
Abstract
In recent years Bamako has been faced with an emerging threat from multidrug resistant TB (MDR-TB). Whole genome sequence analysis was performed on a subset of 76 isolates from a total of 208 isolates recovered from tuberculosis patients in Bamako, Mali between 2006 and 2012. Among the 76 patients, 61(80.3%) new cases and 15(19.7%) retreatment cases, 12 (16%) were infected by MDR-TB. The dominant lineage was the Euro-American lineage, Lineage 4. Within Lineage 4, the Cameroon genotype was the most prevalent genotype (n = 20, 26%), followed by the Ghana genotype (n = 16, 21%). A sub-clade of the Cameroon genotype, which emerged ~22 years ago was likely to be involved in community transmission. A sub-clade of the Ghana genotype that arose approximately 30 years ago was an important cause of MDR-TB in Bamako. The Ghana genotype isolates appeared more likely to be MDR than other genotypes after controlling for treatment history. We identified a clade of four related Beijing isolates that included one MDR-TB isolate. It is a major concern to find the Cameroon and Ghana genotypes involved in community transmission and MDR-TB respectively. The presence of the Beijing genotype in Bamako remains worrying, given its high transmissibility and virulence.
Similar content being viewed by others
Introduction
Tuberculosis is caused by members of the Mycobacterium tuberculosis complex (MTBC). In humans, the main causes include Mycobacterium tuberculosis sensu stricto and Mycobacterium africanum1. M. tuberculosis and M. africanum form seven distinct phylogenetic lineages that are believed to have co-evolved with humans over millennia2,3,4. The East Asian, East-African-Indian and Euro-American lineages (Lineages 2,3 and 4 respectively) form the “modern” clade of tuberculosis5. The East Asian (including the Beijing genotype) and Euro-American lineages are the most widespread lineages globally and are probably more virulent than other lineages of the MTBC6. Recently a new lineage (lineage 7) has been discovered, which is predominantly restricted to Ethiopia in the horn of Africa7,8.
A unique feature of TB in West Africa is the presence of six major lineages (Lineages 1–6). The Euro-American (Lineage 4) and the two M. africanum lineages (Lineages 5 and 6) are the most common causes of human pulmonary TB in West Africa9. The Cameroon genotype of Lineage 4 (also known as LAM-10) is dominant in much of West Africa10,11,12,13,14, although the Ghana genotype of Lineage 4, which often have spoligo patterns that belong to T1 clade, is also disseminated across almost all West African countries9. The two M. africanum lineages are generally restricted to West Africa and cases outside the region are often linked to West African migrants9. Although the prevalence of Beijing strains (lineage 2) is low in West Africa a high prevalence has been reported in Benin and Senegal, prompting concerns about its emergence in the region15,16.
Despite the availability of effective treatment, tuberculosis (TB) remains one of the leading causes of death from infectious disease globally. In 2015, the WHO estimated that there were over ten million new cases of TB globally and 1.5 million deaths17. In sub-Saharan Africa, the fight against TB faces unique challenges due to the combination of the HIV epidemic, the emergence of multidrug-resistant (MDR-TB) strains and inadequate infrastructure for TB case management18,19. A drug resistance survey of MTBC drug resistance in West Africa was conducted between 2008 and 2013 by the West African Nodes of Excellence for AIDS Tuberculosis and Malaria (WANETAM). The survey showed that the estimated prevalence of MDR-TB in West Africa was likely being underestimated and called for urgent measure to tackle the growing threat from MDR-TB in West Africa, one of the poorest regions in the world20.
Bamako, the capital city of Mali, is one of few West African cities that have a well-structured hierarchy for TB case management. However, in recent years, Bamako has been faced with an emerging threat from MDR-TB20,21. Between 2006 and 2014 a surveillance was carried out in Bamako, which included 522 patients treated for pulmonary tuberculosis Bamako22. Phenotypic drug susceptibility testing on 337 unique isolates of MTBC showed resistance to at least one drug was found in 127 (37.7%) of which 75 (22.3%) were multidrug resistance (MDR)22. Spoligotyping confirmed that the most prevalent genotypes over the ten-year study period were MTB T1 (31.9%) and MTB LAM10 (Cameroon Genotype) (15.3%) from lineage 4 and M. africanum 2 (16.8%)23. These proportions were similar to results published by Traore et al. in 2012 where he reported that the most prevalent genotypes causing tuberculosis in Bamako were also MTB T1 (38.9%), M. africanum 2 (MAF2; 26.2%) and MTB LAM 10 (10.3%)21. Over the ten year surveillance period 50% of all cases of MDR-TB were associated with strains belonging to the T123 reinforcing the previous findings that the T1 genotype was associated with MDR-TB in Bamako21.
Genomics has recently become an important tool in clinical management of TB cases24,25,26,27. By studying the genomes of TB isolates, we can infer resistance to anti-TB drugs, unravel transmission chains and gain insights into the evolutionary mechanisms that shape the epidemiology of TB26,28,29,30. Here, we present insights into the evolutionary mechanisms contributing to the rise of MDR-TB in Bamako. Although our sample size was limited, we tried to gain insights into the risk factors associated with MDR-TB in this setting using a logistic regression analysis. Our data highlights how the evolution of drug resistance hampers the fight against TB in a low-resource setting.
Results
Patient characteristics and genomic summary
A subset of randomly selected isolates from patients who had participated in study protocols at the University of Bamako were selected for whole genome sequencing (25). Isolates from 76 patients, who included 21 females and 55 males and individuals aged between 3 to 78 years, were analysed; these included 12 MDR-TB cases (Table 1). Most patients (n = 61, 80%) had not been previously treated for TB. While nine of the 15 retreatment cases (60%) had MDR-TB, only three among sixty-one new cases (5%) were MDR-TB. Sequencing reads mapped to the H37Rv reference genome with an average coverage of 33-fold (range 10 to 93-fold). De novo draft assemblies had an average of 4.4 million bases (range 4294898 to 5984674) and an average N50 of 57958 (range 26213 to 82606).
Phylogeny
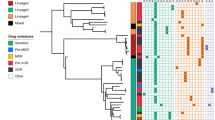
A time dated maximum likelihood phylogeny was reconstructed from 8508 variant core genome sites using BEAST. All the human-associated lineages of MTBC from the “modern” clade (Lineages 2,3 & 4) were detected among patients from Mali (Table 2). The dominant lineage was the Euro-American lineage, Lineage 4, and within Lineage 4 the Cameroon genotype was the most prevalent genotype (n = 20, 26%) (Fig. 1).

A time-dated phylogenetic tree of 76 MTBC isolates from pulmonary tuberculosis patients treated in Bamako, Mali between 2006 and 2013. Phylogenetic tree is shown alongside metadata including genotype, residential province, MDR-TB status based on phenotypic testing, HIV status and treatment history (new case or retreatment). The numbers on the x-axis show the number of years from the root. Closely related isolates from the Ghana genotype and Cameroon genotype are highlighted in blue and pink boxes respectively. White boxes in the metadata indicate missing data. The dashed line is the H37Rv reference genome. Branches are coloured based on genotype and labelled with SNP barcoding genotypes. Heat maps show phenotypic resistance and presence of resistance genotypes for first line drugs.
The Ghana genotype was the second most common genotype isolated from patients in the genomics subset. The Ghana genotype was represented by 16 isolates—seven of these were MDR-TB, therefore, the Ghana genotype represents over half of the 12 MDR-TB isolates in the genomics dataset. Patients infected with the Ghana genotype were over 4 times more likely to have MDR-TB than patients infected with other genotypes but the association was not significant after adjusting for retreatment cases and whether the patients resided in Bamako or not (OR 4.53, 95% CI 0.9–22.7, p value 0.067) (Table 3). Retreatment cases were 30 times more likely to have MDR-TB after adjusting for Ghana genotype and province of residence (OR 29.7, 95% CI 27.9–31.6, p value < 0.001).
Other Euro-American genotypes included the LAM genotype (n = 6), two H37Rv-like isolates and one isolate from the Uganda genotype. Four isolates of the Beijing genotype from Lineage 2, including one MDR-TB, formed a monophyletic clade where isolates differed by an average of 23 core genome SNVs. Our estimates suggest that our Beijing genotype isolates shared a common recent ancestor 40 years ago. Five patients were infected with MAF2 strains, but none were MDR, and another four were infected with the East Asian Indian lineage strains including one MDR-TB retreatment case.
Roles of the Ghana and Cameroon genotypes of MTBC
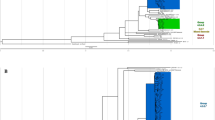
Our phylogeny revealed a sub-clade of Ghana genotype isolates that diverged from a common recent ancestor approximately 30 years ago (95% HPD intervals 24.42–39.44) (Fig. 2). This sub-clade, which is highlighted in blue in Fig. 1, included seven MDR-TB and six non-MDR-TB isolates. On average, strains within this cluster differed by 12 core-genome SNVs. The most divergent pair of strains within this cluster differed by 24 core genome SNVs. The katG Ser315Thr isoniazid resistance mutation was conserved in all the isolates and in one non-MDR isoniazid resistant isolate. Three isoniazid resistant isolates also acquired the fabG1 inhA promoter-region mutation that is known to confer isoniazid and ethionamide resistance. MDR-TB isolates acquired rifampicin resistance through several different mutations in the rpoB gene (Fig. 2).

A time-dated phylogenetic tree of all Ghana genotype isolates in this dataset to highlight the evolution of MDR-TB in this lineage. Phylogeny shown alongside MDR status, treatment status, HIV status, phenotypic resistance profiles and resistance mutations for first line drugs are listed. Heat maps show phenotypic resistance and presence of resistance genotypes for first line drugs. Internal nodes are labelled with age in years.
Phylogenetic dating suggested that the Cameroon genotype isolates diverged from a common recent ancestor approximately 161 years ago (95% HPD intervals 129.52–195.39) to form three clusters (Fig. 3). The cluster of closely related genomes highlighted in pink in Fig. 1 was a sub-clade of the Cameroon genotype clade that encompassed eight non-MDR isolates. This sub clade was highly conserved and isolates within it differed by average pairwise distance of 8 core genome SNVs (range 0 to 13 SNVs). We estimated that this cluster emerged approximately 22 years ago (95% HPD intervals 15.32–29.61) with within-cluster internal node ages ranging from 1.75 years to 16.55 years (Fig. 3).

A time-dated phylogenetic tree of all Cameroon genotype isolates in our dataset. The phylogeny is shown alongside patient metadata and date of isolation to highlight the evolution of this emerging genotype in Bamako. Internal nodes are labelled with age in years.
Evidence of community transmission
Potential transmission clusters were identified as sub clades within the phylogeny that had an average pairwise genetic distance of 12 or less SNVs. Two clusters were identified, a Cameroon genotype cluster and a Ghana genotype cluster, which were illustrated by highlighting the subclades on the phylogenetic tree in Figs. 1–3. The average pairwise genetic differences in the Cameroon genotype (7 SNVs) cluster was significantly lower than the average distance in the Ghana genotype cluster (p value < 0.0001) (Fig. 4). The median pairwise distance between patients’ residential provinces as approximately 11 km in both clusters (Fig. 4) as most patients resided in provinces within Bamako.

An illustration of the pairwise genetic distances between isolates and the pairwise distance between patients’ residential province within the potential transmission clusters. The lines on the charts show mean genetic distance and median distance respectively with error bars indicating the 95% confidence intervals.
Further evidence of community transmission was the identification of the same strain (0 SNV difference) from different patients on four occasions (marked in Fig. 1). In case 1 the strain was a Cameroon genotype strain, which was not from the highlighted transmission cluster. The strain was isolates from two patients in Bamako in 2013 residing in provinces 8 km apart. Case 2 also involved a Cameroon genotype strain, which was part of the transmission cluster. This strain was isolated from a patient in Senou 2012 and a second patient in Daoudabougou, Bamako in 2013 (~20 km apart). Moreover, this strain was closely related (2 SNV difference) to another strain isolated from a resident Kayes (~500 km from Bamako) in 2013. Case 3 involved a strain from the X-type genotype, which was found in two patients in residing in Bamako, Yirimadio and Kalaban-Coura (~ 10 km apart), in 2010. In case 4 the strain was within the Ghana genotype transmission cluster. Both patients, a male and a female aged 18 and 4 years respectively, were resident in Bamako in provinces 7.5 km apart and were both recruited in June 2013. This strain was closely related (3 SNV difference) to two other strains within the transmission cluster, one isolated from resident of Bamako in 2013 and the second one isolated in 2007 from a patient whose provincial domicile was not known.
Discussion
Estimating the burden of drug resistant tuberculosis in Mali has been challenging due to political instability and weak infrastructure. Laboratory based drug resistance surveillance carried out in Bamako between 2006 and 2014 showed that the levels of MDR-TB among patients has remained constant and relatively high22. Our study employs whole genome sequencing to study the evolutionary mechanisms driving the rise of MDR-TB in Mali in recent times and the emergence of virulent lineages of MTBC using a subset of isolates reported in Gehre et al. (25).
Our data shows that an important driver of MDR-TB in Bamako is a sub-clade of the Ghana genotype, which often has spoligo typing patters belonging to the T1 family. This family has previously been implicated with a higher likelihood of resistance compared to other genotypes21. The MDR-TB Ghana genotype sub-clade emerged approximately 30 years ago, most likely through the initial acquisition on the katG Ser415Thr isoniazid resistant mutation. Isoniazid resistance probably presented a selective advantage to the ancestral clone, allowing it to form divergent sub-clones that acquired rifampicin resistance through different mechanisms and is now the leading cause of MDR-TB in Bamako.
The main genotypes of MTBC known to cause tuberculosis in Bamako are the T1 family, Cameroon genotype (LAM10) and the M. Africanum West Africa 2 lineage strains21,23. Our data suggests that the Cameroon genotype is on the rise in Bamako compared to recent estimates21 and there is evidence of community transmission. Although the emerging Cameroon genotype strains are non MDR-TB, the situation should be carefully monitored. In Nigeria, a neighbouring West African country, we have seen evidence of widespread MDR-TB within the Cameroon genotype and signs of community transmission31.
The first report of Beijing strains causing tuberculosis in Bamako was in 2008 when unrelated strains were detected in two patients with active pulmonary tuberculosis32. Here we identified a clade of related Beijing isolates that differed on average by 22 SNVs (range 12 to 30) including one MDR-TB isolate. The Beijing genotype has emerged around the world33,34 and has demonstrated the ability to spread rapidly once introduced into a new population35. Bioinformatics approaches have identified mutations in virulence genes that are unique to the Beijing genotype36 and mutations in immune response associated genes that may aid host-pathogen co-evolution to the detriment of TB control37. The Beijing genotype has also been linked to MDR-TB in other settings38. Tuberculosis surveillance in Mali would be helpful in identifying the extent to which the Beijing and other virulent genotypes have spread in the community.
A disproportionate number of MDR-TB cases end up in retreatment or treatment failure. This is because in Bamako only retreatment or treatment failure patients were subjected to drug susceptibility testing at the National Tuberculosis Reference Laboratory. New cases with MDR-TB would not be detected unless they were enrolled in research protocols, at the SEREFO program where all isolates undergo routine phenotypic or genotypic susceptibility testing22. Implementing drug susceptibility testing across all public hospitals is not cost-effective but the Hain MTBDRplus Line Probe Assay could provide an effective screening tool to detect the most common forms of isoniazid and rifampicin resistance.
Conclusion
MDR-TB is a growing public health concern in Bamako. MDR-TB in Bamako is often caused by a 30-year old sub clade of the Ghana genotype. Introducing molecular susceptibility testing as a screening tool in public hospitals will improve the early detection of MDR-TB, improve treatment outcomes and curb the spread of the MDR-TB including the Ghana genotype MDR-TB sub clade.
Methods
Demographics of study area
The study participants from Mali were recruited in the capital city Bamako, where according to the Ministry of Health more than a third of all TB cases in Mali were managed in 201439. Bamako is divided into six districts and each district has a TB referral health centre that is equipped with diagnosis and treatment facilities. Our isolates were collected at the University Teaching Hospital as part of research protocols, which acts as the principal TB referral centre. Retreatment cases from across Mali are referred to the University Teaching Hospital for further investigation and treatment.
Ethical approval and informed consent
All study protocols were approved by the ethic committee of the faculty of medicine, pharmacy and dentistry of Bamako as well as the MRC Unit The Gambia at LSHTM and Gambia Government joint ethics committee. Signed informed consent was obtained from all patients and from the legal guardian for patients under 18 years of age. All methods were carried out in accordance with relevant guidelines and regulations.
Study isolates
In a surveillance of tuberculosis between 2006 and 2014, which included 522 patients treated in Bamako, co-author Diarra and colleagues22. From this dataset a subset of 208 isolates were sent to MRC Unit The Gambia at LSHTM, as part of the WANETAM survey on drug resistant tuberculosis in West Africa20. This is an ad hoc dataset whereby 85 isolates were chosen at random without any set criteria.
Sub cultured mycobacterial isolates were sent to the MRC Unit The Gambia at LSHTM for viability and antimicrobial susceptibility testing. Isolates were sub-cultured onto MGIT 960 system (Becton Dickinson, Oxford Science Park, Oxford, UK) for viability testing. Positive cultures were tested for purity by inoculation on blood agar and followed up only if there was no growth after 24 hours incubation. The presence of MTBC was confirmed by Ziehl–Neelsen staining. Susceptibility testing for the first-line drugs was performed on the MGIT 960 system (Becton Dickinson, Oxford Science Park, Oxford, UK) according to manufacturer’s instructions40: streptomycin (STR, 1 µg/mL), isoniazid (INH, 0.1 µg/mL), rifampicin (RIF, 1 µg/mL), and ethambutol (EMB, 4.5 µg/mL). Multidrug-resistant strains (resistant to isoniazid and rifampicin) were further tested for susceptibility to the second-line drugs capreomycin (CAP, 2.5 µg/mL), ofloxacin (OFX, 2 µg/mL), and ethionamide (ETH, 5 µg/mL) (Sigma-Aldrich, St. Louis, Mo, USA)20. The MRC Unit The Gambia at LSHTM TB Diagnostics Laboratory participates in external quality assurance from the National External Quality Assessment Service (NEQAS), UK (http://www.ukneqas.org.uk/) and is ISO15189:2012 accredited for’ first- and second-line DST together with other TB diagnostic assays.
Whole genome sequencing
In order to extract whole-genomic DNA isolates were grown in Middlebrooks 7H9 liquid medium (Sigma Aldrich, Gillingham, Dorset, UK) for 3–8 weeks and the presence of MTBC was confirmed by the rapid test CapiliaTM TB-Neo (Sigma Aldrich, Gillingham, Dorset, UK) or BD MGIT™ TBc (Becton, Dickinson and Company, Oxford Science Park, Oxford, UK). To test for purity the culture medium was streaked onto blood agar and incubated at 37 °C for 48 hours. Finally genomic DNA extraction was performed using the cetyl trimethylammonium bromide (CTAB) method41. Genomic DNA libraries were prepared for whole genome sequencing using the Nextera XT library prep kit according to manufacturer’s instructions (Illumina, Little Chesterford, UK) and sequenced on an Illumina MiSeq following the manufacturer’s instructions (Illumina, Little Chesterford, UK).
Genomic and phylogenetic analysis
An adhoc subset of eighty-five isolates of MTBC were selected from 208 archived Malian isolates for whole-genome sequencing. Seven isolates that had low coverage (<10X) and two isolates that had no patient metadata or antimicrobial susceptibility profiles were excluded from the analysis. Sequencing reads were uploaded on the PhyResSe webtool for lineage assignment and antimicrobial susceptibility prediction29. Phylogenetic lineages were inferred based on the whole genome single nucleotide polymorphism typing scheme described by Homolka and colleagues42.
For each isolate de novo contigs were generated from the paired-end sequencing reads using SPAdes (kmers: 21, 33, 55, 77, 99 and 127)43. To correct assemblies, SOAPaligner v2.21 was used to to remap sequencing reads onto each assembly and bases in the assembly were filtered based on a quality score <20 and <10 reads mapped. Each assembly was mapped to the H37Rv reference genome using the nucmer function in MUMmer44 to generate a consensus sequence and all concensus sequences were compiled in a multiple sequence alignment with the reference genome. Using custom Perl scripts all repeat regions identified by Sergeant et al.45 were removed from the alignment and the core genome was inferred from the multiple sequence alignment as sites conserved in all isolates46,47.
Single nucleotide variants (SNVs) were extracted from the core genome multiple sequence alignment using custom Perl scripts and were uploaded on the Bayesian Evolutionary Analysis Sampling Trees (BEAST Geo Shere package version 1.1.2) tool48. A time dated maximum likelihood phylogeny was reconstructed from variable sites in the core genome using a relaxed clock gamma time reversible model with substitution rates estimated from the sequence alignment data and a Yule model for birth rate. Date of sample collection (year and month of collection) and GPS coordinates for patients’ residential province were uploaded as traits. The phylogenetic tree was visualised in the Interactive Tree of Life (iTol)49 and FigTree (Version 1.4.2) and annotated using Inkscape50. A multifasta containing variant core genome sites was uploaded on MEGA 7 and a distant matrix was generated based on the pairwise number of differences between isolates.
MDR-TB risk factor analysis
The associated metadata for each patient was uploaded onto Stata version 12.0 for statistical analysis. A logistic regression analysis using the Chi square test was performed to test whether isolates belonging to the Ghana genotype were more likely to be MDR-TB than isolates belonging to other lineages. We controlled for whether the patients were retreatment cases of new cases and error bars were adjusted for based on the province of origin of the patient.
Study limitations
The limitation of the study is that patients were recruited from the University teaching hospital, which is a referral center. The prevalence of genotypes in our dataset may not be an exact representation of the prevalence of genotypes in the communities within Bamako.
Data declaration
The sequencing reads generated for the analysis presented in this paper have been uploaded onto the European Nucleotide Archives under the project accession number PRJEB27446.
References
Gagneux, S. In Pathogenesis of Mycobacterium tuberculosis and its Interaction with the Host Organism 374, 1–25 (Springer Berlin Heidelberg, 2013).
Comas, I. et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nature genetics 45, 1176–1182 (2013).
Bos, K. I. et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature 514, 494–497 (2014).
Gagneux, S. & Small, P. M. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. The Lancet. Infectious diseases 7, 328–337 (2007).
Gagneux, S. et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America 103, 2869–2873 (2006).
Coscolla, M. & Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Seminars in immunology 26, 431–444 (2014).
Firdessa, R. et al. Mycobacterial Lineages Causing Pulmonary and Extrapulmonary Tuberculosis, Ethiopia. Emerging infectious diseases 19, 460–463 (2013).
Blouin, Y. et al. Significance of the Identification in the Horn of Africa of an Exceptionally Deep Branching Mycobacterium tuberculosis Clade. PloS one 7, e52841 (2012).
Gehre, F. et al. A Mycobacterial Perspective on Tuberculosis in West Africa: Significant Geographical Variation of M. africanum and Other M. tuberculosis Complex Lineages. PLoS neglected tropical diseases 10, e0004408 (2016).
Niobe-Eyangoh, S. N. et al. Genetic biodiversity of Mycobacterium tuberculosis complex strains from patients with pulmonary tuberculosis in Cameroon. Journal of clinical microbiology 41, 2547–2553 (2003).
Thumamo, B. P. et al. Molecular epidemiology and genetic diversity of Mycobacterium tuberculosis complex in the Cross River State, Nigeria. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases 12, 671–677 (2012).
Godreuil, S. et al. First molecular epidemiology study of Mycobacterium tuberculosis in Burkina Faso. Journal of clinical microbiology 45, 921–927 (2007).
Lawson, L. et al. A molecular epidemiological and genetic diversity study of tuberculosis in Ibadan, Nnewi and Abuja, Nigeria. PloS one 7, e38409 (2012).
Yeboah-Manu, D. et al. Genotypic diversity and drug susceptibility patterns among M. tuberculosis complex isolates from South-Western Ghana. PloS one 6, e21906 (2011).
Affolabi, D. et al. First molecular epidemiological study of tuberculosis in Benin. Int. J. Tuberc. Lung Dis. 13, 317–322 (2009).
Diallo, A. B., Walbang, O. G. & Camara, M. Molecular genotypes of Mycobacterium tuberculosis strains circulating in Dakar, Senegal. African Journal of … (2016).
World Health Organization. WHO|Global tuberculosis report 2016. WHO (2016).
Zumla, A., Petersen, E., Nyirenda, T. & Chakaya, J. Tackling the Tuberculosis Epidemic in sub-Saharan Africa – unique opportunities arising from the second European Developing Countries Clinical Trials Partnership (EDCTP) programme 2015–2024. International Journal of Infectious Diseases 32, 46–49 (2015).
Lukoye, D. et al. Variation and risk factors of drug resistant tuberculosis in sub-Saharan Africa: a systematic review and meta-analysis. BMC Public Health 2015 15:1 15, 291 (2015).
Gehre, F. et al. The emerging threat of pre-extensively drug-resistant tuberculosis in West Africa: preparing for large-scale tuberculosis research and drug resistance surveillance. BMC Medicine 14, 160 (2016).
Traore, B. et al. Molecular strain typing of Mycobacterium tuberculosis complex in Bamako, Mali. Int. J. Tuberc. Lung Dis. 16, 911–916 (2012).
Diarra, B. et al. Tuberculosis drug resistance in Bamako, Mali, from 2006 to 2014. BMC infectious diseases 16, 714 (2016).
Togo, A. C. G. et al. The most frequent Mycobacterium tuberculosis complex families in mali (2006–2016) based on spoligotyping. Int J Mycobacteriol 6, 379–386 (2017).
Witney, A. A. et al. Clinical Application of Whole-Genome Sequencing To Inform Treatment for Multidrug-Resistant Tuberculosis Cases. Journal of clinical microbiology 53, 1473–1483 (2015).
Köser, C. U. et al. Routine Use of Microbial Whole Genome Sequencing in Diagnostic and Public Health Microbiology. PLoS pathogens 8, e1002824 (2012).
Köser, C. U. et al. Whole-Genome Sequencing for Rapid Susceptibility Testing of M. tuberculosis, https://doi.org/10.1056/NEJMc1215305 369, 290–292 (2013).
Brown, A. C. et al. Rapid Whole-Genome Sequencing of Mycobacterium tuberculosis Isolates Directly from Clinical Samples. Journal of clinical microbiology 53, 2230–2237 (2015).
Kay, G. L. et al. Eighteenth-century genomes show that mixed infections were common at time of peak tuberculosis in Europe. Nature communications 6, 6717 (2015).
Feuerriegel, S. et al. PhyResSE: a Web Tool Delineating Mycobacterium tuberculosis Antibiotic Resistance and Lineage from Whole-Genome Sequencing Data. Journal of clinical microbiology 53, 1908–1914 (2015).
Hatherell, H.-A. et al. Interpreting whole genome sequencing for investigating tuberculosis transmission: a systematic review. BMC Medicine 14, 21 (2016).
Senghore, M. et al. Whole-genome sequencing illuminates the evolution and spread of multidrug-resistant tuberculosis in Southwest Nigeria. PloS one 12, e0184510 (2017).
Diarra, B. et al. Mycobacterium tuberculosisBeijing Strain, Bamako, Mali. Emerging infectious diseases 16, 361–363 (2010).
European Concerted Action on New Generation Genetic Markers and Techniques for the Epidemiology and Control of Tuberculosis. Beijing/W Genotype Mycobacterium tuberculosisand Drug Resistance. Emerging infectious diseases 12, 736–743 (2006).
Glynn, J. R., Whiteley, J., Bifani, P. J., Kremer, K. & van Soolingen, D. Worldwide Occurrence of Beijing/W Strains of Mycobacterium tuberculosis: A Systematic Review. Emerging infectious diseases 8, 843–849 (2002).
Cowley, D. et al. Recent and Rapid Emergence of W‐Beijing Strains of Mycobacterium tuberculosisin Cape Town, South Africa. Clin Infect Dis. 47, 1252–1259 (2008).
Jia, X. et al. The Bioinformatics Analysis of Comparative Genomics of Mycobacterium tuberculosis Complex (MTBC) Provides Insight into Dissimilarities between Intraspecific Groups Differing in Host Association, Virulence, and Epitope Diversity. Front Cell Infect Microbiol 7, 88 (2017).
Parwati, I., van Crevel, R. & van Soolingen, D. Possible underlying mechanisms for successful emergence of the Mycobacterium tuberculosis Beijing genotype strains. The Lancet. Infectious diseases 10, 103–111 (2010).
Eldholm, V. et al. Armed conflict and population displacement as drivers of the evolution and dispersal of Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America, 201611283, https://doi.org/10.1073/pnas.1611283113 (2016).
la santé et de lHygiene Publique, de, M. Direction nationale de la sante- Rapport d’Activités du Programme National de Lutte contre la Tuberculose, 2013, Mali 2014 (2014).
Ardito, F., Posteraro, B., Sanguinetti, M., Zanetti, S. & Fadda, G. Evaluation of BACTEC Mycobacteria Growth Indicator Tube (MGIT 960) automated system for drug susceptibility testing of Mycobacterium tuberculosis. Journal of clinical microbiology 39, 4440–4444 (2001).
Hosek, J., Svastova, P., Moravkova, M. & Pavlik, I. Methods of mycobacterial DNA isolation from different biological material: a review. Veterinarni medicina (2006).
Homolka, S. et al. High resolution discrimination of clinical Mycobacterium tuberculosis complex strains based on single nucleotide polymorphisms. PloS one 7, e39855 (2012).
Bankevich, A. et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing, https://doi.org/10.1089/cmb.2012.0021 19, 455–477 (2012).
Delcher, A. L., Salzberg, S. L. & Phillippy, A. M. Using MUMmer to Identify Similar Regions in Large Sequence Sets. 10.3.1–10.3.18, https://doi.org/10.1002/0471250953.bi1003s00 (John Wiley & Sons, Inc., 2003).
Chan, J. Z.-M. et al. Metagenomic Analysis of Tuberculosis in a Mummy. New England Journal of Medicine 369, 289–290 (2013).
Zhou, Z. et al. Neutral genomic microevolution of a recently emerged pathogen, Salmonella enterica serovar Agona. PLoS genetics 9, e1003471 (2013).
Zhou, Z. et al. Transient Darwinian selection in Salmonella enterica serovar Paratyphi A during 450 years of global spread of enteric fever. Proceedings of the National Academy of Sciences of the United States of America 111, 12199–12204 (2014).
Bouckaert, R. et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10, e1003537 (2014).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic acids research 44, W242–W245 (2016).
Schöler, U. Inkscape, https://doi.org/10.3139/9783446441972 (Carl Hanser Verlag GmbH & Co. KG, 2014).
Acknowledgements
We wish to acknowledge the University Clinical Research Center (UCRC)-SEREFO-Laboratory, University of Sciences, Techniques and Technologies of Bamako (USTTB). We thank the Medical Research Council Unit the Gambia hosted at the London School of Hygiene and Tropical Medicine for hosting our research. Whole genome sequencing was performed at the University of Warwick, Warwick Medical School, Microbiology and Infection Unit. Whole genome sequencing was funded by a seed fund awarded by the Strategic Tuberculosis Network. The MRC Cloud Infrastructure for Microbial Bioinformatics (CLIMB) remote servers were used in the bioinformatics analysis. This study and the West African Network for TB AIDS and Malaria (WANETAM II) are supported by the European & Developing Countries Clinical Trials Partnership (EDCTP) Grant No: RegNet2015-1049.
Author information
Authors and Affiliations
Contributions
M.A., B.D.J., B.D., J.O. and F.G. designed the drug resistance survey. B.D., M.o.S., B.B., S.O., S.D. and S.e.D. coordinated the recruitment of patients and microbiological identification of MTBC in Bamako. M.S. and G.K. performed the whole genome library preparation and sequencing. M.S. and A.W. performed the bioinformatics analysis. M.S. and A.K.M. performed the statistical analysis. M.S., B.D., M.A., M.P., B.D.J., F.G. and B.K. contributed drafting the manuscript. All authors reviewed the final draft of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Senghore, M., Diarra, B., Gehre, F. et al. Evolution of Mycobacterium tuberculosis complex lineages and their role in an emerging threat of multidrug resistant tuberculosis in Bamako, Mali. Sci Rep 10, 327 (2020). https://doi.org/10.1038/s41598-019-56001-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-56001-0
This article is cited by
-
Whole Genome Sequence Dataset of Mycobacterium tuberculosis Strains from Patients of Campania Region
Scientific Data (2024)
-
Comparative genomics of drug-resistant strains of Mycobacterium tuberculosis in Ecuador
BMC Genomics (2022)
-
Whole genome sequencing of clinical samples reveals extensively drug resistant tuberculosis (XDR TB) strains from the Beijing lineage in Nigeria, West Africa
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
