
Abstract
Low-frequency vibrations are crucial for protein structure and function, but only a few experimental techniques can shine light on them. The main challenge when addressing protein dynamics in the terahertz domain is the ubiquitous water that exhibit strong absorption. In this paper, we observe the protein atoms directly using X-ray crystallography in bovine trypsin at 100 K while irradiating the crystals with 0.5 THz radiation alternating on and off states. We observed that the anisotropy of atomic displacements increased upon terahertz irradiation. Atomic displacement similarities developed between chemically related atoms and between atoms of the catalytic machinery. This pattern likely arises from delocalized polar vibrational modes rather than delocalized elastic deformations or rigid-body displacements. The displacement correlation between these atoms were detected by a hierarchical clustering method, which can assist the analysis of other ultra-high resolution crystal structures. These experimental and analytical tools provide a detailed description of protein dynamics to complement the structural information from static diffraction experiments.
Similar content being viewed by others
Introduction
Trypsin-like serine proteases are activated by heat, which point out that vibrations play a crucial role in their function. Trypsins catalyze the cleavage of peptide bonds in the gut and their catalytic activity increases with temperature (up to an optimal temperature)1. Trypsin is a globular protein and its folded structure is maintained in a wide range of aqueous solutions. This structure forms spontaneously below a melting temperature, but the conversion from the inactive trypsinogen to the active trypsin form requires the cleavage of the Lys-15/Ile-16 peptide bond by an enteropeptidase or an already active trypsin. Premature activation of trypsinogen in the acinar cells or the pancreatic duct leads to tissue damage, which contributes to the development of pancreatitis. The conversion between the active and inactive forms involves a conformational change, which is thermally activated2. Denaturation of serine proteases change their low frequency vibrations3. In addition, vibrations were suggested to influence the catalytic activity of trypsin4,5. Under certain conditions, the low-frequency vibrational modes can be described by a canonical (Boltzmann) probability distribution. Such a distribution is necessary to model enzyme reaction kinetics6,7. The ideal temperature dependence of catalytic rates is usually only observed in a narrow temperature range in enzyme catalyzed reactions8. Fröhlich proposed an alternative to the canonical probability distribution (commonly referred to as Fröhlich condensation)9, which may be more appropriate for non-isolated biological dissipative systems. In this out-of-equilibrium model, the probability distribution is skewed towards the lowest frequency mode in a coupled oscillator system. Different degrees of condensation have been investigated using simulations, which revealed that the rates of excitation, the exchange between the vibrational modes, thermalization and the temperature of the environment are important factors10,11,12,13,14. Collective modes in biological systems are postulated to play a role in long-range protein-protein interactions, the adjustment of biochemical reaction rates and information processing in semicrystalline, supramolecular systems10,15.
Dissipation can facilitate disorder to order transition in different types of condensed matter systems. External terahertz fields were shown to lead to ordering in amorphous sorbitol even at cryogenic temperatures16. In solution phase (poly(3-hydroxybutylate) (PHB)/chloroform mixtures) microcrystal formation was also observed following pulsed terahertz radiation17. Recently, structural changes were observed in lysozyme crystals when pumped with 0.4 THz radiation at room temperature18. In agreement with these findings, continuous excitation of fluorophore-labelled bovine serum albumin with visible light has resulted in a terahertz spectral signal, which evolved over minutes time scales19.
In NMR experiments using the serine protease inhibitor eglin c as model protein, dynamics of amino acid residues were affected by distant, relatively conservative mutations20,21. Site-specific mutations also affected functional motions far away from the mutational site in a kinase recognition loop22. Furthermore, it is frequently observed that a mutation or binding of a small molecule at one site can affect the dynamics at remote sites and computational studies could capture such aspects of correlated motions in proteins23,24. As a general trend, long-range allostery is modeled as a network of interactions from one point to another where the diffusive motions are transmitted stepwise through a chain of local interactions between the distant sites25,26,27. Of note, local interactions still dominate in these network models and hence the influence they exert is expected to decay with increasing distance as they propagate in space. Reactive, near-field interaction of atoms with an oscillating electric field is not assumed to be necessary for explaining allosteric changes. However, electric field pulses can cause reversible, concerted conformational changes at multiple locations in a protein in a similar manner as allosteric effect that connects distant sites together28. Whereas, slow, high-field electric pulses happen infrequently in biological systems near membranes, weak, evanescent terahertz fields are more common in aqueous solutions in the wake of thermal vibrations of partially or fully charged atoms.
The main methods to study low-frequency vibrations in proteins are terahertz spectroscopy, optical Kerr spectroscopy and inelastic/quasielastic neutron scattering. Terahertz spectroscopy29 was particularly successful in separating protein vibrations from those of the bulk solvent and was used to detect long-range vibrations in the protein scaffold30,31. Terahertz spectroscopic measurements also revealed long-range dynamics linked to the functional state of proteins32,33,34. Since the vibrational spectra lack resonant bands a few polynomials are sufficient to describe the entire terahertz region. Polarization varying anisotropic terahertz microscopy breaks this deadlock by recording two dimensional spectra from oriented proteins in crystals yielding dozens of absorption peaks35. Unfortunately, typical proteins contain thousands of atoms and many degrees of freedom. Consequently, the interpretation of the spectra critically depends on the assumed model of protein and solvent dynamics. In contrast, high-resolution X-ray crystallography recovers the three dimensional distribution of atomic displacements described with amplitudes and directions36. If the diffraction experiment is not time-resolved, the displacements are time averaged. Although the displacements are indistinguishable from a static distribution, since atoms move even at 100 K, they have a dynamic component. Nonetheless, the spatial distributions of every atom in the protein are observed, resulting in several thousand parameters. In this study, we demonstrate the use of high-resolution X-ray crystallography to study the terahertz dynamics of proteins. The dynamic component of the distribution was directly investigated by irradiating the crystals with terahertz radiation and compare spatial distributions to the dark state.
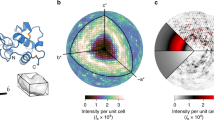
The basis of our analysis involves the six atomic displacement parameter (ADP) tensor components, which can be illustrated by thermal ellipsoids after diagonalization (Fig. 1, Eq. 1). The axis orientations of the ellipsoids represent the eigenvectors of the ADPs and the amplitudes of the axes correspond to the eigenvalues. For a typical protein atom, the amplitudes are not equal (the atom is anisotropic). To simplify the comparison of the ADPs, we adapted the Beq (which are analogous to isotropic B-factors, Eq. 2)37 and anisotropy metrics36 (henceforth ANISO, Eq. 3). ANISO varies between 0 and 1, where 1 indicates perfect isotropy.

Thermal ellipsoids of the carbonyl carbon (left) and amide nitrogen (right) atoms of Thr-125B located close to each other. The carbonyl carbon and amide nitrogen atoms display slightly oblate (pancake shape) and prolate (cigar shape) ellipsoids, respectively. The direction and length of the axes in the ellipsoids are uniquely defined by eigenvectors and the corresponding eigenvalues of the ADP tensors. These two atoms are located in close vicinity to one other, but they display the highest degree of similarity with more distant atoms. The figure was obtained using RASTEP36, numbering of amino acid residues follows the chymotrypsin convention66.
The amplitude and direction of atomic motions are valuable constraints for dynamic models. However, the complexity of protein crystal structures makes it difficult to comprehend the patterns that may correspond to the different dynamical models. We solve this problem by applying an unsupervised machine learning technique: hierarchical clustering. The inclusion of all the information from the ADPs dramatically improved the specificity of the clustering. Groups of atoms that share similar dynamics could be inferred from the similarity of their positional probability distributions.
In this study, the non-thermal effect of 0.5 THz radiation was investigated in bovine trypsin crystals cryo-cooled to 100 K. We analyzed the changes in the ADPs upon terahertz irradiation in order to reveal the dynamical component in the displacements. We discuss the eigenvalue magnitudes of the ADPs on individual amino residue level by averaging the atomic data and we compare those results to simulated data. Finally, we elaborate on the effects on individual atoms related to terahertz dynamics using a clustering method, which analyses all elements of the ADP tensors.
Results
We irradiated orthorhombic bovine trypsin crystals alternately with a 0.5 THz radiation, such that the odd frames correspond to “THz on”-states and the even frames correspond to “THz off”-states. As a reference, we performed the same data analysis on X-ray diffraction data collected from crystals that were not exposed to terahertz radiation. In contrast to an earlier experiment with hen egg white lysozyme18, bovine trypsin crystals were cryo-cooled to 100 K during the entire period of data collection with a dry nitrogen gas stream and suspended in a mylar loop without a surrounding plastic capillary. In total, five terahertz irradiated and four reference crystals were retained for analysis. Data processing statistics are summarized in Table S1.
Comparison of the irradiated and non-irradiated states of the crystals reveal differences between individual residues
An initial comparison of the ADPs was based on the difference in Beq-factor between equivalent atoms determined from the odd and even frames (Beq,odd-Beq,even). The mean and confidence interval (95%) of the difference in Beq-factor for amino acid atoms were determined. Figure 2A shows this type of comparison for the twenty amino acids in the reference (blue) and terahertz irradiated crystals (green). Beq appears to be either unchanged or similar to the references in the terahertz experiments except for the amino acid residue glutamine. The Beq of glycine, glutamine, serine, leucine and methionine atoms appear to decrease in odd data sets in the reference experiments.

(a) Beq-factor change between crystal structures determined from odd and even diffraction images (Beq,odd-Beq,even). (b) The anisotropy value (ANISO) change between crystal structures determined from odd and even diffraction images (ANISOodd-ANISOeven). Odd numbered diffraction images were recorded simultaneously with 0.5 THz irradiation in the THz irradiated crystals. Blue and green indicates reference (no terahertz irradiation at all) and terahertz irradiated crystals, respectively. Each average is calculated from the differences in Beq and ANISO values from the atoms belonging to each amino acid types. The error bars represent 95% CI of the mean.
The comparison of difference ANISO values (Fig. 2b) shows a limited overlap of confidence intervals (CI) between the reference and the terahertz categories. The greatest contrasts between the reference and terahertz categories are observed in atoms from isoleucine, glycine, cysteine, serine, leucine, tryptophan, lysine, asparagine and aspartate residues. In all these cases, the average ANISO of atoms decreased (i.e., they became more anisotropic) upon terahertz irradiation. The position of the amino acid residue in the structure is important, as shown by the ANISO value distribution of individual glycines, tryptophans and histidines are shown in Fig. 3. These amino acids do not have alternative conformations in the structure, which facilitates the comparison between different sites. From this comparison, it is obvious that most of the glycine residues behaved similarly in the terahertz radiation experiments compared to the references (Gly-43 and Gly-78 are exceptions), although in general, the atoms of the glycines in the N-terminal domain had a tendency to become more anisotropic when the crystals were subjected to terahertz radiation. Tryptophan residues became more anisotropic when all atoms were pooled together (Fig. 3) and Trp-51, Trp-215 and Trp-237 had different average anisotropies in relation to the reference, whereas Trp-141 stayed similar. Histidine residues, except catalytic His-57, appeared to be completely unaffected by terahertz radiation. As a general tendency, when there was a reversible change in anisotropy upon terahertz irradiation, it predominantly resulted in more anisotropic behavior in the residue atoms.

Comparison of ANISO change in individual (a) glycine (b) histidine and (c) tryptophan amino acid residues between odd and even diffraction images of the reference (blue) and terahertz irradiated (green) crystals. Each averaged difference is calculated from the differences in ANISO values. The error bars represent 95% CI of the mean.
Comparison to NMA and MD simulations
In order to model ADP tensor components (Uij) we performed NMA on an energy-minimized trypsin structure and generated random samples of the structure using all normal modes. The calculation assumed a temperature of 100 K. The Beqs of predicted atomic Uijs were approximately two orders of magnitude smaller than the experimentally observed values. Since NMA only explores the potential surface around a single minimum, it has a limited ability to follow protein dynamics, which consist of multiple conformations that exchange at slower than picoseconds timescales. Moreover, NMA is performed in a vacuum, which is not the case with solvated forms of proteins. Nevertheless, NMA is useful for inferring protein motions especially when such motions stem from the structure rather than the solvent dynamics, as is the case with cooled crystals, and hence we examined the correlation of NMA predicted Uij metrics with Molecular Dynamics (MD) simulations and also with the experimental data.
An alternative to NMA is MD simulations of solvated protein molecules. Due to the fact that MD force fields are defined at 300 K, and to facilitate conformational changes, we performed the simulations at 300 K. When flash-cooling proteins, conformational variation is locked into a static distribution, but still affects the displacement parameters. Since conformational sampling is more delicate than sampling from an NMA ensemble, we used four MD simulations to predict the Beq and anisotropy of the protein atoms and compared the values obtained to the corresponding parameters of the refined crystal structure (Fig. 4). The predicted Beq values were approximately on the same scale as the experimental values. They appeared to be underestimated for the well-ordered parts of the structure, but overestimated for the flexible regions. In particular, the Beq of Tyr, Asn, Gln, Phe and Lys residues were greatly overestimated compared to the experimental data. There was a 0.53 Pearson correlation between the calculated (from segments of four times 10 ns simulations) and the experimental means of Beq per residue (calculated for all equivalent atoms from the reference crystals). The mean ANISO values in amino acid residues showed a smaller correlation (0.39), and MD simulations generally overestimated the anisotropy. By contrast, ANISO values derived from NMA simulations were more isotropic and agreed better with the experimental data. MD simulations predicted Asn residues to be much more anisotropic relative to other amino acid types as well (Fig. 4d).

(a) Comparison of experimentally determined and simulated average Beq-factor in individual amino acid residues. (b) Comparison of average ANISO in individual amino acid residues. (c) Comparison of experimentally determined and simulated average Beq-factor in different amino acid types. (d) Comparison of average ANISO in different amino acid types. Blue and green correspond to the reference and terahertz irradiated crystals, respectively. Orange and purple indicate the predictions from molecular dynamics simulations and normal mode analysis, respectively.
Perhaps it is unrealistic to expect good agreement of Uij derived values between experiments and predictions on an absolute scale. Beq was found to be on approximately the same scale in MD simulations. Nevertheless, one expects that Beq will be estimated self-consistently and reproducibly by MD simulations. Since Pearson correlation coefficients (CC) are not affected by the absolute magnitudes of compared values, they are also a suitable measure for comparing crystal structure and predicted values (self- and cross-correlations). As Table S2 shows, the MD Beq estimations are not stable and their correlation with the estimated Beq decays over time. It does not matter which reference time point is chosen. For example, the simulated trajectory from MD1 had a Beq CC of 0.83, 0.71 and 0.64 when compared to MD2, 3 and 4, respectively. Choosing MD2 as a starting point also shows a decreasing CC trend. NMA Beq shows a substantially lower correlation with any of the MD predictions (the best correlation is 0.45). Moreover, when compared to the Beq of crystal structures, earlier trajectories show higher correlations than later ones irrespective of the crystal structure to which they are compared (Table S3). NMA Beq correlations with crystals is on par with some of the MD predictions (the best correlation is 0.45 to crystal x28 and x30). Reference crystal structures had Beq CC in the range of 0.99–0.82 among themselves due to experimental and modelling variation (Table S4).
Anisotropy values showed a smaller correlation within the experimental data and also between the theoretical predictions, although the predictions seem to become more stable over time (Tables S5, S6) than the CCs for Beq. Nevertheless, the combined experimental and modeling errors make predicted and experimental anisotropy less correlated (CC < 0.4 for MD and CC < 0.3 for NMA, Table S7). As such, we also have to consider the mismatch of the absolute level of anisotropy values between experiments and model predictions (Fig. 4b and 4d). We provide additional discussion about the NMA and MD modelling in the Supporting Online Material.
Metric-free clustering of atoms based on pairs of Uij matrices
Hierarchical clustering is an exploratory, unsupervised machine learning technique, which we use for identifying similar atoms (in a large pool of atoms) described by many features (elements of ADP tensors). Hierarchical clustering was used in the crystallographic analysis, for example by grouping isomorphous crystals together38 or detecting structural similarity39. In the context of analyzing ADPs, clustering algorithms have not been used before. Our atomic clustering method was based on similarities between the elements of ADP tensors, quantified by the Euclidean distance between the elements of the ADP tensors (Figure S1, Eq. 4). This way, atoms with similarly shaped ellipsoids, similar size and displacement directionality are grouped together. The heat map represents the magnitude of the tensor elements, providing a quick visual feedback about the similarity and the nature of the remaining differences. By changing the definition of distance, it is possible to emphasize different aspects of ADPs, but these alternative measures should converge to zero between identical ADPs. Different distance metrics capture small changes from ADP identity similarly. We focused our attention on the tips of the branches where the similarity between atoms is the highest and the type of distance is the least important. Another reason why we focused on the highest similarity between atoms is that they occur less likely by chance.
Completely ignoring certain aspects of ADPs for example considering only the mean displacement, related to the sum of the diagonal elements of ADP tensor (Beq) yields a very coarse clustering. However, slight differences in shape and orientation are enough to direct atoms to specific clusters. In Figure S1, the atoms from terahertz-radiated crystals are represented by green regions and these appear to cluster together. These clustered atoms tend to be located in the core regions of the protein structure.
In order to illustrate different types of formed clusters, Figs. 5–7 highlights different regions of the cluster map, separated by thousands of atoms. Figure 5 zooms in on the top highlighted region in Fig. S1. Here, we examine 7 out of 9 observations of the Nε atom of Gln-135 on one branch. The immediately adjacent branch on the dendrogram consists of the Nδ atom of Asn-223, where all of the observations of this atom in all crystals can be found. It is worth emphasizing that no other information was used other than the 2 × 6 elements of the Uij matrices. Despite this, it is possible to uniquely identify up to 9 identical atoms from a total pool of 15921 atoms. Thus, ADPs are highly reproducible and weakly affected by experimental and modeling errors. It is important to note that these two atoms were not close to one other in the crystal structure, but they shared a similar position in a chemical group. The thermal ellipsoids these atoms are illustrated in Figure S2.

Clustering appears to identify amide nitrogen atoms of Gln-135 and Asn-223 residues in multiple crystals (red). The two amino acids do not make intra- or intermolecular contacts. The trypsin structure is illustrated by a ribbon diagram. Those amino acid residues to which the clustered atoms belong are represented by sticks.

A cluster consisting of atoms from terahertz irradiated crystals that are involved in the catalytic process of the protein (Ser-195, His-57, Ser-214, Trp-215) and a high proportion of atoms originating from asparagine and tryptophan residues. The trypsin structure is illustrated by a ribbon diagram. Those amino acid residues to which the clustered atoms belong are represented by sticks.

A cluster consisting of atoms from mostly terahertz irradiated crystals that are involved in the catalysis and activation of trypsin (His-57, Asp-102, Asp-194 and Asp-189). A high proportion of atoms originates from tryptophan residues and all histidine residues are represented by atoms in the cluster. The trypsin structure is illustrated by a ribbon diagram. Those amino acid residues to which the clustered atoms belong are represented by sticks.
Figure 6 focuses on a central region highlighted in Fig. S1, where most of the clustered atoms share the fact that they were observed in terahertz pumping experiments. The highlighted cluster contains three observations of Cβ of Ser-195 and two observations of Cβ of His-57. Ser-195 carries out the nucleophilic attacks and His-57 accepts the protons generated during the nucleophilic attacks. The ADPs of the two Cβ atoms are illustrated as thermal ellipsoids in Fig. S3. Atoms from Trp-215 and Ser-214 were also observed in this region. The backbone of these two amino acids form the antiparallel β-sheet with the substrate peptide in the Michaelis-complex and acyl-enzyme, and in addition the hydroxyl group of Ser-214 is hydrogen bonded to the third catalytic amino acid, Asp-102, which is not part of this cluster. The residue Trp-237 is represented with multiple atoms in the branch and this residue appears to be affected by terahertz radiation.
Figure 7 shows the lower marked region in Fig. S1, where the link between His-57 and Asp-102 appears to be stronger. In particular, there is a similarity between Cε2 of His-57 and Oδ2 of Asp-102. In addition, other Oδ2 atoms of functionally important aspartates seem to be spatially tuned together: Asp-194, which forms a salt bridge with N-terminal Ile-16 (a step necessary to convert the enzyme into its active form)40, and Asp-189, which (in general) forms the salt bridge with the P1 residue of the substrate. In this crystal structure, Asp-189 forms a salt bridge with the amidinium group of the inhibitor benzamidine. It is important to mention that atoms belonging to Trp-237 and Trp-51 also appear in these trees, as the anisotropy of these tryptophanes is affected by terahertz radiation.
Discussion
ADP similarity between chemically similar atoms
Our analysis revealed that atoms in functionally important residues appear repeatedly together in local clusters. Atoms in separate branches are often predominantly observed in either terahertz radiated or non-radiated crystals, respectively. In Fig. 6, one such terahertz radiated branch (green) is highlighted. The large physical distance between these atoms does not appear to be a hindrance, whereas the chemical nature of the atoms clearly does have an influence. For example, not all carbon atoms are preferentially associated with other carbon atoms, rather carbon atoms in carbonyl, α and β positions in the amino acid residues are different enough to sort them to different clusters. (Fig. S3–S5) The association between the chemical nature of atoms and their ADPs is an unexpected result and requires an explanation.
One could thus consider the ADP similarity to be an effect of the atom types having a similar partial charge. For example, such an assumption could be applied for the amide side chain nitrogen of asparagine and glutamine (Fig. 5), which are assumed to have a strong and similar negative partial charge in most empirical force fields. In a protein, there is a wide palette of atom types, and we approximate each as having a specific static partial charge between approximately −0.8 and + 0.9. Consequently, each of these atom types may have a slightly different response to oscillating EM fields. The partial charges are distributed alternately in the protein structure forming a mosaic of partial charges where positively charged atoms are usually bonded to negatively charged atoms. As a result, polar vibrations (optical phonons) could emerge when exposed to oscillating terahertz EM fields. Since the wavelength of terahertz (millimeter) EM waves is much longer than the size of protein molecules these fields are virtually uniform across the protein molecule at any given moment. Terahertz EM fields could originate from the thermal motions in the protein (crystal) and the solvent and these together generate evanescent fields. We also periodically applied an external terahertz field, but the combination of these field vectors also spatially uniform over a protein molecule and the surrounding solvent molecules. The association between the chemical nature of atoms and their ADPs is not fully explained by the arguments above. For example, Cβ of Ser-195 is assumed partially positive, because of its bonding to the more electronegative hydroxyl group, whereas Cβ of His-57 is assumed to be more neutral, because it is attached to another carbon of an imidazole ring. Nonetheless, these two Cβ atoms have extremely similar ADP in several crystals (Fig. 6). If the two atoms of the same position (Cβ) are cluster together either they have a similar charge, which is not in agreement with the force field, but may be true due to local effects or it is not only the charge that is important.
Do protein domain structures emerge from entrainment of atomic displacements
In architecture, one models the dynamics of an arbitrary structure (a house or a bridge) and then alters the structure until the dynamical properties are acceptable. Molecular NMA follows this same conventional logic, except that making planned changes in a protein structure is not a trivial process. In contrast to conventional engineering, a protein structure is not created by placing the atoms into arbitrary positions; rather, it emerges spontaneously from a quite dynamic polymer under suitable conditions (protein folding). ADPs of atoms do not (exclusively) depend on where they are located in the structure, which suggests that most atoms are not passive passengers in their structural elements.
We showed that in the folded, crystalline form of a protein, atomic fluctuations are characteristic, stationary (reproducibly seen in multiple crystal structures) and coordinated across long distances connecting together different structural regions. At first glance, intriguingly, atoms of a sharply defined chemical nature align themselves with similar atoms in other identical chemical groups. Another non-random aspect is that atoms cluster together when they originate from terahertz pumped crystals (Figs. 6, 7 and S4). Terahertz radiation induced ordering is not specific to proteins and aqueous solutions, but it has been observed in amorphous solids and non-aqueous solutions16,17. Trypsin crystals are well ordered even before terahertz irradiation, but the dissipation of applied terahertz radiation enhances the clustering of atoms. Atoms from terahertz irradiated crystals more likely cluster together. Terahertz irradiation also increases the anisotropy of atoms in a large fraction of tryptophan residues. On the other hand, the ADPs in our reference crystals already provide substantial information about the long-range pattern: the pattern is not exclusively present in an over pumped condition.
In a protein crystal structure some, but not all, chemically similar atoms have similar ADPs (Figs. 5–7 and S4). The mechanism for selecting from the many chemically similar atoms needs to be investigated further. It appears that protein atoms form an intercalating mosaic of coexisting lattices (Fig. 6d), where it is common to see atoms from the same amino acid residues ending up in different clusters. Perhaps the most noticeable, but certainly not exclusive, example is the group of tryptophan residues, where the indole ring is often approximated as a rigid aromatic plane with torsional fluctuations around its two axes as the most important motions. Complicating this picture, the atoms within the indole ring cluster differently and share clusters with atoms in other tryptophan residues. Tryptophan residues are often difficult to replace by mutagenesis and play a key role in maintaining the thermodynamic stability of protein structures. They are one of the strongest promoters of ordered states41,42.
The disorder-to-order transition is also facilitated by asparagine and threonine residues42. In particular asparagine residues were poorly described by MD simulations, which predict larger Beq and lower ANISO in absolute terms and relative to other amino acid types (Fig. 4C,B). It appears that in the folded form, the side chain amide of asparagine residues undergoes very limited fluctuations even in the absence of local interactions. Given the limited local contacts, MD simulations predict more positional freedom than is experimentally observed in our measurements. In the terahertz experiments, some of the asparagine atoms also appear to clusters together with catalytic residues (Fig. 6). A preliminary proposal is presented on how ADP similarity might be linked to structural constraints and to the symmetry between chemical groups in a protein in the Supporting Online Material, Figs. S4 and S5.
Delocalized polar vibrations are important for structure and function prediction
Protein dynamics incorporates diffusive motions of folded and unfolded domains relative to one other (Fig. S6), conformational changes, elastic stretching (Fig. S6A), bending (Fig. S6C) and torsions. Rigid body approximation even at the level of small structural moieties such as aromatic side chains does not always provide natural clusters for ADP similarities. Our study demonstrates that within a folded unit polar terahertz vibrations can be identified (Fig. S6D). Importantly, the entrainment of these polar vibrations may have the ability to steer the orientation of involved atoms at great distances and the electron dynamics of covalent bonds provide additional local constraints for orienting the attached functional groups.
Physical proximity and local complementarity have been and still are the primary guiding principles in protein engineering. Local shape complementarity is necessary, but not sufficient condition for structure formation. Directed engineering of protein interfaces is less than straightforward due to the elusive (allosteric) influence of the rest of the structure. The similarity of parameters that describe fluctuations are not routinely analyzed. Biso-factors and NMR relaxation parameters by themselves do not contain much information: two atoms can easily have similar parameters, just by coincidence. Experimental and modeling errors make the comparison even more difficult. It requires series of observations43,44 in order to recognize the covariance of these parameters and assign multiple atoms to a specific functional role. With anisotropic ADPs, the chances of coincidental similarities are dramatically reduced. While multiple related structures help (as demonstrated in this study), it is possible to detect similarities between atoms with good confidence even in a single crystal structure. The examples described here show that the anisotropy of these fluctuations appears to have a functional relevance with two or more atoms with similar ADPs being responsible for a similar function (catalysis, conformational change between active and inactive form of the enzyme). These similarities can be revealed faster than time-consuming mutagenesis studies, which are also impossible when main chain atoms are concerned. While the extracellular enzyme trypsin is not typically seen as an allosteric enzyme, its substrate specificity, activation, and inhibitor binding involve diffusely distributed, concerted action of distant amino acid atom positions suggesting that the basis of allostery is a universal, emergent and dynamic process.
The identification of similar ADPs in a macromolecular structure is just the first essential step. The second and possibly more difficult step is to find and understand the geometric patterns between atoms belonging to the same ADP cluster. Since similar ADPs seem to be linked to the chemical nature of atoms, empirical geometric rules may be recognized or applied in the absence of ADP observations. These patterns would then assist the development of long-range ADP restraints, complementing restraints based on bonding connectivity and positional proximity. Improved ADP restraints may provide better electron density maps and more detailed structural models at lower crystallographic resolutions. More importantly, long-range geometric patterns between chemically similar atoms coupled to effective conformational sampling (simulated annealing) may be able to assist the de novo prediction of protein structures.
Conclusions
Our study focused on two inseparable questions: the effect of terahertz radiation on protein crystals and the terahertz dynamics of folded proteins. We used crystallographic ADPs to measure the effect of terahertz radiation on crystals cooled to 100 K. Locally, we observed an increase in anisotropy and focused our attention on glycine and tryptophan residues, and the catalytic residue His-57, where the most obvious changes in anisotropy occur. The clustering of atoms based on their ADPs provided a relational map, which complemented the positional analysis of atoms based on their proximity. We performed NMA and MD simulations to predict ADPs and achieved only limited success. The clustering revealed that chemically similar atoms tend to have ADPs that are more similar. These atoms are distributed sparsely in the protein structure and belong to functionally connected amino acid residues.
Methods
Protein crystallization and flash cooling
Bovine trypsin (Sigma) was dissolved in 30 mM HEPES pH 7.0, 3 mM CaCl2 and 6 mg/ml benzamidine to obtain 60 mg/ml protein solution. Trypsin was crystallized using the hanging drop vapor diffusion method, by mixing 5 μl protein solution and 5 μl of precipitant solution (18% PEG8000, 50 mM HEPES pH 7.0, 0.2 M Ammonium sulfate, 3 mM CaCl2 and 6 mg/ml benzamidine). After 3 days, orthorhombic crystals appeared with approximate dimensions of 100–300 µm. The crystals were harvested and soaked in cryoprotectant solution (additional 33% glycerol) for 1–2 seconds prior to flash cooling in liquid nitrogen.
X-ray diffraction data collection
X-ray diffraction data were collected at the EMBL P14 beamline in Hamburg. (Fig. S7) The incident X-ray beam was focused to a 10 µm horizontal × 50 µm vertical rectangular area at the sample position. The photon energy of the beam was 14 keV and the beam was attenuated to a photon flux of 4 × 1011 photons/s. Optimization of the experimental geometry allowed us to collect diffraction to higher resolution (wider angles) than previously18. The central panels of the Pilatus 6M-F detector were used (2463 × 2527 pixels) and the detector was placed at the distance of 130.4 mm from the crystal. We discuss the effect of X-ray radiation damage on ADPs in the Supporting Online Material and Fig. S8.
The crystals were oriented with the minikappa goniometer such that the normal vector of a clean crystal surface was aligned with the spindle direction. The beam cross section was positioned less than 100 μm from the crystal surface and 20 μm offset from the spindle axis. During X-ray exposure, the crystals were continuously rotated with 0.4 °/s while the Pilatus 6M-F was continuously recording images at 25 ms intervals. During each image recording period, the detector spent 22 ms with photon counting and 3 ms with I/O operations (read out).
The terahertz radiation was generated by an amplifier / frequency multiplier chain (X32 stage AMC, manufactured by Viginia Diodes Inc., Charlottesville, VA), which was driven by a MG3692c microwave signal generator (Anritsu). The input microwave radiation was set to 15.625 GHz (10 mW), which resulted in 0.5 THz radiation (1 mW). The diagonal horn antenna (WR-2.2) was positioned approximately at 0.5 cm from the crystals. Under the assumption of a Gaussian beam profile, the THz spot size was approximately 4 mm. This estimate assumes a waist radius of 1.3 mm and that the beam originates from approximately 1/3 inside the antenna. We examine the thermal effect of the terahertz radiation in the Supporting Online Material.
To reduce systematic differences associated with thermal effects, the terahertz radiation source was operated alternately. A DG645 pulse (delay) generator (Stanford Research Systems) was triggered on the raising edge of the EN OUT signal of the Pilatus 6M-F detector. Upon triggering, the pulse generator was programmed to send a 23.5 ms pulse to the terahertz radiation source (THzon period). Simultaneously, the pulse generator was set to ignore subsequent triggers for 40 ms. Consequently, every odd numbered image was taken from the crystal in the terahertz irradiated state whereas the even numbered images recorded the diffraction image of the crystal in the non-irradiated state.
Odd and even numbered diffraction images still contained sufficiently super sampled measurements to integrate their total intensity independently18,45. This applies even for the widest-angle reflections recorded for this study, observed far away from the spindle axis.
X-ray diffraction data analysis
The first diffraction image was always affected by the slow opening of the millisecond shutter and this image had systematically lower recorded scattering intensity. The data processing program XDS46 uses the first image of each data set for initial scale, which introduced a systematic difference between the data sets based on odd and even images. Therefore, the first odd and first even images of each data set were removed from subsequent analysis. In addition, the kappa goniometer shadow was identified in each data set and the affected images were removed. An even number of images were removed in order to maintain the equal number of odd and even numbered diffraction images.
The sets of diffraction images from the odd and even frames of each crystal were processed separately. By comparing the data and refinement statistics, we selected the crystals with generally high data quality, shown in Table S1. This pre-selection yielded 18 data sets, recorded from four reference and five terahertz radiated crystals. Moreover, since the frames affected by the goniometer shadow differed from crystal to crystal, the unaffected rotation wedges were integrated individually. The indexing and integration steps were carried out with the default settings in XDS. The subsequent scaling and merging were performed using the programs XSCALE and XDSCONV of the XDS package, respectively. In both programs, the standard settings for non-anomalous diffraction data were used.
Model building and structural refinement
The subsequent model building and structural refinement were performed on each individual state (the odd and the even frames) of the individual crystals, using Refmac5 of the CCP4 package47. A single starting model was refined against the 18 data sets (9 crystals, recorded on odd and even diffraction images). Refinement statistics is shown in Table S1. This initial structure was based on pdb entry 4I8G, and the structure was solved by molecular replacement48. The starting model included 2067 non-hydrogen atoms, 288 of which belonged to water molecules, 20 were heteroatoms and 1759 were part of amino acid residues. Riding hydrogen atoms were included during refinement.
The individual crystal refinement was carried out with Refmac5, in three steps: rigid body, restrained isotropic and restrained anisotropic refinement procedures. Each refinement procedure was performed with the default settings for 100 cycles with an automatic weighting of restraints.
ADP analysis and clustering
The diagonalization of the ADP tensor is defined as:
where Uij is the ADP tensor, P is the eigenvector matrix from the ADP tensor and E1, E2, E3 are eigenvalues of the ADP tensor. The eigenvalues and eigenvector are obtained by transforming the ADP tensor to its row echelon form: i.e by solving the equation Ux = 0, where x is a vector in R3 and 0 is the zero vector. Each atom in each crystal is described with two Uij matrices: in the “odd” and “even” state of the crystals. Odd and even states correspond to a “THz on” and a “THz off” state for THz irradiated crystals, respectively.
The equivalent B factors (Beq) are defined as:
where tr(Uij) is defined as the trace of the ADP tensor (i.e. U11 + U22 + U33). The anisotropy value (ANISO) is defined as:
i.e. the ratio of the minimum and maximum eigenvalue.
Through hierarchical clustering, all the observed atoms were iteratively grouped using the distance between their unmodified Uij matrices. By having two observations of each atom in the same crystal, we obtained two symmetric Uij matrices with six unique elements each.
We defined the Euclidian distance between atoms u and v using their experimental Uij matrices:
The clustering was thus performed in this twelve-dimensional feature space. For clustering, Ward’s method was used49. Ward’s method groups the input data into individual clusters and selectively reorganize the clusters to minimize the total within-cluster variance. A smaller within-cluster variance groups the clusters into a closer branch. The calculations and visualization were performed with the help of the python libraries numpy, scipy, pandas50, cctbx51, seaborn and NGLView52.
Apparent X-ray dose calculations
In order to compare the apparent absorbed doses in the different crystal structures first we estimated the cumulative absorbed X-ray dose corresponding to each diffraction image. The absorbed dose was calculated using the program RADDOSE 3D53. The apparent dose was calculated as the product of the dose per frame, multiplied with the weighted arithmetic mean of the number of frames for each crystal.
Normal mode analysis and molecular dynamics simulations
Normal mode analysis (NMA) was performed using the Gromacs simulation package54,55,56 version 2018–2, compiled with double precision. The structure was first energy minimized rigorously by employing several cycles of Steepest Descents and Conjugate Gradients energy minimization cycles. The CHARMM3657,58,59 force field was used in all calculations. Electrostatics and van der Waals forces were truncated at 1.8 nm with a shift cutoff from 1.5 nm. After energy minimization, the Hessian matrix was diagonalized and all eigenvalues and eigenvectors were used except the first six rotational and translational modes. An ensemble of structures was generated at 100 K by the Gromacs program gmx nmens and ADPs were calculated by the Gromacs program gmx rmsf for all non-hydrogen protein atoms.
Molecular dynamics simulations were carried out using energies and forces as implemented in the OPLS-AA/L force field60. The protein was solvated in a cubic box so that the minimum distance between any protein atom and the edge of the box was 1.0 nm. The water molecules were approximated with the TIP3P model61. Cl- ions were added in order to neutralize the charge. Before the MD simulation, internal constraints were relaxed by energy minimization. After the minimization, a restrained MD run was performed for 20 ps. During the restrained simulations, protein heavy atoms were fixed to their initial positions with a force constant of 1000 kJ mol−1 nm−2. The restraints were released and the system was equilibrated for 60 ns before data collection for analysis.
The temperature was kept constant (T = 300 K) by use of the velocity‐rescaling algorithm (τT = 0.1 ps)62. The pressure was coupled to an external bath with Berendsen’s coupling algorithm63 (Pref = 1 bar, τP=1 ps) during the equilibration and with the Parrinello–Rahman algorithm64 afterwards. Van der Waals forces were truncated at 1.0 nm with a plain cutoff. Long‐range electrostatic forces were treated using the particle mesh Ewald method65. Dispersion correction was applied for the energy and pressure.
After equilibration, the remaining 40 ns of simulations were divided into 10 ns segments and the ADPs were calculated with the Gromacs program gmx rmsf from the trajectory segments.
Accession code
Crystal structures and X-ray diffraction data is available at the Protein Data Bank under entries: THz irradiated crystals (odd frames/even frames): 6SV0/6SUX, 6SV8/6SV6, 6SVB/6SV9, 6SVG/6SVD, 6SVJ/6SVI, Reference crystals (odd frames/even frames): 6SVR/6SVN, 6SVV/6SVU, 6SVX/6SVW, 6SW0/6SVZ.
References
Toth, J. et al. Thermodynamic analysis reveals structural rearrangement during the acylation step in human trypsin 4 on 4-methylumbelliferyl 4-guanidinobenzoate substrate analogue. J. Biol. Chem. 281, 12596–12602, https://doi.org/10.1074/jbc.M512301200 (2006).
Toth, J. et al. Site directed mutagenesis at position 193 of human trypsin 4 alters the rate of conformational change during activation: role of local internal viscosity in protein dynamics. Proteins 67, 1119–1127, https://doi.org/10.1002/prot.21398 (2007).
Chou, K. C. Biological Functions of Low-Frequency Vibrations (Phonons).3. Helical Structures and Microenvironment. Biophysical Journal 45, 881–890, https://doi.org/10.1016/S0006-3495(84)84234-4 (1984).
Ma, W. Z., Tang, C. & Lai, L. H. Specificity of trypsin and chymotrypsin: Loop-motion-controlled dynamic correlation as a determinant. Biophysical Journal 89, 1183–1193, https://doi.org/10.1529/biophysj.104.057158 (2005).
Havsteen, B. H. A New Principle of Enzyme Catalysis - Coupled Vibrations Facilitate Conformational-Changes. Journal of theoretical biology 140, 101–127, https://doi.org/10.1016/S0022-5193(89)80032-3 (1989).
Petersson, G. A. Perspective on “The activated complex in chemical reactions” - Eyring H (1935) J Chem Phys 3: 107. Theor Chem Acc 103, 190–195, https://doi.org/10.1007/s002149900102 (2000).
Kamerlin, S. C. & Warshel, A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins 78, 1339–1375, https://doi.org/10.1002/prot.22654 (2010).
van der Kamp, M. W. et al. Dynamical origins of heat capacity changes in enzyme-catalysed reactions. Nature communications 9, 1177, https://doi.org/10.1038/s41467-018-03597-y (2018).
Fröhlich, H. Long-range coherence and energy storage in biological systems. Int J Quantum Chem 2, 641–649 (1968).
Reimers, J. R., McKemmish, L. K., McKenzie, R. H., Mark, A. E. & Hush, N. S. Weak, strong, and coherent regimes of Frohlich condensation and their applications to terahertz medicine and quantum consciousness. Proc. Natl. Acad. Sci. USA 106, 4219–4224, https://doi.org/10.1073/pnas.0806273106 (2009).
Preto, J. Semi-classical statistical description of Frohlich condensation. Journal of biological physics 43, 167–184, https://doi.org/10.1007/s10867-017-9442-y (2017).
Salari, V., Tuszynski, J., Rahnama, M. & Bernroider, G. Plausibility of quantum coherent states in biological systems. J Phys Conf Ser 306, Artn 012075, https://doi.org/10.1088/1742-6596/306/1/012075 (2011).
Mesquita, M. V., Vasconcellos, A. R. & Luzzi, R. Near-dissipationless coherent excitations in biosystems. Int J Quantum Chem 60, 689–697 (1996). 10.1002/(Sici)1097-461x(1996)60:2<689::Aid-Qua7>3.0.Co;2-T.
Zhang, Z., Agarwal, G. S. & Scully, M. O. Quantum Fluctuations in the Frohlich Condensate of Molecular Vibrations Driven Far From Equilibrium. Phys Rev Lett 122, 158101, https://doi.org/10.1103/PhysRevLett.122.158101 (2019).
Preto, J., Pettini, M. & Tuszynski, J. A. Possible role of electrodynamic interactions in long-distance biomolecular recognition. Phys Rev E 91, 052710 (2015).
Ruggiero, M. T. et al. The significance of the amorphous potential energy landscape for dictating glassy dynamics and driving solid-state crystallisation. Physical Chemistry Chemical Physics 19, 30039–30047, https://doi.org/10.1039/c7cp06664c (2017).
Hoshina, H. et al. Polymer Morphological Change Induced by Terahertz Irradiation. Sci Rep 6, 27180, https://doi.org/10.1038/srep27180 (2016).
Lundholm, I. V. et al. Terahertz radiation induces non-thermal structural changes associated with Fröhlich condensation in a protein crystal. Structural Dynamics 2, 054702, https://doi.org/10.1063/1.4931825 (2015).
Nardecchia, I. et al. Out-of-Equilibrium Collective Oscillation as Phonon Condensation in a Model Protein. Phys Rev X 8, 031061, https://doi.org/10.1103/PhysRevX.8.031061 (2018).
Clarkson, M. W. & Lee, A. L. Long-range dynamic effects of point mutations propagate through side chains in the serine protease inhibitor eglin c. Biochemistry 43, 12448–12458, https://doi.org/10.1021/bi0494424 (2004).
Clarkson, M. W., Gilmore, S. A., Edgell, M. H. & Lee, A. L. Dynamic coupling and allosteric behavior in a nonallosteric protein. Biochemistry 45, 7693–7699, https://doi.org/10.1021/bi060652l (2006).
Bobay, B. G., Thompson, R. J., Hoch, J. A. & Cavanagh, J. Long range dynamic effects of point-mutations trap a response regulator in an active conformation. FEBS Lett 584, 4203–4207, https://doi.org/10.1016/j.febslet.2010.08.051 (2010).
DuBay, K. H., Bowman, G. R. & Geissler, P. L. Fluctuations within Folded Proteins: Implications for Thermodynamic and Allosteric. Regulation. Accounts Chem Res 48, 1098–1105, https://doi.org/10.1021/ar500351b (2015).
Verma, D., Jacobs, D. J. & Livesay, D. R. Changes in Lysozyme Flexibility upon Mutation Are Frequent, Large and Long-Ranged. Plos Comput Biol 8, https://doi.org/10.1371/journal.pcbi.1002409 (2012).
Whitley, M. J. & Lee, A. L. Frameworks for Understanding Long-Range Intra-Protein Communication. Curr Protein Pept Sc 10, 116–127, https://doi.org/10.2174/138920309787847563 (2009).
Leitner, D. M. Energy flow in proteins. Annual review of physical chemistry 59, 233–259, https://doi.org/10.1146/annurev.physchem.59.032607.093606 (2008).
Ermakova, E. & Kurbanov, R. Effect of ligand binding on the dynamics of trypsin. Comparison of different approaches. J Mol Graph Model 49, 99–109, https://doi.org/10.1016/j.jmgm.2014.02.001 (2014).
Hekstra, D. R. et al. Electric-field-stimulated protein mechanics. Nature 540, 400–405, https://doi.org/10.1038/nature20571 (2016).
Mittleman, D. M. Perspective: Terahertz science and technology. J Appl Phys 122, https://doi.org/10.1063/1.5007683 (2017).
Xu, Y. & Havenith, M. Perspective: Watching low-frequency vibrations of water in biomolecular recognition by THz spectroscopy. J Chem Phys 143, https://doi.org/10.1063/1.4934504 (2015).
Acbas, G., Niessen, K. A., Snell, E. H. & Markelz, A. G. Optical measurements of long-range protein vibrations. Nature communications 5, 3076, https://doi.org/10.1038/ncomms4076 (2014).
Niessen, K. A., Xu, M. & Markelz, A. G. Terahertz optical measurements of correlated motions with possible allosteric function. Biophys Rev 7, 201–216, https://doi.org/10.1007/s12551-015-0168-4 (2015).
Niessen, K. A. et al. Moving in the Right Direction: Protein Vibrational Steering Function. Biophys. J. 112, 933–942, https://doi.org/10.1016/j.bpj.2016.12.049 (2017).
Turton, D. A. et al. Terahertz underdamped vibrational motion governs protein-ligand binding in solution. Nature communications 5, 3999, https://doi.org/10.1038/ncomms4999 (2014).
Niessen, K. A. et al. Protein and RNA dynamical fingerprinting. Nature communications 10, 1026, https://doi.org/10.1038/s41467-019-08926-3 (2019).
Merritt, E. A. Expanding the model: anisotropic displacement parameters in protein structure refinement. Acta Cryst D 55, 1109–1117, https://doi.org/10.1107/S0907444999003789 (1999).
Merritt, E. A. Some B-eq are more equivalent than others. Acta Cryst A 67, 512–516, https://doi.org/10.1107/S0108767311034350 (2011).
Foadi, J. et al. Clustering procedures for the optimal selection of data sets from multiple crystals in macromolecular crystallography. Acta Cryst D 69, 1617–1632, https://doi.org/10.1107/S0907444913012274 (2013).
Ortiz, A. R., Strauss, C. E. M. & Olmea, O. MAMMOTH (Matching molecular models obtained from theory): An automated method for model comparison. Protein Science 11, 2606–2621, https://doi.org/10.1110/ps.0215902 (2002).
Walter, J. et al. On the disordered activation domain in trypsinogen. chemical labelling and low-temperature crystallography. Acta Crystallogr. B 38, 1462 (1982).
Dunker, A. K. et al. Intrinsically disordered protein. J Mol Graph Model 19, 26–59, https://doi.org/10.1016/S1093-3263(00)00138-8 (2001).
Dunker, A. K. et al. The unfoldomics decade: an update on intrinsically disordered proteins. Bmc Genomics 9, https://doi.org/10.1186/1471-2164-9-S2-S1 (2008).
Graf, L., Molnar, T., Kardos, J., Gaspari, Z. & Katona, G. The Role of Structural Flexibility and Stability in the Interaction of Serine Proteases with their Inhibitors. Curr Protein Pept Sci 16, 521–531 (2015).
Akke, M. Conformational dynamics and thermodynamics of protein-ligand binding studied by NMR relaxation. Biochem Soc T 40, 419–423, https://doi.org/10.1042/Bst20110750 (2012).
Casanas, A. et al. EIGER detector: application in macromolecular crystallography. Acta Cryst D 72, 1036–1048, https://doi.org/10.1107/S2059798316012304 (2016).
Kabsch, W. X. Acta Cryst D 66, 125–132, https://doi.org/10.1107/S0907444909047337 (2010).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Cryst D 67, 235–242, https://doi.org/10.1107/S0907444910045749 (2011).
Liebschner, D., Dauter, M., Brzuszkiewicz, A. & Dauter, Z. On the reproducibility of protein crystal structures: five atomic resolution structures of trypsin. Acta Cryst D 69, 1447–1462, https://doi.org/10.1107/S0907444913009050 (2013).
Ward, J. H. Hierarchical Grouping to Optimize an Objective Function. J Am Stat Assoc 58, 236, https://doi.org/10.2307/2282967 (1963).
McKinney, W. In Proceedings of the 9th Python in Science Conference (ed Stefan van der Walt and Jarrod Millman) 51 - 56 (2010).
Grosse-Kunstleve, R. W., Sauter, N. K., Moriarty, N. W. & Adams, P. D. The Computational Crystallography Toolbox: crystallographic algorithms in a reusable software framework. J. Appl. Crystallogr. 35, 126–136, https://doi.org/10.1107/S0021889801017824 (2002).
Nguyen, H., Case, D. A. & Rose, A. S. NGLview-interactive molecular graphics for Jupyter notebooks. Bioinformatics 34, 1241–1242, https://doi.org/10.1093/bioinformatics/btx789 (2018).
Bury, C. S., Brooks-Bartlett, J. C., Walsh, S. P. & Garman, E. F. Estimate your dose: RADDOSE-3D. Protein Science 27, 217–228, https://doi.org/10.1002/pro.3302 (2018).
Berendsen, H. J. C., Vanderspoel, D. & Vandrunen, R. Gromacs - a Message-Passing Parallel Molecular-Dynamics Implementation. Comput Phys Commun 91, 43–56, https://doi.org/10.1016/0010-4655(95)00042-E (1995).
Van Der Spoel, D. et al. GROMACS: fast, flexible, and free. J Comput Chem 26, 1701–1718, https://doi.org/10.1002/jcc.20291 (2005).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25, https://doi.org/10.1016/j.softx.2015.06.001 (2015).
Best, R. B. et al. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone phi, psi and Side-Chain chi(1) and chi(2) Dihedral Angles. J Chem Theory Comput 8, 3257–3273, https://doi.org/10.1021/ct300400x (2012).
MacKerell, A. D. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616, https://doi.org/10.1021/jp973084f (1998).
MacKerell, A. D. Jr., Feig, M. & Brooks, C. L. 3rd Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 126, 698–699, https://doi.org/10.1021/ja036959e (2004).
Kaminski, G. A., Friesner, R. A., Tirado-Rives, J. & Jorgensen, W. L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. Journal of Physical Chemistry B 105, 6474–6487, https://doi.org/10.1021/jp003919d (2001).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys 79, 926–935, https://doi.org/10.1063/1.445869 (1983).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J Chem Phys 126, https://doi.org/10.1063/1.2408420 (2007).
Berendsen, H. J. C., Postma, J. P. M., Vangunsteren, W. F., Dinola, A. & Haak, J. R. Molecular-Dynamics with Coupling to an External Bath. J Chem Phys 81, 3684–3690, https://doi.org/10.1063/1.448118 (1984).
Parrinello, M. & Rahman, A. Polymorphic Transitions in Single-Crystals - a New Molecular-Dynamics Method. J Appl Phys 52, 7182–7190, https://doi.org/10.1063/1.328693 (1981).
Darden, T., York, D. & Pedersen, L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J Chem Phys 98, 10089–10092, https://doi.org/10.1063/1.464397 (1993).
Shotton, D. M. & Hartley, B. S. Evidence for Amino-Acid Sequence of Porcine Pancreatic Elastase. Biochemical Journal 131, 643–675, https://doi.org/10.1042/bj1310643 (1973).
Acknowledgements
This work was supported by the Swedish Research Council (2014-04168), the Röntgen-Ångström Framework (2015–06099) and the Knut and Alice Wallenberg Foundation (KAW 2012.0284). The diffraction experiments were performed at the P14 beamline of Petra III. Open access funding provided by University of Gothenburg.
Author information
Authors and Affiliations
Contributions
J.S. and G.K. conceived the experiment. I.L., M.J.G., G.B., T.S., V.Z. and G.K. performed the experiments. V.A.G., H.R., J.S. and G.K. wrote the main manuscript text. G.K. performed molecular dynamics simulations. R.F. performed normal mode analysis. V.A.G. and G.K. developed the analysis software. V.A.G. prepar ed Figs. 1–7, S1, S7 and S8. G.K. prepared Figures S2–S6. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gagnér, V.A., Lundholm, I., Garcia-Bonete, MJ. et al. Clustering of atomic displacement parameters in bovine trypsin reveals a distributed lattice of atoms with shared chemical properties. Sci Rep 9, 19281 (2019). https://doi.org/10.1038/s41598-019-55777-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-55777-5
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
