
Abstract
Both high activity and mass production potential are important for bifunctional electrocatalysts for overall water splitting. Catalytic activity enhancement was demonstrated through the formation of CoS2 nanoparticles with mono-phase and extremely porous structures. To fabricate porous structures at the nanometer scale, Co-based metal-organic frameworks (MOFs), namely a cobalt Prussian blue analogue (Co-PBA, Co3[Co(CN)6]2), was used as a porous template for the CoS2. Then, controlled sulfurization annealing converted the Co-PBA to mono-phase CoS2 nanoparticles with ~ 4 nm pores, resulting in a large surface area of 915.6 m2 g−1. The electrocatalysts had high activity for overall water splitting, and the overpotentials of the oxygen evolution reaction and hydrogen evolution reaction under the operating conditions were 298 mV and −196 mV, respectively, at 10 mA cm−2.
Similar content being viewed by others


Introduction
To replace depleting fossil fuels, the development of environmentally-friendly and sustainable energy sources has been strongly urged. Among alternative energy sources, hydrogen-based energy sources are one of the most promising candidates due to their outstanding energy density, high energy conversion efficiency, and environmental friendliness1,2,3,4. Although there are a variety of technologies to produce hydrogen energy, electrochemical water splitting is one of the most promising choices due to commercial aspects5,6,7,8,9,10.
Electrochemical water splitting consists of two half reactions, namely the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER). Although noble metal electrocatalysts, such as Pt (for HER) and Ru- or Ir-based materials (for OER), show excellent catalytic performance, they are difficult to apply on a large scale because of cost issues. Thus, active electrocatalysts based on earth-abundant elements, such as transition metal compounds, have been extensively studied to replace noble metals11,12. Transition metal oxides13,14, hydroxides15,16, carbide17, nitride18, phosphide19, and sulfides20,21,22 have been studied as OER electrocatalysts under alkaline conditions, and transition metal sulfides20,21,22, phosphides6,7, carbonitride23, and selenides24 have been studied as HER electrocatalysts under acidic conditions. In regard to OER and HER performance, the effects of activation and optimization of electrocatalysts have recently been reported17,18,23. Furthermore, with a growing demand for simplicity and cost effectiveness, the need for bifunctional electrocatalysts, which operate in the same electrolyzer, is rapidly increasing21,25,26.
Cubic pyrite-phase transition metal dichalcogenides, such as FeS227,28, NiS229, and CoS226, have been proposed as candidates for bifunctional electrocatalysts30. Among them, cobalt disulfide (CoS2) has been reported to exhibit high electrical conductivity31 and excellent activity for both the OER and the HER9,26,32. Since water splitting involves hydrogen and oxygen gas evolution reactions, porosity in the electrocatalysts is very important for improving their performance by facilitating gas emission and exposing active sites26. Therefore, to further increase the catalytic activity of CoS2, it is necessary to synthesize porous CoS2 with nanometer scale pores.
Metal-organic frameworks (MOFs), which comprise metal ions and organic linkers, can provide a large surface area with open structures, so they are used in many electrochemical applications, such as batteries, capacitors, and catalysts33,34,35. However, MOFs have a low electrical conductivity and are not suitable as electrocatalysts, which require a large current density for electrical efficiency.
Here, we fabricated mono-phase CoS2 with ~ 4 nanometer-scale pores as bifunctional water splitting electrical catalysts. Cobalt-based MOFs, namely a cobalt Prussian blue analogue (Co-PBAs, Co3[Co(CN)6]2), were used as a starting material. The Co-PBA was then sulfurized in thermodynamically controlled conditions to remove the organic linkers in the MOFs, resulting in pure and mono-phase CoS2. The synthetic process was predicted based on thermodynamics because cobalt sulfide has been reported in various phases36, where the phase depends on the S/Co ratio, such as CoS37, Co9S838, and CoS239,40. Thermodynamic calculations control the sulfur vapor pressure depending on the amount of cobalt-based starting materials before sulfurization. Such MOF-driven CoS2 nanoparticles still preserve the nonporous structure in the starting MOFs, so high electrocatalytic activities are achieved.
Result and Discussion
Designing of MOF-driven CoS2 electrocatalyst
Figure 1 shows the synthetic process before and after completion. In the first step, the amount of sulfur was controlled through the thermodynamic prediction, and then the sulfurization of the Co-PBAs was conducted under each condition using a thermal treatment. Before sulfurization, the morphology of the synthesized Co-PBAs was confirmed through field-emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM) images, as shown in Fig. S1. An angular shape is typically confirmed in PBAs, whose size distribution is usually from tens of nanometers to micrometers. Additionally, PBA particles were uniformly synthesized by adding sodium citrate. As proved in previous reports, dissolved citrate in the solution suppressed the nucleation of PBAs41.

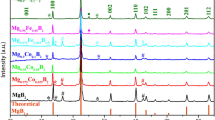
Summary of the fabrication of porous CoS2 and related thermodynamics calculations and its structural characterization. (a) Schematics of the synthesis of porous CoS2 through the sulfurization of Co-PBAs. (b) Ternary phase diagram at 500 °C for predicting the required composition to form the target phase and expected products depending on the amount of sulfur through thermodynamic prediction. (c–e) X-ray diffraction (XRD) analysis for verification of sulfurization under each condition. (c) Annealing for only Co-PBAs, (d) sulfurization of Co-PBAs with 3 mg sulfur (0.1 weight ratio of sulfur/Co-PBAs), and (e) sulfurization of Co-PBAs with 15 mg sulfur (0.5 weight ratio of sulfur/Co-PBAs).
For thermodynamically controlled sulfurization of Co-PBAs, components of the material and process parameters should be determined before the experiment. The synthesis of MOF-driven CoS2 is schematically shown in Fig. 1a. As a cobalt precursor, a Co-PBA was selected, and sulfur powder was used as a sulfur source. A closed system, comprising a sealed glass ampoule (Fig. S2), was used to ensure the loaded solid sulfur source was in a fully vaporized state for the thermodynamic calculations. Both were inserted into the glass ampoule, and the ampoule was fused to create a seal while the pressure inside was maintained at 0.1 Torr by rotary pump. Next, the ampoule was heated to 500 °C, which is higher than the boiling point of sulfur (445 °C). The sulfur inside was completely vaporized, and a reaction between the solid Co-PBA and gaseous sulfur took place. However, during such sulfurization processes, reaction products are dependent on the composition.
The Co-S-O ternary phase diagram at 500 °C was used to find the mole fraction of cobalt and oxygen for the sulfurization of the cobalt-based MOFs, and the determined value was set as the sulfur amount (left of Fig. 1b). Since C and N in Co-PBAs are vaporized to CN at 500 °C, the ternary phase diagram consists of Co-S-O components (Fig. S3). As the amount of sulfur content is increased, a cobalt sulfide phase with an increased S/Co ratio is synthesized. To set the exact sulfur amount, the ratio of each element (Co and O) was calculated. The moles of Co and O were fixed to the amount corresponding to 30 mg of Co-PBA in air 0.1 Torr in a 5 ml ampoule, illustrated as the dotted line in the phase diagram. Then, the amount of sulfur was the only parameter. Three experimental conditions for sulfur content were selected: 0 (0 mg), 0.1 (3 mg), and 0.5 (15 mg) weight ratios of sulfur/Co-PBAs, as marked in Fig. 1b as a circle, triangle, and star, respectively. The amount of reaction products at 500 °C according to the sulfur loading was calculated and is illustrated on the right in Fig. 1b. As the sulfur content increased, Co9S8 was produced at first, and then the amount of CoS2 increased. For a sulfur content with a ratio of more than 0.5 (15 mg), a CoS2 single phase can be obtained. However, when the sulfur content exceeded 0.9 (27 mg), the liquid sulfur phase remained.
Figure 1c–e shows the phase analysis of annealed Co-PBAs after the different processing conditions. XRD was conducted for phase analysis after each thermodynamically controlled condition was held for 2 hours. When a 0.5 ratio (15 mg) of sulfur was loaded with mono-phase CoS2, the target phase was synthesized, while a 0 ratio (0 mg) of sulfur synthesized the Co single phase and a 0.1 ratio (3 mg) of sulfur synthesized the Co9S8 phase. As a result of the XRD analysis, it was determined that annealing Co-PBAs without sulfur corresponds to the Co reference card in Fig. 1c, and sulfurization of the Co-PBAs in the condition represented by a triangle corresponds to the Co9S8 reference card in Fig. 1d. The XRD analysis in Fig. 1e clarified that CoS2 is the mono-phase.
Structural characterization of MOF-driven CoS2
Figure 2 shows the results of microstructural, chemical, and spectroscopic analyses to characterize MOF-driven CoS2 nanoparticles. The SEM (Fig. S4a) and TEM images (Fig. 2a) after sulfurization show that the MOF-driven porous CoS2 had uniform particle sizes of approximately 25 nm. Using high-resolution TEM, as shown in Fig. 2b, it was confirmed that the MOF-driven CoS2 had uniform crystallinity for each nanoparticle with a d-spacing of 0.28 nm, which corresponds to the (002) planes. The same result was obtained from the FFT pattern in the inset of Fig. 2b, which revealed that the MOF-driven CoS2 was highly crystalline with an (002) preferred orientation. The energy dispersive X-ray spectroscopy (EDX) was shown that the Co and S of synthesized MOF-driven CoS2 were uniformly distributed (Fig. S4b). Microstructural characterization of MOF-driven Co, Co9S8 (0, 0.1 ratio of sulfur/Co-PBAs), and commercial CoS2 were displayed in Fig. S5. It is confirmed that the higher S/Co ratio, the smaller the particle size. The electron energy loss spectroscopy (EELS) was conducted due to verification of charge distribution of MOF-driven cobalt compounds (Fig. S6). The chemical shift of Co L23 edge to higher energy was observed depending on higher S/Co ratio of cobalt sulfide42.

Microstructural and chemical characterization of MOF-driven porous CoS2 nanoparticles. (a) TEM image after sulfurization of Co-PBAs. (b) High-resolution TEM image and fast Fourier transformation (FFT) analysis (inset) of MOF-driven CoS2. Brunauer-Emmett-Teller (BET) analysis of (c) isothermal plot with N2 adsorption/desorption and (d) pore diameter distribution of MOF-driven Co, Co9S8, CoS2, and commercial CoS2 (inset: magnification of the small pore diameter area). (e,f) X-ray photoelectron spectroscopy (XPS) analysis for observation of bonding of CoS2. (e) Cobalt 2p spectrum and (f) sulfur 2p spectrum of MOF-driven CoS2.
To further verify the formation of porous structures in the MOF-driven CoS2, shown in Fig. 2c,d, BET analysis was completed, where the BET specific surface area and pore diameter were obtained. In the case of MOF-driven CoS2, the BET specific surface area in Fig. 2c was 915.6 m2 g−1, compared with 6.1, 46.0, and 60.1 m2 g−1 for commercial CoS2, MOF-driven Co, and Co9S8, respectively (Co-PBAs: 43.2 m2 g−1 in Fig. S7). This result implies that MOF-driven CoS2 had 150 times higher porosity than that of commercial CoS2. Furthermore, the Barrett-Joyner-Halenda (BJH) desorption was measured to quantitatively express the pore diameter shown in Fig. 2d. Two main peaks were observed in the BJH results, and the peaks appeared at approximately 3.5 nm in MOF-driven CoS2 (red line) and at ~20 nm (gradually increasing from 6 nm). A gradually increasing peak includes the surface of particles because there is no selectivity (pore and surface) for the adsorption/desorption of N2 during BET measurements. Although relatively low intensity, the peak of MOF-driven Co, Co9S8 were also detected at about 3.5 nm. Increasing the sulfur ratio leads to increasing the sulfur base gas (Fig. S3), which can be related to change of particle size and pore distribution. However, the pore size was not nearly detected in the commercial CoS2.
The porous structures were also observed in the TEM images, which confirmed the BET analysis. In Fig. 2a, there are bright parts in the TEM image that are highlighted with dotted yellow circles43,44,45,46. As shown in the TEM image, the size of the bright area was approximately 3 to 4 nm. Additionally, the results of electrochemical surface area (ECSA) were shown in Fig. S8. The linear slopes of commercial CoS2 on Ni foam electrode and MOF-driven CoS2 on Ni foam electrode are 0.7 and 1.5 mF cm−2 respectively, confirmed that the ECSA difference was about 2.1-fold (MOF-driven Co: 0.7, Co9S8: 1.1 mF cm−2). The ECSA of each electrode was calculated in Fig. S8.
XPS was conducted to observe the chemical and electronic states of MOF-driven CoS2 as shown in Fig. 2e,f. The broad scan in Fig. S9a shows peaks at binding energies of approximately 780, 530, 400, 286, and 163 eV, which were indexed to Co 2p, O 1 s, N 1 s, C 1 s, and S 2p, respectively. Specifically, in Fig. 2e, the Co 2p spectrum consists of peaks at 794 and 779 eV, which correspond to Co 2p1/2 and Co 2p3/2 respectively. Each peak can split into three sub-peaks, which are located at 796.7, 795.1 and 793.7 eV for the Co 2p1/2 spectrum and 781, 779.7 and 778.5 eV for the Co 2p3/2 spectrum. As indicated in Fig. 2e, the sub-peaks are known as satellites corresponding to Co2+ and Co3+ 6,10,26,47,48,49. The S 2p spectrum shown in Fig. 2f can typically be split two sub-parts with peaks at 168 eV and 163 eV. The 163 eV peak generally indicates the ‘−1’ valence state of sulfur, with sub-peaks indexed as 2p1/2 (163.6 eV) and 2p3/2 (162.5 eV). The peak at 168 eV is typically indexed as a sulfur oxide32,50. In this result, the sulfur oxide peak has a negligibly low intensity compared with other previous reports that describe hydrothermally synthesized CoS2. The results of the physical and chemical property analysis through XRD and XPS show that the synthesized CoS2 had high purity.
Investigation of electrochemical characterization
As shown in Fig. 3, to examine the bifunctional electrocatalytic activity of the MOF-driven porous CoS2 nanoparticles on the Ni foam electrode, linear sweep voltammetry (LSV) was conducted for the bare Ni foam, Ir foil, Pt electrode, commercial CoS2, and MOF-driven CoS2. The OER and HER were performed with a conventional three-electrode system in 1.0 M KOH (see for methods section for details). Polarization measurements were conducted in the potential range between 1.2 and 1.7 V (vs. reversible hydrogen electrode (RHE)) for the OER at a scan rate of 1 mV s−1 and between −0.4 and 0.0 V (vs. RHE) for the HER at a scan rate of 5 mV s−1. First, the OER activity of the electrocatalysts was measured as the overpotential at a current density of 10 mA cm−2 and the Tafel slope. Figure 3a displays the polarization curves of the OER range. The MOF-driven CoS2 electrode shows a low overpotential requirement of 298 mV at 10 mA cm−2, while the others show a higher overpotential. Under the same conditions, the overpotentials of the MOF-driven Co, MOF-driven Co9S8, commercial CoS2, IrO2, and MOF-driven Co3O4 were 411, 362, 379, 374, and 359 mV, respectively. The LSVs of Ir foil, Pt electrode, and Ni foam were conducted (Fig. S10a). The activity of MOF-driven Co3O4 was investigated for comparison with MOF-driven CoS2. This Co3O4 was fabricated with annealed Co-PBAs at same temperature condition in air, which was confirmed by XRD (Fig. S11).

Electrochemical characterization of MOF-driven porous CoS2 nanoparticles in 1.0 M KOH. (a) OER polarization curves of electrodes with a scan rate of 1 mV s−1. (b) Tafel slopes of polarization curves in OER. (c) Chronoamperometric stability test of fabricated CoS2 at constant voltage. (d) HER polarization curves of electrodes with a scan rate of 5 mV s−1. (e) Tafel slopes of polarization curves in HER. (f) Chronoamperometric stability test of fabricated CoS2 at constant voltage.
Additionally, the oxidation peak in the polarization curve was confirmed to be from the Co(III) to Co(IV) transition51. The MOF-driven CoS2 shows a larger oxidation peak in this location than that in previously reported studies. The larger the specific surface area, the greater the intensity of the redox peak.
The Tafel plot was plotted based on the polarization curves, and the Tafel slope was calculated (Fig. 3b). As with many other reported results, the tendency of the Tafel slope and overpotential were similar. The synthesized CoS2 electrode, which had the lowest overpotential in the polarization curves, showed a low Tafel slope value (94 mV dec−1).
In Fig. S12, electrochemical impedance spectroscopy (EIS) of commercial CoS2, 0, 0.1, and 0.5 ratios of sulfur/Co-PBAs (MOF-driven Co, Co9S8, and CoS2, respectively) was conducted at 300 mV (vs. RHE). Equivalent circuit model was shown in inset of Fig. S12. According to the EIS result, MOF-driven CoS2 was shown the lowest charge transfer resistance.
To verify the stability of the MOF-driven porous CoS2 nanoparticles, a long-term durability test was conducted using the chronoamperometric method (Fig. 3c). At a constant voltage of 1.53 V (vs. RHE), the synthesized CoS2 electrode delivered a relatively stable current density of 12 mA cm−2 over 20 hours. As oxygen evolution occurs during the test, fluctuations were observed due to gas bubbling.
Next, HER electrocatalytic activity was assessed as the overpotential at a current density of 10 mA cm−2 and the Tafel slope. As shown Fig. 3d, the HER overpotential was confirmed through LSV. The MOF-driven CoS2 electrode displays a low overpotential requirement of −196 mV at 10 mA cm−2, while the commercial CoS2 shows a large overpotential. The overpotentials of the MOF-driven Co, MOF-driven Co9S8, IrO2, and Pt electrode are −214, −213, −198, and −67 mV, respectively. The LSVs of Ir foil, Ni foam were conducted (Fig. S10c). The synthesized CoS2 electrode also shows high activity in the HER despite the presence of a strong alkaline electrolyte.
As shown in Fig. 3e, the Tafel slope was calculated based on the HER polarization curves, as in the OER results. Under the overpotential results, Pt and synthesized CoS2 electrodes exhibit Tafel slopes of 38 and 113 mV dec−1, respectively. Therefore, the synthesized CoS2 electrode shows the Volmer-Heyrovsky mechanism behavior52.
To confirm the stability in HER activity, a long-term durability test was also conducted with chronoamperometry (Fig. 3f). At a constant voltage of −0.2 V (vs. RHE), the synthesized CoS2 electrode delivered a relatively stable current density of −11 mA cm−2 over 20 hours. As hydrogen evolution occurs during the test, fluctuations were also observed due to gas bubbling.
In Fig. 4, to further investigate the change of MOF-driven CoS2 upon electrochemical test, structural and chemical characterization was performed after OER and HER durability test. As shown in Fig. 4b,f, although slight change of morphology was shown in FE-SEM images after OER and HER durability test, the MOF-driven CoS2 nanoparticles well-retained on the Ni foam compared with Fig. 4a. This slight change is inevitable, since O2 and H2 gas was vigorously emitted during OER and HER durability test, respectively10,53. The XRD pattern of MOF-driven CoS2 electrode had hardly changed after HER, while peak of CoOOH was slightly detected in the electrode after OER (Fig. S13).

Microstructural characterization of MOF-driven CoS2 electrode after durability test. (a) FE-SEM image of the bare Ni foam. (b–e) Electron microscopy images of MOF-driven CoS2 electrode after OER durability at 1.53 V (vs. RHE). (b) FE-SEM (inset: low magnification) (c) TEM, and (d,e) high-resolution TEM (inset: FFT analysis) images. (f,g) Electron microscopy images of MOF-driven CoS2 electrode after HER durability at −0.2 V (vs. RHE). (f) FE-SEM (inset: low magnification) (g) TEM, and (h) high-resolution TEM (inset: FFT analysis) images.
As shown in Fig. 4c–e, TEM analysis after OER durability was performed. The lattice fringes corresponding to CoS2 (Fig. 4d) and CoOOH (Fig. 4e) were observed. However, these lattice fringes featured low crystallinity, because of the crystalline to amorphous transition in electrocatalysts of transition metal compound6. EDX mapping after OER durability test of MOF-driven CoS2 electrode was conducted (Fig. S14a). Although the Co and O of MOF-driven CoS2 after OER durability test were uniformly distributed, signal of S was relatively weak due to the formation of surface CoOOH54. XPS analysis was conducted (Fig. S14b–d). The peak of CoOOH (783 eV) was observed in Co 2p spectrum54,55. In S 2p spectrum, only S-O peaks were featured6,56. The intensity of total O 1 s after OER was relatively increased compared to O 1 s before test. Furthermore, new peak was also observed at 529.9 eV (cobalt oxide)51.
After carrying out the HER durability test, TEM analysis was also conducted (Fig. 4f–h). Similar to the OER part, lattice fringe of low crystalline CoS2 was observed (Fig. 4h). EDX mapping and XPS analysis after HER test (Fig. S15). In EDX mapping, relatively weak signal of O and S was detected due to formation of transition chalcogenide hydrides during HER10,57. In XPS analysis, the peak of intensity at 781 eV (Co 2p3/2) was increased, which this phenomenon was also reported in other work6.
Figure 5 shows the performance of bifunctional electrocatalytic activity, which was conducted through a full-cell test of electrochemical water splitting for a practical two-electrode system in 1.0 M KOH. Both the anode and cathode were assembled from the same electrode material, which was MOF-driven CoS2 on Ni foam. The potential range was from 0.0 to 2.0 V. As shown in Fig. 5a, the assembled full-cell exhibited an overpotential of 1.65 V at 10 mA cm−2. The durability test was also conducted for symmetric full-cells (Fig. 5b). At a constant voltage of 1.65 V, the synthesized CoS2 electrode delivered a relatively stable current density of 11 mA cm−2 over 20 hours.

Overall water splitting performance of MOF-driven porous CoS2 nanoparticles. (a) Polarization curve and (b) chronoamperometric stability test of symmetric full cell.
As shown above, it was verified with structural characterization, chemical characterization and physical adsorption/desorption analysis that the synthesized CoS2 had a highly porous structure, including nanoscale pores. According to these results, the MOF-driven CoS2 nanoparticles provided facile diffusion kinetics as well as enough active sites. For this reason, the synthesized CoS2 electrode had a high OER and HER catalytic activities despite no additional treatment.
Conclusion
Highly porous structures including nanoscale pores were effective at enhancing the OER and HER electrocatalytic activities of the mono-phase CoS2 nanoparticles and reached values of 298 and −196 mV at 10 mA cm−2, respectively. Thermodynamic calculations were performed for the predictive synthesis of CoS2 from a PBA, one of the MOFs. The structural characterization of the synthesized MOF-driven CoS2 confirmed the uniformity and porous structure. From the investigation of the electrochemical characterization of the MOF-driven CoS2 electrode, the electrocatalytic performance of water splitting was high due to the kinetically favorable porous structure. Furthermore, the possibility of a bifunctional electrocatalyst was confirmed with a full-cell system.
Methods
Preparation of porous CoS2 nanoparticles
First, the cobalt Prussian blue analogue (Co-PBA), namely Co3[Co(CN)6]2 powder, was used as a starting material before sulfurization. The Co-PBA was synthesized through a facile precipitation method. Two types of solutions were used for precipitation. The recipe for the type 1 solution was 30 mM cobalt acetate (Co(CH3COO)2∙4H2O, Aldrich) and 17 mM sodium citrate (Na3C6H5O7∙2H2O, Sigma-Aldrich) in deionized water (DIW). The concentration of potassium hexacyanocobaltate (III) (K3[Co(CN)6], Sigma-Aldrich) was 20 mM in DIW41. Then, the type 1 solution was dropwise added into the type 2 solution through a syringe pump at 400 ml/hr and vigorously stirred for 10 min. Subsequently, the mixed solution was aged for 18 hours at room temperature. The dispersed precipitate was filtered through a vacuum pump and washed with DIW several times to remove residues. Finally, the obtained powder was dried in an oven at 60 °C for 12 hours.
Second, the cobalt disulfide (CoS2) nanoparticles were synthesized through thermal treatment. The 30 mg of the synthesized Co-PBA and certain amounts of sulfur (S, Sigma-Aldrich) were loaded in a 5 ml ampoule. After that, the prepared ampoule was sealed through a vacuum pump under 0.1 Torr. Finally, the sealed ampoule was thermally treated in a furnace at 500 °C for 2 hours at a ramp rate of 5 °C/min7.
Preparation of catalyst samples
To estimate the electrochemical performance, an ink was prepared. The 4 mg of synthesized CoS2 nanoparticles were dispersed in a mixture of DIW (800 μl), ethanol (200 μl, Sigma-Aldrich), and Nafion (80 μl, Aldrich) and sonicated for 30 min. After dispersion, 50 μl of the mixed ink was coated on the Ni foam (Aldrich) by drop casting through a micropipette. Finally, the coated electrode was dried in an oven at 60 °C for 12 hours. The ink preparation for the Co, Co9S8, Co3O4, commercial CoS2 powder (Aldrich), and IrO2 powder (Aldrich) was similar.
Thermodynamic calculation
The ternary phase diagram, and analysis graphs of expected products depending on sulfur amount were drawn with a thermochemical database program (FACTSAGE software). The database used in this study was the FACT pure substances database (FactPS)58.
Microstructural characterization
Phase analysis of the synthesized nanoparticles was performed using XRD (New D8 Advance, Bruker). Microstructural analysis of the synthesized CoS2 nanoparticles, including the Co-PBA, and acquisition of the FFT patterns was conducted using TEM (JEM-2100F, JEOL Ltd.). Additionally, the microstructure, including the size and morphologies of the synthesized nanoparticles, was observed using FE-SEM (SIGMA, Carl Zeiss). The surface area was conducted using a BET analyzer with N2 adsorption/desorption at 77.3 K (ASAP2420, Micromeritics Instruments).
Electrochemical characterization
LSV of each electrode was performed in a three-electrode system using a potentiostat (ZIVE MP2A, Wonatech, Korea). The working electrode was the prepared catalyst electrode. The counter and reference electrodes were Pt foil and a saturated calomel electrode (SCE) (Qrins, Korea), respectively, in 1.0 M KOH (Sigma-Aldrich). The LSV was conducted in the potential range between 1.2 and 1.7 V (vs. RHE) with a scan rate of 1 mV s-1 for the OER and between −0.4 and 0.0 V (vs. RHE) for the HER. In the case of overall water splitting, both electrodes were prepared using a potential range between 0.0 and 2.0 V. In HER and overall water splitting LSVs, the scan rate was fixed at 5 mV s−1. Electrochemical surface area (ECSA) was conducted using cyclic voltammetry (CV). CV was conducted in the voltage range between 0.18 and 0.28 V (vs. RHE) at a scan rate of 5, 10, 25, 50, 100, 200, 400, 600, 800, and 1000 mV s−1. The linear slope was calculated by plotting ΔJ at 0.23 V (vs. RHE) depending on scan rates59. EIS was performed within a frequency range from 100,000 Hz to 0.01 Hz at 300 mV (vs. RHE)60.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
References
Xia, B. Y. et al. A metal–organic framework-derived bifunctional oxygen electrocatalyst. Nature Energy 1, https://doi.org/10.1038/nenergy.2015.6 (2016).
Tahir, M. et al. Electrocatalytic oxygen evolution reaction for energy conversion and storage: A comprehensive review. Nano Energy 37, 136–157, https://doi.org/10.1016/j.nanoen.2017.05.022 (2017).
Pei, C., Chen, H., Dong, B., Yu, X. & Feng, L. Electrochemical oxygen evolution reaction efficiently catalyzed by a novel porous iron-cobalt-fluoride nanocube easily derived from 3-dimensional Prussian blue analogue. Journal of Power Sources 424, 131–137, https://doi.org/10.1016/j.jpowsour.2019.03.089 (2019).
Suen, N. T. et al. Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives. Chem Soc Rev 46, 337–365, https://doi.org/10.1039/c6cs00328a (2017).
Swesi, A. T. et al. Textured NiSe2 Film: Bifunctional Electrocatalyst for Full Water Splitting at Remarkably Low Overpotential with High Energy Efficiency. Sci Rep 7, 2401, https://doi.org/10.1038/s41598-017-02285-z (2017).
Song, H. J., Yoon, H., Ju, B., Lee, G.-H. & Kim, D.-W. 3D Architectures of Quaternary Co-Ni-S-P/Graphene Hybrids as Highly Active and Stable Bifunctional Electrocatalysts for Overall Water Splitting. Advanced Energy Materials 8, 1802319, https://doi.org/10.1002/aenm.201802319 (2018).
Kim, J.-Y. et al. Predictive fabrication of Ni phosphide embedded in carbon nanofibers as active and stable electrocatalysts. Journal of Materials Chemistry A 7, 7451–7458, https://doi.org/10.1039/c9ta00455f (2019).
Caban-Acevedo, M. et al. Efficient hydrogen evolution catalysis using ternary pyrite-type cobalt phosphosulphide. Nat Mater 14, 1245–1251, https://doi.org/10.1038/nmat4410 (2015).
Zheng, M., Ding, Y., Yu, L., Du, X. & Zhao, Y. In Situ Grown Pristine Cobalt Sulfide as Bifunctional Photocatalyst for Hydrogen and Oxygen Evolution. Advanced Functional Materials 27, 1605846, https://doi.org/10.1002/adfm.201605846 (2017).
Sivanantham, A., Ganesan, P. & Shanmugam, S. Hierarchical NiCo2S4 Nanowire Arrays Supported on Ni Foam: An Efficient and Durable Bifunctional Electrocatalyst for Oxygen and Hydrogen Evolution Reactions. Advanced Functional Materials 26, 4661–4672, https://doi.org/10.1002/adfm.201600566 (2016).
Xu, H., Ci, S., Ding, Y., Wang, G. & Wen, Z. Recent advances in precious metal-free bifunctional catalysts for electrochemical conversion systems. Journal of Materials Chemistry A 7, 8006–8029, https://doi.org/10.1039/c9ta00833k (2019).
Lu, F., Zhou, M., Zhou, Y. & Zeng, X. First-Row Transition Metal Based Catalysts for the Oxygen Evolution Reaction under Alkaline Conditions: Basic Principles and Recent Advances. Small 13, https://doi.org/10.1002/smll.201701931 (2017).
Rong, F. et al. Highly active water oxidation on nanostructured biomimetic calcium manganese oxide catalysts. Journal of Materials Chemistry A 4, 6585–6594, https://doi.org/10.1039/c5ta08217j (2016).
Wu, L.-K. et al. A nanostructured nickel–cobalt alloy with an oxide layer for an efficient oxygen evolution reaction. Journal of Materials Chemistry A 5, 10669–10677, https://doi.org/10.1039/c7ta02754k (2017).
Tang, C., Wang, H.-F., Wang, H.-S., Wei, F. & Zhang, Q. Guest–host modulation of multi-metallic (oxy)hydroxides for superb water oxidation. Journal of Materials Chemistry A 4, 3210–3216, https://doi.org/10.1039/c6ta00328a (2016).
Cai, Z. et al. Recent advances in layered double hydroxide electrocatalysts for the oxygen evolution reaction. Journal of Materials Chemistry A 7, 5069–5089, https://doi.org/10.1039/c8ta11273h (2019).
Kou, Z. et al. 2D carbide nanomeshes and their assembling into 3D microflowers for efficient water splitting. Applied Catalysis B: Environmental 243, 678–685, https://doi.org/10.1016/j.apcatb.2018.11.008 (2019).
Kou, Z. et al. Rational Design of Holey 2D Nonlayered Transition Metal Carbide/Nitride Heterostructure Nanosheets for Highly Efficient Water Oxidation. Advanced Energy Materials 9, 1803768, https://doi.org/10.1002/aenm.201803768 (2019).
Xue, Z.-H. et al. Janus Co/CoP Nanoparticles as Efficient Mott-Schottky Electrocatalysts for Overall Water Splitting in Wide pH Range. Advanced Energy Materials 7, 1602355, https://doi.org/10.1002/aenm.201602355 (2017).
Xu, P. et al. (Fe0.2Ni0.8)0.96S tubular spheres supported on Ni foam as an efficient bifunctional electrocatalyst for overall water splitting. Sci Rep 8, 9425, https://doi.org/10.1038/s41598-018-27477-z (2018).
Xiong, Q. et al. One-step synthesis of cobalt-doped MoS2 nanosheets as bifunctional electrocatalysts for overall water splitting under both acidic and alkaline conditions. Chem Commun (Camb) 54, 3859–3862, https://doi.org/10.1039/c8cc00766g (2018).
Xin, W. et al. NiS2 nanodotted carnation-like CoS2 for enhanced electrocatalytic water splitting. Chem Commun (Camb) 55, 3781–3784, https://doi.org/10.1039/c9cc01235d (2019).
Kou, Z. et al. Twinned Tungsten Carbonitride Nanocrystals Boost Hydrogen Evolution Activity and Stability. Small 15, e1900248, https://doi.org/10.1002/smll.201900248 (2019).
Liu, T., Liu, Q., Asiri, A. M., Luo, Y. & Sun, X. An amorphous CoSe film behaves as an active and stable full water-splitting electrocatalyst under strongly alkaline conditions. Chem Commun (Camb) 51, 16683–16686, https://doi.org/10.1039/c5cc06892d (2015).
Yan, Y., Xia, B. Y., Zhao, B. & Wang, X. A review on noble-metal-free bifunctional heterogeneous catalysts for overall electrochemical water splitting. Journal of Materials Chemistry A 4, 17587–17603, https://doi.org/10.1039/c6ta08075h (2016).
Guan, C. et al. Metal–organic framework derived hollow CoS2 nanotube arrays: an efficient bifunctional electrocatalyst for overall water splitting. Nanoscale Horiz. 2, 342–348, https://doi.org/10.1039/c7nh00079k (2017).
Kuo, T. R. et al. Improving Hydrogen Evolution Activity of Earth-Abundant Cobalt-Doped Iron Pyrite Catalysts by Surface Modification with Phosphide. Small 13, https://doi.org/10.1002/smll.201603356 (2017).
Li, Z. et al. Pyrite FeS2/C nanoparticles as an efficient bi-functional catalyst for overall water splitting. Dalton Trans 47, 14917–14923, https://doi.org/10.1039/c8dt02927j (2018).
Liu, H. et al. Electronic Structure Reconfiguration toward Pyrite NiS2 via Engineered Heteroatom Defect Boosting Overall Water Splitting. ACS Nano 11, 11574–11583, https://doi.org/10.1021/acsnano.7b06501 (2017).
Ganesan, P., Sivanantham, A. & Shanmugam, S. CoS2–TiO2 hybrid nanostructures: efficient and durable bifunctional electrocatalysts for alkaline electrolyte membrane water electrolyzers. Journal of Materials Chemistry A 6, 1075–1085, https://doi.org/10.1039/c7ta09096j (2018).
Faber, M. S., Lukowski, M. A., Ding, Q., Kaiser, N. S. & Jin, S. Earth-Abundant Metal Pyrites (FeS2, CoS2, NiS2, and Their Alloys) for Highly Efficient Hydrogen Evolution and Polysulfide Reduction Electrocatalysis. J Phys Chem C Nanomater Interfaces 118, 21347–21356, https://doi.org/10.1021/jp506288w (2014).
Faber, M. S. et al. High-performance electrocatalysis using metallic cobalt pyrite (CoS(2)) micro- and nanostructures. J Am Chem Soc 136, 10053–10061, https://doi.org/10.1021/ja504099w (2014).
Furukawa, H., Cordova, K. E., O’Keeffe, M. & Yaghi, O. M. The chemistry and applications of metal-organic frameworks. Science 341, 1230444, https://doi.org/10.1126/science.1230444 (2013).
Lee, J.-H., Ali, G., Kim, D. H. & Chung, K. Y. Metal-Organic Framework Cathodes Based on a Vanadium Hexacyanoferrate Prussian Blue Analogue for High-Performance Aqueous Rechargeable Batteries. Advanced Energy Materials 7, 1601491, https://doi.org/10.1002/aenm.201601491 (2017).
Xia, W., Mahmood, A., Zou, R. & Xu, Q. Metal–organic frameworks and their derived nanostructures for electrochemical energy storage and conversion. Energy & Environmental Science 8, 1837–1866, https://doi.org/10.1039/c5ee00762c (2015).
Wang, Y. et al. Phase-controlled synthesis of cobalt sulfides for lithium ion batteries. ACS Appl Mater Interfaces 4, 4246–4250, https://doi.org/10.1021/am300951f (2012).
Zhou, J. et al. MOF Template-Directed Fabrication of Hierarchically Structured Electrocatalysts for Efficient Oxygen Evolution Reaction. Advanced Energy Materials 7, 1602643, https://doi.org/10.1002/aenm.201602643 (2017).
Feng, L. L. et al. Carbon-armored Co9S8 nanoparticles as all-pH efficient and durable H2-evolving electrocatalysts. ACS Appl Mater Interfaces 7, 980–988, https://doi.org/10.1021/am507811a (2015).
Yang, J. et al. Highly efficient oxygen evolution from CoS2/CNT nanocomposites via a one-step electrochemical deposition and dissolution method. Nanoscale 9, 6886–6894, https://doi.org/10.1039/c7nr01293d (2017).
Zhang, H. et al. A metallic CoS2 nanopyramid array grown on 3D carbon fiber paper as an excellent electrocatalyst for hydrogen evolution. Journal of Materials Chemistry A 3, 6306–6310, https://doi.org/10.1039/c5ta00707k (2015).
Zhang, H. et al. Facile preparation of Prussian blue analogue Co3[Co(CN)6]2 with fine-tuning color transition temperature as thermochromic material. CrystEngComm 19, 2057–2064, https://doi.org/10.1039/c7ce00384f (2017).
Ma, D. et al. Highly active nanostructured CoS2/CoS heterojunction electrocatalysts for aqueous polysulfide/iodide redox flow batteries. Nat Commun 10, 3367, https://doi.org/10.1038/s41467-019-11176-y (2019).
Wang, X.-F. et al. Prussian Blue analogue derived porous NiFe2O4 nanocubes for low-concentration acetone sensing at low working temperature. Chemical Engineering Journal 338, 504–512, https://doi.org/10.1016/j.cej.2018.01.072 (2018).
Ren, J., Meng, Q., Xu, Z., Zhang, X. & Chen, J. CoS2 hollow nanocubes derived from Co-Co Prussian blue analogue: High-performance electrode materials for supercapacitors. Journal of Electroanalytical Chemistry 836, 30–37, https://doi.org/10.1016/j.jelechem.2019.01.049 (2019).
Hu, L. et al. Foamlike porous spinel Mn(x)Co(3-x)O4 material derived from Mn3[Co(CN)6]2nH2O nanocubes: a highly efficient anode material for lithium batteries. Chemistry 18, 15049–15056, https://doi.org/10.1002/chem.201200412 (2012).
Zhang, L. et al. Rational Design of High-Performance DeNOx Catalysts Based on MnxCo3–xO4 Nanocages Derived from Metal–Organic Frameworks. ACS Catalysis 4, 1753–1763, https://doi.org/10.1021/cs401185c (2014).
Peng, S. et al. Hollow nanospheres constructed by CoS2 nanosheets with a nitrogen-doped-carbon coating for energy-storage and photocatalysis. ChemSusChem 7, 2212–2220, https://doi.org/10.1002/cssc.201402161 (2014).
Xie, J., Liu, S., Cao, G., Zhu, T. & Zhao, X. Self-assembly of CoS2/graphene nanoarchitecture by a facile one-pot route and its improved electrochemical Li-storage properties. Nano Energy 2, 49–56, https://doi.org/10.1016/j.nanoen.2012.07.010 (2013).
Qiu, W., Jiao, J., Xia, J., Zhong, H. & Chen, L. A self-standing and flexible electrode of yolk-shell CoS2 spheres encapsulated with nitrogen-doped graphene for high-performance lithium-ion batteries. Chemistry 21, 4359–4367, https://doi.org/10.1002/chem.201405821 (2015).
Ouyang, C., Wang, X. & Wang, S. Phosphorus-doped CoS2 nanosheet arrays as ultra-efficient electrocatalysts for the hydrogen evolution reaction. Chem Commun (Camb) 51, 14160–14163, https://doi.org/10.1039/c5cc05541e (2015).
Sivanantham, A. et al. A Stable Graphitic, Nanocarbon-Encapsulated, Cobalt-Rich Core-Shell Electrocatalyst as an Oxygen Electrode in a Water Electrolyzer. Advanced Energy Materials 8, 1702838, https://doi.org/10.1002/aenm.201702838 (2018).
Mahmood, N. et al. Electrocatalysts for Hydrogen Evolution in Alkaline Electrolytes: Mechanisms, Challenges, and Prospective Solutions. Adv Sci (Weinh) 5, 1700464, https://doi.org/10.1002/advs.201700464 (2018).
Lv, X. et al. Hollow mesoporous NiCo2O4 nanocages as efficient electrocatalysts for oxygen evolution reaction. Dalton Trans 44, 4148–4154, https://doi.org/10.1039/c4dt03803g (2015).
Zhang, Q. et al. Self-Interconnected Porous Networks of NiCo Disulfide as Efficient Bifunctional Electrocatalysts for Overall Water Splitting. ACS Appl Mater Interfaces 10, 27723–27733, https://doi.org/10.1021/acsami.8b04386 (2018).
Kwak, I. H. et al. CoSe(2) and NiSe(2) Nanocrystals as Superior Bifunctional Catalysts for Electrochemical and Photoelectrochemical Water Splitting. ACS Appl Mater Interfaces 8, 5327–5334, https://doi.org/10.1021/acsami.5b12093 (2016).
Hu, C. et al. Synergism of Geometric Construction and Electronic Regulation: 3D Se-(NiCo)Sx(OH)x Nanosheets for Highly Efficient Overall Water Splitting. Adv Mater 30, e1705538, https://doi.org/10.1002/adma.201705538 (2018).
Jervis, R. et al. Hydrogen Oxidation on PdIr/C Catalysts in Alkaline Media. Journal of The Electrochemical Society 161, F458–463, https://doi.org/10.1149/2.037404jes (2014).
Nam, D. H. et al. Controlled Molybdenum Disulfide Assembly inside Carbon Nanofiber by Boudouard Reaction Inspired Selective Carbon Oxidation. Adv Mater 29, https://doi.org/10.1002/adma.201605327 (2017).
Fan, X. et al. Defect-enriched iron fluoride-oxide nanoporous thin films bifunctional catalyst for water splitting. Nat Commun 9, 1809, https://doi.org/10.1038/s41467-018-04248-y (2018).
Ahn, I. K. et al. Improved Battery Performance of Nanocrystalline Si Anodes Utilized by Radio Frequency (RF) Sputtered Multifunctional Amorphous Si Coating Layers. ACS Appl Mater Interfaces 10, 2242–2248, https://doi.org/10.1021/acsami.7b17890 (2018).
Acknowledgements
This research was supported by Creative Materials Discovery Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2017M3D1A1040688). The SEM and TEM analysis was supported by the Research Institute of Advanced Materials (RIAM) at Seoul National University.
Author information
Authors and Affiliations
Contributions
I.-K.A. and Y.-C.J. conceived the designed the experiments. W.J. and S.-Y.L. contributed to the thermodynamic calculation and XPS analysis. I.-K.A. and J.-H.L. synthesized the MOF and conducted the electrochemical characterization. I.-K.A. and J.-Y.K. contributed to the sulfurization of MOF. H.G.K., G.-B.L., and M.K. conducted the TEM measurements. I.-K.A. performed the structural characterization. Y.J. contributed to the correction of the manuscript. All authors contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahn, IK., Joo, W., Lee, JH. et al. Metal-organic Framework-driven Porous Cobalt Disulfide Nanoparticles Fabricated by Gaseous Sulfurization as Bifunctional Electrocatalysts for Overall Water Splitting. Sci Rep 9, 19539 (2019). https://doi.org/10.1038/s41598-019-56084-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-56084-9
This article is cited by
-
Electrodeposited nanostructured flakes of cobalt, manganese and nickel-based sulfide (CoMnNiS) for electrocatalytic alkaline oxygen evolution reaction (OER)
Journal of Materials Science: Materials in Electronics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
