 mm) with phenylephrine hydrochloride (2.5%) and tropicamide (1%). Horizontal foveal SD-OCT scans and SW-AF (488 nm excitation) were acquired with the Spectralis HRA + OCT (Heidelberg Engineering, Heidelberg, Germany). The SW-AF images were acquired with either a 55 or 30-degree field of view such that the entire hyperAF ring could be appreciated within the image.
Image analysis
The SW-AF images from the patients that met the inclusion criteria for the study were analyzed independently by two different graders (RJ and LC). The hyperAF rings were grouped into two categories based on morphology: 1) regular and 2) irregular. Regular rings were defined as closed rings with an ellipsoid/round shape and regular borders, while irregular rings included any ring morphologies that deviated from the above, including open rings, closed rings with irregular borders, and closed rings with non-ellipsoid shapes. Measurements of the horizontal and vertical diameters of the hyperautofluorescent ring, along with the width of the ellipsoid zone (EZ) line from the SD-OCT scans, were performed by the two graders on closed rings only, as not all of these parameters were always well-defined in open rings. A total of 151 patients (302 eyes) from the total cohort of 168 presented with closed rings. The measurements on both eyes of each patient were performed using a built-in measurement tool in the Spectralis HRA + OCT software. The horizontal diameter was defined as the longest distance between the nasal and temporal borders of the ring, while the vertical diameter was perpendicular to the defined horizontal diameter. The external boundary of the ring, which is better defined than the internal boundary, was used as the borderline for the diameter measurements.
Statistical analysis
The statistical analyses were performed using Stata 12.1 (StataCorp, College Station, Texas, USA) software. The Pearson correlation was calculated for the measurements of both independent graders. Given the high correlation between the two graders (r = 0.99, p < 0.001 for all parameters measured), the average of the two values obtained from the graders was calculated and used for subsequent analysis. For each parameter, the difference between both eyes was calculated and descriptive statistics for the horizontal, vertical diameters, and EZ line width were calculated (see Supplementary Table S1). Given the categorical nature of our data, chi-squared tests were used to compare disease asymmetry and ring morphology among the different modes of inheritance and to assess for an association between categorical variables.
Asymmetry analysis
Patients were defined to have asymmetric disease if they exhibited a difference greater than the 95th percentile cut-off in one or more of the parameters. Additionally, if patients exhibited a different ring morphology in each eye, they were also considered to have asymmetric disease. For patients with open rings, only the best-defined parameter was considered in the analysis. Patients were analyzed as an entire cohort and as sub-cohorts based on mode of inheritance of the disease (autosomal dominant, autosomal recessive, and X-linked recessive). Patients were also divided into sub-cohorts based on the identified disease-causing gene, and only those cohorts with ten or more patients were analyzed.
Results
Patients
In total, 168 patients with RP were analyzed for this study. Among the 168 patients, 57 (34%) presented with adRP, 100 (60%) with arRP, and 11 (6%) with XLRP. From the arRP patient cohort, 22 patients presented with syndromic disease: 6 with Usher syndrome type I, 13 with Usher type II, 2 with Usher type III, and 1 with Bardet-Biedl syndrome. The average age of the patients was 40 years old, which was similar to the average age of the sub-cohorts, except for XLRP, where the average age was 25 years. The most common disease-causing gene was RHO for adRP (37%), USH2A for arRP (42%), and RPGR for XLRP (100%). Patient demographics and genetic characterization are summarized in Table 1 (for more complete demographic and genetic characterization, see Supplementary Table S2).
Morphology of the hyperautofluorescent rings
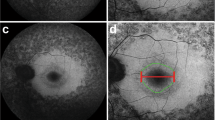
When analyzing the morphology of the hyperAF rings, the majority of patients presented with regular shapes (86% of eyes), while the rest presented with irregular shapes (Fig. 1). When segregating the patients by mode of inheritance, we observed that irregular ring morphology is more common in the autosomal dominant (21%) as compared to the autosomal recessive (10%, P = 0.007) and X-linked (9%, P = 0.191) forms. We also analyzed the occurrence of regular and irregular ring shapes by segregating patients into cohorts by disease-causing gene. We found that there is an association between the disease-causing gene and whether the ring presents with regular or irregular morphology (P = 0.004). We observed that disease caused by EYS, RP1, and RHO presented with the highest proportions of irregular rings (29%, 23%, and 21%, respectively), whereas PDE6A/B, USH2A, and RPGR have the highest proportions of regular rings (100%, 96%, and 91%, respectively). This information is summarized in Table 2.

Regular and irregular hyperautofluorescent rings on patients with retinitis pigmentosa. Short-wavelength fundus autofluorescence (SW-AF) imaging reveals hyperautofluorescent rings with the regular, ellipsoid shape in the majority of patients, as seen in Patients. (A–D) A minority of patients, however, can present with irregular ring shapes in a variety of forms as observed in Patients. (E–L) Disease-causing genes for these patients are USH2A (A,B,D,J and L), PDE6B (C), SNRNP200 (E), MAK (F,I), PRPF8 (G), BBS1 (H), and SPATA7 (K).
Patients with disease asymmetry
From our patient cohort, we found that 23 patients (14%) presented with asymmetric disease (Fig. 2). Autosomal dominant RP presented with the highest proportion of asymmetric disease (23%) as compared to autosomal recessive (9%, P = 0.017) and X-linked (9%, P = 0.303). Furthermore, the majority of patients with asymmetric disease presented with irregular ring shapes (70%, P < 0.001). When segregating by disease-causing gene, we observed that RP1-adRP, RHO-adRP, and PRPF31-adRP presented with the highest proportion of patients with asymmetric disease (31%, 24%, and 10%, respectively), whereas PDE6A/B-arRP and USH2A-arRP presented with the lowest proportion (0% and 7%, respectively). This information is summarized in Table 3.

Asymmetric disease on observed in retinitis pigmentosa patients. Short-wavelength fundus autofluorescence (SW-AF) imaging of patients with disease asymmetry between both eyes. Disease-causing genes for these patients are SPATA7 (Patient 1), USH2A (Patient 2), EYS (Patient 3), and BBS1 (Patient 4).
Discussion
With the recent advances of ocular gene therapy as a promising treatment modality for retinal dystrophies, it is important to study the level of asymmetry in these diseases, as the contralateral eye is often used as a control for the treatment eye due to the assumption of disease symmetry16,17,18,19. Although some studies have studied asymmetry in RP, they are either limited to a certain population of RP patients or are lacking in genetic characterization, a crucial aspect in a patient’s diagnosis due to the mutation-specific nature of gene therapy. In a study by Fakin et al., for example, 54 patients with Usher Syndrome type I and II were characterized with SD-OCT and SW-AF, and they report asymmetry in 10% of their patients. A different study by Sujirakul et al. used SW-AF to measure the horizontal and vertical diameters of the hyperAF ring, and they reported asymmetry in approximately 14% of their patients12. Nevertheless, genetic characterization was only available for 30 out of the 88 (34%) patients they analyzed12. In our study, we observed asymmetry in 14% of our patients, which is similar to what the above two studies reported. Moreover, the complete genetic characterization of our patient cohort allowed us to not only analyze asymmetry in RP, but to also correlate asymmetry with mode of inheritance.
We observed that adRP presents with both higher proportions of patients with irregular hyperAF ring shapes (24%) and asymmetry (23%), as compared to arRP (10% and 9%, respectively) and XLRP (2% and 9%, respectively). We theorize that this higher proportion of disease asymmetry in adRP might be related to genetic factors and variable expressivity of the diseased allele. As compared with other forms of inheritance, adRP is known to frequently present with variations in expressivity, and multiple studies have analyzed variable expressivity and incomplete penetrance in genes that cause adRP such as PRPF8 and PRPF312,20,21. A similar study to ours analyzed asymmetry in the disease progression of Stargardt disease, where the authors report that lower inter-eye correlations are more likely to be found on late-onset Stargardt disease22. Similar to adRP as compared to arRP or XLRP, late-onset Stargardt is milder than the other forms of the disease, such as early-onset23. Thus, these results suggest that disease asymmetry might be associated with mild disease severity. Of note, despite observing a higher proportion of patients with asymmetry and irregular AF ring shapes in adRP as compared to XLRP, we did not observe a statistically significant difference when comparing the two. We believe that this is due to the low number of XLRP patients in our cohort, as asymmetry is uncommon in RP and our cohort only contains 11 (6%) XLRP patients.
We categorized the hyperAF rings into two groups: regular and irregular ring shapes. The vast majority of patients presented with regularly shaped rings (86% of eyes analyzed). Similar to the analysis of disease asymmetry, adRP also presented with the greatest proportion of rings with irregular shapes. Currently, studies using quantitative autofluorescence (qAF) suggest that the mechanism for ring formation involves accelerated bisretinoid formation in actively degenerating receptor cells24. Clinically, we also know how the hyperAF ring relates to a patient’s vision. The signal for SW-AF (488 nm excitation) is derived mostly from RPE lipofuscin, which is formed in the photoreceptors as a byproduct of all-trans-retinal reactions25,26,27. The inner border of the hyperAF in RP patients corresponds to the lateral end of the EZ line on SD-OCT, and as disease progresses, the EZ line shortens along with constriction of the hyperAF ring24,28. Previous studies have shown that the point at which the EZ line disappears corresponds to the edge of the patient’s visual field and marks the boundary between healthy and unhealthy retina28,29,30. The results from these studies help us to conclude that not only is asymmetry observed in SW-AF and SD-OCT images, but also in functional vision parameters such as visual fields. Visual acuity, however, should not be affected by the asymmetric process, as RP starts on the periphery and affects central vision during the later stages of the disease. In fact, we observed no difference in the degree of asymmetry in visual acuity between fellow eyes in our cohort of patients with asymmetric disease as compared to those with symmetric disease (P = 0.255). The presentation of patients with asymmetric RP was also similar to patients with symmetric RP in regards to age of disease onset and severity of disease. It has been widely reported that XLRP is more severe than adRP31. This was also observed in our patient cohort, regardless of whether the patient presented with asymmetric disease or not. As expected, patients with RPGR-XLRP, for example, presented with more aggressive disease and earlier onset as compared to patients with RHO-adRP.
In addition to mode of inheritance, we also decided to analyze disease asymmetry and ring morphology when stratifying by the disease-causing gene. From our patient cohort, we observe that although every gene exhibits a regular ring shape more frequently, genes like EYS, RP1, and RHO have a higher proportion of patients with irregular ring morphology (29%, 23%, and 21% of eyes, respectively). This is in contrast to genes where irregular ring morphologies were observed infrequently, such as the PDE6A/B family, USH2A, and RPGR (0%, 4%, and 9%, respectively). Similarly, we observed disease asymmetry proportions were the highest in RP1 (31%) and RHO (24%), which generally have milder presentation than disease caused by the PDE6A/B family, USH2A, and RPGR.
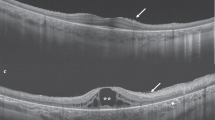
We were able to characterize asymmetry based on the hyperAF rings observed in SW-AF imaging. Similarly, hyperAF rings are also observed in near-infrared autofluorescence imaging (NIR-AF)14,32. Previous studies from our group have reported that the hyperAF rings appear larger in SW-AF imaging and that similar rates of disease progression are observed in both modalities14,32. In the patient cohort we present for this study, only a few patients had been imaged with both modalities. Yet, in those patients, we were able to observe disease asymmetry in both NIR-AF and SW-AF imaging (Fig. 3). Future studies with larger cohorts of patients with both SW-AF and NIR-AF imaging should analyze the extent to which NIR-AF imaging demonstrates asymmetry as compared to SW-AF.

Asymmetric disease with irregular rings as observed in short-wavelength (SW-AF) and near-infrared (NIR-AF) fundus autofluorescence. Patient 1 presented with RHO-autosomal dominant retinitis pigmentosa (A,B) and Patient 2 presented with USH2A-autosomal recessive retinitis pigmentosa. (C,D) Disease asymmetry and irregular rings are observed in both patients on SW-AF and NIR-AF images.
In conclusion, our study suggests genotype-phenotype correlations that can help the clinician in the diagnosis and management of RP patients. Based on our results, there is a relationship between symmetry of disease and ring morphology. Thus, if a patient presents with asymmetric disease or irregular rings on SW-AF, a diagnosis of adRP is more likely. Among the different methods of genetic sequencing, such as clinical exome or clinical genome, targeted sequencing has greater availability to patients, as its price is lower and results are obtained in less time. If either clinical exome or clinical genome sequencing cannot be obtained for a patient with asymmetric disease, emphasis should be placed in targeted sequencing of genes known to cause adRP, such as RP1 and RHO. Furthermore, given the higher likelihood of adRP disease, emphasis should be placed in screening other family members. Our results also beget the important question of whether disease progression is asymmetric between fellow eyes in those patients with asymmetric disease, which future studies should address.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809, https://doi.org/10.1016/S0140-6736(06)69740-7 (2006).
Hamel, C. Retinitis pigmentosa. Orphanet J Rare Dis 1, 40, https://doi.org/10.1186/1750-1172-1-40 (2006).
Narayan, D. S., Wood, J. P., Chidlow, G. & Casson, R. J. A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol 94, 748–754, https://doi.org/10.1111/aos.13141 (2016).
Schuerch, K., Marsiglia, M., Lee, W., Tsang, S. H. & Sparrow, J. R. Multimodal Imaging of Disease-Associated Pigmentary Changes in Retinitis Pigmentosa. Retina 36(Suppl 1), S147–S158, https://doi.org/10.1097/IAE.0000000000001256 (2016).
Farrell, D. F. Unilateral retinitis pigmentosa and cone-rod dystrophy. Clin Ophthalmol 3, 263–270 (2009).
Lotery, A. J., McBride, M. O., Larkin, C. & Sharkey, J. A. Pseudoretinitis pigmentosa due to sub-optimal treatment of neurosyphilis. Eye (Lond) 10(Pt 6), 759–760, https://doi.org/10.1038/eye.1996.182 (1996).
Laatikainen, L. & Mustonen, E. Asymmetry of retinitis pigmentosa-related to initial optic disc vasculitis. Acta Ophthalmol (Copenh) 70, 543–548 (1992).
Sekimoto, M., Hayasaka, S., Noda, S. & Setogawa, T. Pseudoretinitis pigmentosa in patients with systemic lupus erythematosus. Ann Ophthalmol 25, 264–266 (1993).
Cogan, D. G. Pseudoretinitis pigmentosa. Report of two traumatic cases of recent origin. Arch Ophthalmol 81, 45–53 (1969).
Biro, I. Symmetrical development of pigmentation as a specific feature of the fundus pattern in retinitis pigmentosa. Am J Ophthalmol 55, 1176–1179 (1963).
Massof, R. W. et al. Bilateral symmetry of vision disorders in typical retinitis pigmentosa. Br J Ophthalmol 63, 90–96 (1979).
Sujirakul, T. et al. Bilateral Concordance of the Fundus Hyperautofluorescent Ring in Typical Retinitis Pigmentosa Patients. Ophthalmic Genet 36, 113–122, https://doi.org/10.3109/13816810.2013.841962 (2015).
Fakin, A. et al. Fundus autofluorescence and optical coherence tomography in relation to visual function in Usher syndrome type 1 and 2. Vision Res 75, 60–70, https://doi.org/10.1016/j.visres.2012.08.017 (2012).
Jauregui, R., Park, K. S., Duong, J. K., Sparrow, J. R. & Tsang, S. H. Quantitative Comparison of Near-infrared Versus Short-wave Autofluorescence Imaging in Monitoring Progression of Retinitis Pigmentosa. Am J Ophthalmol 194, 120–125, https://doi.org/10.1016/j.ajo.2018.07.012 (2018).
Cabral, T. et al. Retrospective Analysis of Structural Disease Progression in Retinitis Pigmentosa Utilizing Multimodal Imaging. Sci Rep 7, 10347, https://doi.org/10.1038/s41598-017-10473-0 (2017).
Russell, S. et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860, https://doi.org/10.1016/S0140-6736(17)31868-8 (2017).
Lam, B. L. et al. Choroideremia Gene Therapy Phase 2 Clinical Trial: 24-Month Results. Am J Ophthalmol, https://doi.org/10.1016/j.ajo.2018.09.012 (2018).
Dimopoulos, I. S. et al. Two-Year Results After AAV2-Mediated Gene Therapy for Choroideremia: The Alberta Experience. Am J Ophthalmol 193, 130–142, https://doi.org/10.1016/j.ajo.2018.06.011 (2018).
MacLaren, R. E. et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet 383, 1129–1137, https://doi.org/10.1016/S0140-6736(13)62117-0 (2014).
Maubaret, C. G. et al. Autosomal dominant retinitis pigmentosa with intrafamilial variability and incomplete penetrance in two families carrying mutations in PRPF8. Invest Ophthalmol Vis Sci 52, 9304–9309, https://doi.org/10.1167/iovs.11-8372 (2011).
Rose, A. M. & Bhattacharya, S. S. Variant haploinsufficiency and phenotypic non-penetrance in PRPF31-associated retinitis pigmentosa. Clin Genet 90, 118–126, https://doi.org/10.1111/cge.12758 (2016).
Lambertus, S. et al. Asymmetric Inter-Eye Progression in Stargardt Disease. Invest Ophthalmol Vis Sci 57, 6824–6830, https://doi.org/10.1167/iovs.16-20963 (2016).
Westeneng-van Haaften, S. C. et al. Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology 119, 1199–1210, https://doi.org/10.1016/j.ophtha.2012.01.005 (2012).
Schuerch, K. et al. Quantifying Fundus Autofluorescence in Patients With Retinitis Pigmentosa. Invest Ophthalmol Vis Sci 58, 1843–1855, https://doi.org/10.1167/iovs.16-21302 (2017).
Delori, F. C. et al. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci 36, 718–729 (1995).
Sparrow, J. R., Wu, Y., Kim, C. Y. & Zhou, J. Phospholipid meets all-trans-retinal: the making of RPE bisretinoids. J Lipid Res 51, 247–261, https://doi.org/10.1194/jlr.R000687 (2010).
Katz, M. L. & Robison, W. G. Jr. What is lipofuscin? Defining characteristics and differentiation from other autofluorescent lysosomal storage bodies. Arch Gerontol Geriatr 34, 169–184 (2002).
Hood, D. C. et al. Method for deriving visual field boundaries from OCT scans of patients with retinitis pigmentosa. Biomed Opt Express 2, 1106–1114, https://doi.org/10.1364/BOE.2.001106 (2011).
Cai, C. X., Locke, K. G., Ramachandran, R., Birch, D. G. & Hood, D. C. A comparison of progressive loss of the ellipsoid zone (EZ) band in autosomal dominant and x-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci 55, 7417–7422, https://doi.org/10.1167/iovs.14-15013 (2014).
Hood, D. C., Lazow, M. A., Locke, K. G., Greenstein, V. C. & Birch, D. G. The transition zone between healthy and diseased retina in patients with retinitis pigmentosa. Invest Ophthalmol Vis Sci 52, 101–108, https://doi.org/10.1167/iovs.10-5799 (2011).
Jauregui, R. et al. Multimodal structural disease progression of retinitis pigmentosa according to mode of inheritance. Sci Rep 9, 10712, https://doi.org/10.1038/s41598-019-47251-z (2019).
Duncker, T. et al. Comparison of near-infrared and short-wavelength autofluorescence in retinitis pigmentosa. Invest Ophthalmol Vis Sci 54, 585–591, https://doi.org/10.1167/iovs.12-11176 (2013).
Acknowledgements
This work was supported by the National Institutes of Health [P30EY019007, R01EY024091, R01EY018213, R01EY024698, R01EY026682, R21AG050437], National Cancer Institute Core [5P30CA013696], Foundation Fighting Blindness [TA-NMT-0116-0692-COLU], the Research to Prevent Blindness (RPB) Physician-Scientist Award, unrestricted funds from RPB, New York, NY, USA.
Author information
Authors and Affiliations
Contributions
R.J., J.R.S. and S.H.T. conceived the study design. R.J. and L.C. performed the image analyses, analyzed, and interpreted the data. R.J., J.K.O. and A.C. wrote the manuscript text. S.H.T. and J.R.S. supervised the study and provided resources. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jauregui, R., Chan, L., Oh, J.K. et al. Disease asymmetry and hyperautofluorescent ring shape in retinitis pigmentosa patients. Sci Rep 10, 3364 (2020). https://doi.org/10.1038/s41598-020-60137-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60137-9
This article is cited by
-
Allelic hierarchy for USH2A influences auditory and visual phenotypes in South Korean patients
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
