
Abstract
Amlodipine, a L-type calcium channel blocker, has been reported to have a neuroprotective effect in brain ischemia. Mitochondrial calcium overload leads to apoptosis of cells in neurologic diseases. We evaluated the neuroprotective effects of amlodipine camsylate (AC) on neural stem cells (NSCs) injured by oxygen glucose deprivation (OGD) with a focus on mitochondrial structure and function. NSCs were isolated from rodent embryonic brains. Effects of AC on cell viability, proliferation, level of free radicals, and expression of intracellular signaling proteins were assessed in OGD-injured NSCs. We also investigated the effect of AC on mitochondrial structure in NSCs under OGD by transmission electron microscopy. AC increased the viability and proliferation of NSCs. This beneficial effect of AC was achieved by strong protection of mitochondria. AC markedly enhanced the expression of mitochondrial biogenesis-related proteins and mitochondrial anti-apoptosis proteins. Together, our results indicate that AC protects OGD-injured NSCs by protecting mitochondrial structure and function. The results of the present study provide insight into the mechanisms underlying the protective effects of AC on NSCs.
Similar content being viewed by others

Introduction
Oxygen gluco se deprivation (OGD) leading to free radical attack against neural cells contributes to neuronal cell death1. Hypertension is a major risk factor for ischemic stroke and is associated with OGD in the brain2. A long acting L-type calcium channel blocker, amlodipine, is one of the drugs most commonly used to treat hypertension3,4. Amlodipine has been reported to have a neuroprotective effect in the ischemic brain5,6,7,8,9,10,11. Previous studies have shown that amlodipine reduces stroke size, oxidative stress, enhances survival signals, and inhibits death signals5,6,7,8,9,10,11. However, to our knowledge, there are no published studies regarding the association between amlodipine and mitochondrial ultrastructure and function in neural stem cells (NSCs). In this study, instead of neuronal cultures or brain slices, we have used NSCs to investigate the association between amlodipine and NSCs injured by OGD; this could help in predicting the effect of amlodipine on regeneration and recovery after stroke12.
It is well documented that mitochondrial calcium overload induces activation of the mitochondrial permeability transition (MPT) and that this process leads to loss of mitochondrial membrane potential, swelling of the mitochondrial matrix, and outer membrane rupture, followed by cytochrome c release, activation of downstream cascades, and ultimately apoptosis of cells in neurologic diseases13,14. In light of this knowledge, we hypothesized that amlodipine, via its calcium inhibitory properties, would reduce both cellular and mitochondrial calcium levels and subsequently inhibit activation of the MPT, and that this process would help maintain mitochondrial membrane potential and structure with reduction of cytochrome c release, leading to protection of NSCs from OGD.
In the present study, we evaluated the neuroprotective effects of amlodipine camsylate (AC) on NSCs exposed to OGD with a focus on mitochondrial structure and function. We also examined the effects of AC on intracellular survival signaling proteins and mitochondrial biogenesis-related proteins in NSCs.
Results
Effects of OGD and AC on the viability and toxicity of NSCs
To measure the alteration in neural stem cell (NSC) viability after exposure to OGD, NSCs were incubated with OGD for different exposure times. Cell viability and death were measured with trypan blue staining (TBS) (0, 2, 4, 8, and 24 hours) and LDH assay (0,1,2,3,4,6,8, and 24 hours); results are shown in Fig. 1A. A significant decrease in NSC viability and increase in cell death were observed after exposure to hypoxic conditions in a time-dependent manner. The cytotoxicity of AC was evaluated by treating cells with different concentrations of AC (Fig. 1B). Treatment of cells with above 100 μM AC significantly reduced NSC viability. To identify the effect of AC on cell viability in the context of OGD, we co-treated cells with OGD and three different concentrations of AC for 8 hours (0.1,1 and 10 μM) (Fig. 1C). Cells treated with 1 μΜ AC under OGD for 8 hours showed significantly higher NSC viability and lower NSC toxicity than the cells treated with the other two concentrations of AC and OGD. Treatment of cells with 10 μΜ AC decreased the viability of NSCs exposed to OGD.

Effect of OGD and AC on viability of neural stem cells. (A) Neural stem cells (NSCs) were treated with oxygen–glucose deprivation (OGD) for different periods of time. OGD induced cytotoxicity and decreased the viability of NSCs in a time-dependent manner. (B) To evaluate the effect of AC itself on NSCs, cells were treated with several concentrations of AC (0, 0.1, 1, 10, 100, or 1000 μM) for 8 h. AC (up to 10 μM) did not affect viability and was not cytotoxic to NSCs. (C) To measure the effects of AC on OGD-induced neurotoxicity, NSCs were simultaneously treated with several concentrations of AC (0, 0.1, 1, or 10 μM) and OGD for 8 h. AC up to 1 μM protected NSCs from OGD, but 10 μM AC was not protective. DAPI and TUNEL staining (D) and Annexin V/PI FACS analysis (E) showed that AC effectively blocked apoptotic cell death of NSCs induced by OGD. (F) AC restored the levels of each of these signaling proteins in NSCs under OGD. Data are means (% of control) ± SD from five independent experiments. Treatment groups were compared with the control group using Tukey’s test after one-way ANOVA. *p < 0.05 and **p < 0.01 (vs. control group), #p < 0.05 and ##p < 0.05 (vs. the group treated only with OGD). Full-length blots/gels are presented in Supplementary Fig. S1.
Anti-apoptotic effect of AC on NSCs under OGD
TUNEL and DAPI staining were performed to examine the effect of AC on OGD-induced apoptosis of NSCs (Fig. 1D). Percentage of apoptotic cells increased significantly after treatment with OGD for 8 hours. However, the percentage apoptotic cells decreased significantly upon treatment with AC in a concentration-dependent manner. We also measured the apoptosis of NSCs using fluorescence-activated cell sorting (FACS) and observed a significant reduction in the percentage of apoptotic cells upon treatment with 1 μΜ AC under OGD for 8 hours (Fig. 1E).
Effect of AC on NSC intracellular signaling
To investigate the effects of AC on signaling proteins associated with the survival of NSCs, we analyzed PI3K, phospho-Akt (Ser473), phospho-GSK3β (Ser9), and phospho-p44/42 MARK (Erk1/2) expression by western blotting (Fig. 1F). Immunoreactivities of PI3K, phospho-Akt (Ser473), phospho-GSK3β (Ser9), and phospho-p44/42 MARK (Erk1/2) were higher in NSCs co-treated with AC than in NSCs treated with OGD alone. To determine whether NSCs were affected by OGD, we also measured levels of HIF-1α in the NSCs by western blotting. HIF-1α expression was significantly increased in NSCs treated with OGD alone, but it was markedly decreased in NSCs co-treated with AC and OGD.
Effect of AC on NSC proliferation under OGD
NSC proliferation was assessed by colony formation assay. Compared with NSCs treated with OGD alone for 8 hours, co-treatment with AC significantly increased NSC proliferation in a concentration-dependent manner (Fig. 2A,B). In addition, Ki67 immunoreactivity (proliferation marker) was increased in NSCs co-treated with AC compared to cells treated with OGD alone (Fig. 2C). Furthermore, NSCs treated with AC under OGD showed significantly higher cell migration activities than NSCs treated with OGD alone for 8 hours (Fig. 2D).

Effect of OGD and AC on NSC proliferation. (A) Colony forming unit (CFU) assays were performed. The OGD-induced decrease in proliferative activity of NSCs was significantly recovered by treatment with AC. BrdU assay (B) and Ki67 staining (C) also showed that AC effectively increased proliferation of NSCs subjected to OGD. (D) A cell migration assay showed that the migratory activity of NSCs was reduced by OGD but strongly increased by AC treatment. Data are means (% of control) ± SD from five independent experiments. Treatment groups were compared with the control group using Tukey’s test after one-way ANOVA. *p < 0.05 and **p < 0.01 (vs. control group), #p < 0.05 (vs. the group treated only with OGD). Full-length blots/gels are presented in Supplementary Fig. S2.
Protective effect of AC on mitochondrial cristae ultrastructure and function in NSCs injured by OGD
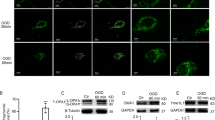
Transmission electron microscopy (TEM) was used to evaluate ultrastructural changes of the cristae in the mitochondria of NSCs subjected to OGD and/or AC treatment (Fig. 3A). Structural enlargement of the mitochondrial matrix and deformation of the mitochondrial cristae were observed in NSCs treated with OGD for 8 hours compared with the control. However, co-treatment of NSCs with AC plus OGD for 8 hours resulted in significant preservation of mitochondrial cristae ultrastructure in a concentration-dependent manner. Low magnification TEM images are presented in Supplementary Fig. S3. In addition, mitochondrial function was evaluated in NSCs treated with OGD and/or AC by western blotting using mitochondrial biogenesis-related marker antibodies (Fig. 3B). Significantly higher expression of mitochondrial biogenesis-related proteins was observed in NSCs co-treated with AC and OGD than in cells treated with OGD alone.

Effect of OGD and AC on NSC mitochondria. (A) Representative transmission electron microscopy (TEM) images of NSCs treated with OGD only (left) or OGD and AC (right). Mitochondrial damage was characterized by the loss of cristae (top) and abnormal enlargement (bottom). OGD-induced mitochondrial damage was prevented by AC treatment. (B) Western blot showing that expression levels of proteins associated with mitochondria biogenesis of NSCs, such as phospho-p38, PGC1α, NRF-1, Drp1, Mfn 1, and Mfn 2, were decreased by OGD treatment but increased with AC treatment. Data are means (% of control) ± SD from five independent experiments. Treatment groups were compared with the control group using Tukey’s test after one-way ANOVA. *p < 0.05 (vs. control group), #p < 0.05 (vs. the group treated only with OGD). Full-length blots/gels are presented in Supplementary Fig. S4.
Effect of AC on OGD-induced free radical production and mitochondrial damage in NSCs
Oxidative damage was evaluated by detecting production of reactive oxygen species (ROS) in NSCs. NSCs were treated with re-oxygenation for 4 hours following OGD for 8 hours and ROS levels were assessed using DCF fluorescence (Fig. 4A). Treatment with OGD for 8 hours followed by re-oxygenation for 4 hours resulted in significantly increased free radical production in NSCs compared with the control group. However, co-treatment with AC (0.1 and 1 μM) resulted in a significant decrease in ROS production in a concentration-dependent manner. Additional analyses were conducted to evaluate mitochondrial changes in NSCs treated with OGD and/or AC. Intracellular calcium level and mitochondrial membrane potential were measured, and an ATP assay was performed. Intracellular calcium levels were significantly lower in NSCs co-treated with AC than in cells treated with OGD alone (Fig. 4B). In addition, mitochondrial membrane potential and ATP concentrations were significantly higher in NSCs co-treated with AC and OGD than cells treated with OGD alone (Fig. 4C,D). Using FACS, lower JC-1 monomer expression was also confirmed in NSCs co-treated with AC compared to NSCs treated with OGD alone (Supplementary Fig. S5).

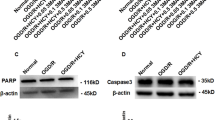
Effect of AC on mitochondrial oxidative stress in NSCs injured by OGD. (A) NSCs were treated with OGD for 30 m and free radical generation was measured using the fluorescent probe DCFH-DA. OGD significantly increased levels of intracellular reactive oxygen species (ROS). However, co-treatment with AC dramatically decreased ROS levels in NSCs. (B) Measurement of intracellular Ca2+ levels suggested that intracellular Ca2+ influx into NSCs was increased by OGD but is decreased by AC treatment. (C) Mitochondrial membrane potential was measured in NSCs under diverse conditions and it decreased significantly upon OGD treatment, whereas AC effectively restored membrane potential to almost normal. (D) ATP level decreased with OGD while AC increased ATP level. (E) Western blots show that expression of proteins associated with cell survival, such as Bcl-2 and mitochondrial cytochrome c, decreased in response to OGD treatment but increased with AC treatment. Proteins related to cell death, including Bax, cytosolic cytochrome c, and cleaved PARP increased with OGD treatment but decreased with AC treatment. Data are means (% of control) ± SD from five independent experiments. Treatment groups were compared with the control group using Tukey’s test after one-way ANOVA. *p < 0.05 and **p < 0.01 (vs. control group), #p < 0.05 (vs. the group treated only with OGD). Full-length blots/gels are presented in Supplementary Fig. S6.
Effect of AC on mitochondrial apoptosis-related proteins in NSCs
The effect of AC on mitochondrial apoptosis-related proteins was evaluated in NSCs that were injured by OGD using western blotting (Fig. 4E). Expression of the survival-related protein Bcl-2 and mitochondrial cytochrome c was significantly increased in NSCs co-treated with AC than NSCs treated with OGD alone. However, the expression of the apoptosis-related protein Bax, cytosolic cytochrome c, and cleaved PARP decreased in response to co-treatment with AC compared to cells treated with OGD alone.
The PI3K/Akt pathways play a critical role in mediating AC-induced protective effects and restoration of mitochondrial damage in NSCs injured by OGD
The roles of the PI3K/Akt pathways in mediating the protective effect of AC on the NSCs were investigated. NSCs were pre-treated with LY294002, a PI3K inhibitor, before OGD and/or AC treatment. LY294002 blocked the protective effect of AC on NSCs damaged by OGD, and cell viability was decreased by approximately 18%, compared to NSCs that were not pre-treated with the PI3K inhibitor (Fig. 5A). The levels of the PI3k/Akt pathway related signaling proteins, and mitochondrial structure and function related proteins, were also measured. The expression of PI3K p110, phospho-Akt (Ser473), Drp1, Mfn1 and Mfn2 was significantly decreased in NSCs pre-treated with LY294002 (Fig. 5B,C).

Role of the PI3K/Akt pathway in mediating the protective effect of AC in NSCs injured by OGD. (A) NSCs were pre-treated with LY294002, a PI3K inhibitor, (10 µM) and the graph shows significant inhibition of the protective effect of AC on NSCs injured using OGD. (B) Western blots show that expression of the PI3K/Akt pathway associated proteins increased in response to AC treatment in NSCs injured using OGD, and treatment with LY294002 decreased expression. (C) LY294002 also inhibited the effect of amlodipine on the expression of the mitochondrial structure and function-related proteins such as Drp1, Mfn1, and Mfn2 in NSCs injured using OGD. Data are means (% of control) ± SD from five independent experiments. Treatment groups were compared with the control group using Tukey’s test after one-way ANOVA. *p < 0.05 and **p < 0.01 (vs. control group), #p < 0.05 and ##p < 0.01 (vs. the group treated only with OGD). Full-length blots/gels are presented in Supplementary Figs S7 and S8.
Discussion
Previously, we reported that amlodipine had possible neuroprotective effects on oxidative stress-injured cortical neurons and NSCs5,6. In the present study, we focused on the effects of AC on mitochondrial structure and function in OGD-injured NSCs. AC increased cell viability, PI3K survival signaling, and NSC proliferation (Figs 1 and 2). AC also effectively preserved the mitochondrial structure of NSCs based on TEM findings (Fig. 3A). In addition, AC enhanced the expression of mitochondrial biogenesis-related proteins (Fig. 3B) and inhibited the expression of mitochondrial anti-apoptosis proteins (Fig. 4). Based on these findings, we hypothesize that AC improves cell survival by protecting mitochondria and inhibiting apoptosis in OGD-injured NSCs. Although previous studies have reported possible neuroprotective effects of amlodipine, this study is the first to identify the protective effects of AC on mitochondria in OGD-injured NSCs7,8,10,11. In addition, we identified that the PI3K/Akt pathway plays an important role in mediating the AC-induced protective effects, and restoration of mitochondrial damage in NSCs injured by OGD (Fig. 5)15.
Neurotoxicity is mediated by elevation of cytosolic Ca2+, and acute neural cell death correlates with the absolute amount of intracellular calcium16,17. Mitochondria are a major calcium-regulating organelle and mitochondrial calcium uptake and release mechanisms are key regulators of cell life or death18,19. Calcium overload in mitochondria and subsequent dysfunction leads to cell death after ischemic and traumatic brain injury20,21. The MPT pore is located in the inner mitochondrial membrane and allows solutes with molecular masses of up to 1500 Da to enter or exit the mitochondrial matrix14. Activation of the MPT pore is the most frequently observed consequence of extensive Ca2+ accumulation in the mitochondria20. Opening of the MPT pore results in osmotic swelling of the mitochondrial matrix that is associated with a decrease in membrane potential and release of cytochrome c with other apoptotic factors14,20. Elevated Ca2+ in mitochondria is thought to be a critical determinant of whether cells undergo oxidative stress-induced apoptosis22. Via its calcium inhibitory properties, amlodipine, a calcium channel blocker used as an antihypertensive drug23, can block calcium influx and reduce calcium storage in cells. Similar to our findings, a previous study reported that administration of azelnidipine, a L-type calcium antagonist, reduced not only cellular Ca2+ accumulation but also mitochondrial Ca2+ accumulation in renal cells24. In addition to stabilizing cellular and mitochondrial Ca2+ homeostasis, azelnidipine protects renal cells from apoptosis induced by hypoxic injury through inhibition of the MPT, cytochrome c release, and downstream signaling cascades. Amlodipine is also an L-type calcium antagonist25. Therefore, we hypothesized that AC would inhibit intracellular calcium influx and reduce Ca2+ accumulation in both the cellular cytoplasm and mitochondria of NSCs. This reduction in mitochondrial Ca2+ accumulation could potentially inhibit MPT pore opening, thereby preventing cytochrome c release, the downstream cascade, and, apoptosis of NSCs, as we demonstrated in this study.
We found that AC effectively enhanced the expression of PI3K pathway proteins, ERK, and anti-apoptotic protein Bcl-2 in OGD-injured NSCs. Similar to our findings, a previous study showed that amlodipine markedly activated the PI3K/Akt pathway in neonatal rat cardiac muscle4. It is well documented that the PI3K/Akt signaling pathway plays a survival role and is involved in neuroprotection by increasing levels of downstream survival-related proteins including phosphorylated Akt, phosphorylated GSK-3β, and Bcl-2, and by decreasing levels of apoptosis-related proteins including cytosolic cytochrome c and cleaved caspase 326,27,28,29,30. A previous study also reported that pretreatment of human umbilical venous endothelial cells with amlodipine resulted in an increase in the Bcl-2/Bax ratio31. Bcl-2-family proteins can modulate intracellular Ca2+ transport systems at the endoplasmic reticulum, mitochondria, and plasma membrane32. In addition, Bcl-2-family proteins finely regulate mitochondrial calcium uptake and can maximize the positive effects of Ca2+ on mitochondrial bioenergetics while protecting the mitochondria from Ca2+ overload leading to cell death32. A previous study reported that Bcl-2 proteins reduce Ca2+ at the endoplasmic reticulum, leading to reduction of mitochondrial calcium and consequently, inhibition of cell apoptosis19.
We also observed in the present study that AC upregulated the expression of biogenesis-related proteins. Both PGC-1 and NRF-1 participate in mitochondrial biogenesis and respiration33,34. Phosphorylated dynamin-related protein (Drp) 1 reduces mitochondrial fission and promotes cell survival35. Mfn1 and 2 play important roles in mitochondrial fusion, and disruption of mitochondrial fusion results in mitochondrial heterogeneity and dysfunction36.
Based on our results, we propose the following mechanism for the protective effects of AC in NSCs under OGD (Fig. 6). The mechanisms that were not established in this study, were referenced from above studies33,34,35,36. Under OGD, extracellular calcium influx occurs in NSCs with an increase in ROS and hypoxia/ischemia related protein HIF-1α. This process causes mitochondrial structural changes and activates the apoptosis-related protein Bax in the mitochondrial membrane. Mitochondrial cytochrome c is then released from the mitochondria to the cytosol and this process increases caspase-3 activity. Ultimately, caspase-3 induces cleavage of PARP and cell apoptosis. In contrast, when NSCs under OGD are treated with AC, calcium influx is blocked and the PI3K cell survival signaling pathway is upregulated. In addition, the survival-related protein Bcl-2, which is activated by AC, may contribute to the maintenance of the mitochondrial membrane potential and inhibit release of mitochondrial cytochrome c. Furthermore, AC increases the expression of mitochondrial biogenesis-related proteins such as mitofusin (Mfn) which may serve to maintain mitochondrial morphology and function in NSCs under OGD. Together, these findings indicate that AC protects NSCs from OGD and further oxidative damage and promotes cell survival via mitochondrial protection and inhibition of apoptosis.

Summary of the effects of AC on NSCs under OGD. OGD disrupts mitochondria in NSCs, but AC can protect mitochondria against OGD through diverse mechanisms including inhibition of calcium influx, activation of the PI3K/Akt pathway, and mitochondria biogenesis.
The present study had several limitations. First, because this study was an in vitro study, the neuroprotective effects of AC need be re-confirmed in experimental animal models. Second, this study was conducted with AC only, which limits the generalizability of our findings. Therefore, neuroprotective effects of other amlodipine salt formulations should be investigated under the same conditions. However, considering our previous report that amlodipine besylate and AC have similar neuroprotective effects5, it is likely that other amlodipine salts might have similar protective effects.
In conclusion, our results provide evidence that AC protects OGD-injured NSCs by inhibiting cellular and mitochondrial calcium influx, activating the PI3K pathway, and enhancing the expression of mitochondrial biogenesis-related proteins. In addition, AC has a preservative effect on mitochondrial structure and the functions of NSCs injured by OGD. The results of the present study provide insight into the mechanisms underlying the protective effects of amlodipine on NSCs; namely, AC protect NSCs from OGD by protecting mitochondria.
Methods
Materials
AC was a kind gift from Hanmi Pharmaceutical Corporation (Seoul, South Korea). AC was dissolved in dimethyl sulfoxide with a final concentration in culture medium of 1% (vol/vol).
Culture of neural stem cells and production of a hypoxic environment
All procedures involving animals were performed in accordance with the Hanyang University guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee (IACUC) of Hanyang University. Every effort was made to minimize the number of animals used and to limit animal suffering. Each animal was used only once.
NSCs were isolated from rodent embryonic brains, cultured, and expanded. NSC culture was performed as described previously37,38,39,40. Briefly, rat embryos were decapitated at embryonic day 13 (E13). Brains were rapidly removed and placed in a petri dish half full of ice-cold Hank’s balanced salt solution (HBSS; 137 mM NaCl, 5.4 mM KCl, 0.3 mM Na2HPO4, 0.4 mM KH2PO4, 5.6 mM glucose, and 2.5 mM HEPES; GIBCO BRL, Grand Island, NY, USA). Single cells were dissociated from the whole cerebral cortex, lateral ganglionic eminence, and ventral midbrain of the fetal rats. The resulting cells were plated at a density of 2 × 104 cells/cm2 on culture dishes pre-coated with poly-L-ornithine/fibronectin in Ca2+/Mg2+-free phosphate-buffered saline (PBS; GIBCO) and cultured in N2 medium (Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12, 25 mg/L insulin, 100 mg/L transferrin, 30 nM selenite, 100 μM putrescine, 20 nM progesterone, 0.2 mM ascorbic acid, 2 nM L-glutamine, 8.6 mM D(+) glucose, and 20 nM NaHCO3; Sigma, St. Louis, MO, USA) supplemented with basic fibroblast growth factor (BFGF; 10 ng/ml, R&D Systems, Minneapolis, MN, USA). Cultures were maintained at 37 °C under a humidified 5% CO2 atmosphere for 4–6 days.
OGD was achieved in a Forma anaerobic chamber (Thermo Fisher Scientific, Waltham, MA USA). A gas mixture containing CO2 (5%), O2 (0.2%), and N2 (94.8%) was flushed through the chamber for 1, 2, 3, 4, 6, 8, and 24 h. This procedure maintained a non-fluctuating hypoxic environment below 1% O2. We measured hypoxic-inducible factor-1alpha (HIF-1α) (which is known to be induced by OGD) using western blotting to determine whether NSCs were affected by OGD delivered by this procedure.
To evaluate the effect of AC on NSCs, cells were treated with several concentrations of AC alone for 1 and 8 h. Finally, to measure the protective effect of AC against OGD, we treated NSCs with several concentrations of AC under OGD for 1 and 8 h, as described above.
To investigate whether the PI3K/Akt pathway plays a critical role in mediating the neuroprotection afforded by AC against OGD in NSCs, we also treated NSCs with 10 μM LY294002, a PI3K inhibitor, 30 min before treatment with 1 μM AC in the background of OGD. Cell viability and expression of proteins were assessed using TBS and western blot, respectively. NSCs were separated into 4 groups as follows: control (group 1), 1 or 8 h OGD alone (group 2), 1 or 8 h OGD + 1 μM AC (group 3), 10 μM LY294002 + 1 or 8 h OGD + 1 μM AC (group 4).
Trypan blue staining and the lactate dehydrogenase (LDH) release assay to measure cell viability
For trypan blue staining, 10 μl aliquots of cells were incubated with 10 μl of trypan blue solution for 2 m. Unstained live cells were counted with a hemocytometer. A colorimetric assay kit (Roche Boehringer–Mannheim, IN, USA) was used to quantify LDH released from cultured NSCs according to the manufacturer’s instructions. Cell viability was assessed using an ELISA plate reader (Synergy H1 Hybrid reader, BioTek Instruments, Winooski, VT, USA) at 490 nm with a reference wavelength of 690 nm. All results were normalized to the OD of an identical well without cells41.
DAPI and TUNEL staining to evaluate apoptosis
NSCs were seeded on collagen-coated 13 mm diameter glass cover slips and treated with AC without exposure to OGD, without AC but with exposure to OGD for 8 h, or with exposure to OGD in the presence of AC (0.1 and 1 μM) for 8 h. Cells were then rinsed twice with PBS, air-dried, and fixed with 4% paraformaldehyde in PBS for 1 h at room temperature.
Apoptotic cell death was identified by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) (Roche Boehringer–Mannheim, IN, USA). To monitor intact, condensed, and fragmented nuclei, TUNEL-stained cells were counterstained with 4’,6-diamidino-2-phenylindole (DAPI, Vector Laboratories, Burlingame, CA, USA) for 20 m, washed several times with PBS, and mounted on glass slides with mounting medium (Merck, Kenilworth, NJ, USA). Cells were then observed under an Olympus Bx53 microscope (Olympus, Tokyo, Japan).
Annexin V and propidium iodide (PI) staining and detection
Annexin V/PI staining was measured using a FITC Annexin V Apoptosis Detection Kit I (Beckton Dickinson, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions. Briefly, cortical neurons were exposed to OGD with AC (0.1 and 1 µM) and incubated for 8 h at 37 °C. Cells were then rinsed twice with cold PBS and resuspended in 1× Binding Buffer. They were transferred (100 μl of the solution; 1 × 105 cells) to a 1.5 ml culture tube followed by application of 5 μl of FITC Annexin V and 10 μl PI. Samples were gently mixed and incubated for 15 m at room temperature in the dark. Finally, 400 μl of 1× Binding Buffer was added to each tube and samples were analyzed by flow cytometry (Accuri C6 Flow cytometer, BD Biosciences). Data were acquired and analyzed with BD Accuri C6 software.
Colony-forming unit assay
Proliferation of NSCs was measured via a colony-forming unit (CFU) assay. Approximately 0.5 × 104 cells were seeded in a 60-mm grid plate and treated with OGD and AC (0.1 and 1 µM) for 8 h. Cells were then washed with DPBS, and the culture medium was changed. After 14 days, cells were washed again with DPBS and stained with 0.5% crystal violet (Sigma) in methanol for 30 m at room temperature. After staining, plates were washed with DPBS and allowed to dry. Colonies were counted under a dissecting microscope and colonies less than 2 mm in diameter or faintly stained were excluded.
Migration assays
Cell migration assays were performed using a QCM 24-well colorimetric cell migration assay kit (Chemicon, Temecula, CA, USA) according to the manufacturer’s instructions. Briefly, OGD and AC (0.1 and 1 µM) were added into the upper chamber of the plate followed by incubation for 24 h at 37 °C. Cells that had migrated through the membrane were stained, and the number of migrated cells was determined by measuring the absorbance at 560 nm using a spectrophotometric plate reader (Synergy H1, BioTek Instruments)42.
BrdU cell proliferation assay
NSCs were incubated in BrdU-labeling medium (10 μM BrdU) for 5 h, and cell proliferation was measured using a BrdU Labeling and Detection Kit (Roche Boehringer–Mannheim, IN, USA) according to the manufacturer’s instructions. Cell proliferation was assessed using an ELISA plate reader (Synergy H1, BioTek Instruments) at 370 nm with a reference wavelength of 492 nm. All results were normalized to the OD of an identical well without cells.
Electron microscopy
NSCs (1 × 106) were seeded on 150 mm dishes and exposed to OGD and AC (0.1 and 1 µM) for 8 h. Cells were then washed twice with PBS and fixed with EM fixing buffer for 1 h at room temperature. Twenty-four hours after MPP + (1-methyl-4-phenylpyridinium) treatment, cells were washed with DMEM and fixed in 0.1 M cacodylate buffer (pH 7.0, TED PELLA INC., Redding, CA, USA) containing 2% paraformaldehyde (Merck) and 0.5% glutaraldehyde (EMS, Hatfield, PA, USA) at 37 °C for 15 m, after which fixation was allowed to proceed at room temperature. Cells were washed with the same buffer three times, post-fixed with 1% osmium tetroxide (Heraeus, Hanau, Germany) for 30 m, and EM bloc-stained with 0.2% uranyl acetate (EMS) solution for 1 h. Subsequently, cells were dehydrated through an ascending ethanol series and embedded in an Epon mixture. Thin sections of 80 nm were cut using a Reichert-Jung Ultracut E ultramicrotome (Leica, Wetzlar, Germany) and mounted on a 200-mesh grid. Electron microscopy observation was performed with a Hitachi H-7500 transmission electron microscope (Hitachi, Ltd., Tokyo, Japan) with 80 kV acceleration voltage.
Determination of free radical production
To measure free radical production, NSCs were exposed to OGD for 30 m and incubated with the fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) (Molecular Probes Inc., Eugene, OR, USA) for 15 m. Then, NSCs treated with different concentrations of AC were exposed to OGD for 8 h followed by sample reoxygenation for 4 h. After incubation with 10 µM H2DCF-DA at 37 °C for 15 m, cells were washed with PBS three times. H2DCF-DA freely crosses cell membranes and is hydrolyzed by cellular esterases to 2′,7′-dichlorodihydrofluorescein (H2DCF), which is oxidized to 2′7′-dichlorofluorescein (DCF), which fluoresces in the presence of peroxides. Therefore, DCF fluorescence indicates the level of intracellular hydrogen peroxide, not superoxide. Accumulation of DCF in cells was assessed using an ELISA plate reader (Synergy H1 reader, BioTek Instruments) by measuring fluorescence (excitation 488 nm; emission 530 nm). All results were normalized to the OD of an identical well without cells.
Mitochondrial membrane potential assay
NSCs were seeded at a density of 5 × 105/ml per well of a 96-well culture plate. Cells were exposed to OGD with AC (0.1 and 1 µM) and incubated for 8 h at 37 °C. Mitochondrial membrane potential was measured using a JC-1 Mitochondrial Membrane Potential Assay Kit (Abnova, Taipei Taiwan) according to the manufacturer’s instructions. Mitochondrial membrane potential was assessed using an ELISA plate reader (Synergy H1 reader, BioTek Instruments). Apoptotic or unhealthy cells with mainly JC-1 monomers were detected with settings designed to detect FITC (excitation/emission = 485/535 nm).
ATP assay
ATP concentrations were measured using an ATP assay kit (Abcam, Cambridge, UK) according to the manufacturer’s instructions and fluorimetrically assessed using an ELISA plate reader (Synergy H1 reader, BioTek Instruments). ATP concentration was detected with an excitation/emission setting of 535/587 nm.
Determination of Ca2+ level
Intracellular calcium levels in NSCs was measured via a Fura 2-AM. Approximately 1–2 × 105 cells were seeded in a 24 well plate and treated with OGD and AC (0.1 and 1 µM) for 4 h. Cells were incubated with 10 mM Fura-2-AM (Sigma) for 1 h at 25–37 °C, in Hank’s balanced salt solution (HBSS), HEPES buffer saline (87 mM NaCl, 50 mM KCl, 1 mM MgCl2, 12 mM HEPES, 10 mM Glucose and 5 mM CaCl2 (pH 7.4). After 1 h of incubation, cells were washed with HEPES buffer saline for 30 min at room temperature. Fura-2 loaded cells were illuminated with dual excitation wave lengths of 340 nm and 380 nm, while capturing the emission signal at 510 nm with the help of appropriate excitation and emission filters (Synergy H1, BioTek Instruments).
NSCs were treated for 8 h, the culture medium was replaced with new medium containing 5 μM Fluo-4 AM (Life Technologies, Carlsbad, IL, USA), and cells were incubated at 37 °C for 1 h. Cells were washed in PBS and incubated for 30 m to allow complete de-esterification of intracellular acetomethyl (AM) esters for fluorescence analysis. Accumulation of fluo-4AM in cells was assessed using an ELISA plate reader (Synergy H1 reader, BioTek Instruments) by measuring fluorescence (excitation 505 nm; emission 530 nm).
Fluo-4 AM intensity was quantified within a region of interest (ROI) for each cell and expressed as the relative change in fluorescence: ΔF/F0 = (F − F0)/F0, where F0 is the fluorescence level at the start of the experiment after subtracting the background fluorescence. Peak amplitudes of ΔF/F0 were determined on stimulation and cumulative increases in ΔF/F0 were calculated over a 420 s post-stimulus time period using Microsoft Excel.
Western blot analyses
Levels of phosphatidylinositol 3-kinase (PI3K), phosphorylated Akt (pAkt) (Ser473), phosphorylated glycogen synthase kinase-3β (pGSK-3β) (Ser9), phospho-p44/42 MARK (Erk1/2), HIF-1α, Ki67, phosphor-p38 (Thr180/Thr182), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1 α), nuclear respiratory factor 1 (NRF-1), dynamin-related protein 1 (Drp1), mitofusin 1 (Mfn 1), mitofusin 2 (Mfn 2), Bcl-2, Bax, cytochrome-C and cleaved PARP were analyzed by western blotting43. Western blotting was performed immediately after treatment for 1 or 8 h. Briefly, 5 × 106 cells were washed twice in cold PBS, incubated for 30 m on ice in lysis buffer [RIPA II cell lysis buffer 1× with Triton, without EDTA; 1 mM phenylmethylsulfonyl fluoride (PMSF); 1 mM sodium fluoride (NaF); 1 mM sodium orthovanadate (Na3VO4); and 0.5% protease inhibitor cocktail 1x]. Next, cells were sonicated (Sonoplus, Bandelin Electronics, Berlin, Germany) and incubated for 30 m on ice. To evaluate cytosolic and mitochondria cytochrome c levels, mitochondrial and cytosolic fractions were isolated using the Mitochondria/Cytosol Fractionation Kit (Abcam, UK) according to the manufacturer’s instructions. Briefly, after OGD for 8 h with different concentrations of AC treatment, NSCs were harvested, washed once with ice-cold PBS, and resuspended in 1.0 mL of 1× cytosol extraction buffer mix containing dithiothreitol (DTT) and protease inhibitors. After a 10-m incubation on ice, the cell suspension was sonicated (Sonoplus) 5–10 times on ice. Samples were centrifuged at 3,000 rpm at 4 °C for 10 m. Supernatants were centrifuged again at 13,000 rpm for 30 m to separate the mitochondrial fraction (pellet) and the cytosolic fraction (supernatant). The mitochondria pellet was washed once with the isolation buffer, and then lysed in mitochondrial extraction buffer containing DTT and protease inhibitors. Samples containing equal amounts (30 μg) of protein were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, Buckinghamshire, UK). Membranes were blocked with 5% skim milk and then incubated with specific primary antibodies. The antibodies used were as follows: anti-p85α PI3K (1:1000, Millipore), anti-PI3K p110 (1:1000, Cell signaling, Beverly, MA, USA), anti-pAkt (1:500, Cell Signaling), anti-Akt (1:1000, Cell Signaling), anti-pGSK-3β (Ser9) (1:500, Cell Signaling, Beverly, MA, USA), anti- p-p44/42 MARK (1:1000, Cell Signaling), anti-HIF-1 (1:1000, Santa Cruz, CA, USA), anti-Ki67 (1:200, Abcam, UK), anti-p-p38 (1:1000, Cell Signaling), anti-PGC1α (1:1000, Abcam), anti-NRF-1 (1:1000, Abcam), Drp1 (1:500, Cell Signaling), anti-Mfn 1 (5 μg/ml, Abcam), anti-Mfn 2 (5 μg/ml, Abcam), anti-Bcl-2 (1:500, Abcam), anti-Bax (1:1000, Abcam), anti-cytochrome c (1:200, Cell Signaling), and anti-PARP (1:400, Cell Signaling). Membranes were washed with Tris-buffered saline containing 0.1% Tween-20 (TBST), and then processed using HRP-conjugated anti-rabbit antibody or anti-mouse antibody (Amersham Pharmacia Biotech, Piscataway, NJ, USA) followed by ECL detection (Amersham Pharmacia Biotech). Blots were quantified with an image analyzer (GE Healthcare, ImageQuant LAS 4000).
Statistical analysis
All data are presented as means ± standard deviations of five or more independent experiments. Viabilities of different treatment groups were compared with Tukey’s test after one-way analysis of variance (ANOVA). Levels of apoptosis and free radicals in different treatment groups and the western blotting results were compared using Tukey’s test after two-way ANOVA. P-values less than 0.05 were considered statistically significant.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information Files).
References
Zhao, L.-P. et al. Oxygen glucose deprivation (OGD)/re-oxygenation-induced in vitro neuronal cell death involves mitochondrial cyclophilin-D/P53 signaling axis. Neurochem. Res. 38, 705–713 (2013).
Hsieh, Y. S., Kwon, S., Lee, H. S. & Seol, G. H. Linalyl acetate prevents hypertension-related ischemic injury. PLoS ONE 13 (2018).
Shakoor, A. et al. Effect of L-type calcium channel blocker (amlodipine) on myocardial iron deposition in patients with thalassaemia with moderate-to-severe myocardial iron deposition: protocol for a randomised, controlled trial. BMJ Open 4, e005360 (2014).
Li, X.-Q. et al. Amlodipine inhibits TNF-alpha production and attenuates cardiac dysfunction induced by lipopolysaccharide involving PI3K/Akt pathway. Int. Immunopharmacol. 9, 1032–1041 (2009).
Lee, Y. J., Park, H.-H., Koh, S.-H., Choi, N.-Y. & Lee, K.-Y. Amlodipine besylate and amlodipine camsylate prevent cortical neuronal cell death induced by oxidative stress. J. Neurochem. 119, 1262–1270 (2011).
Choi, N.-Y. et al. Neuroprotective effects of amlodipine besylate and benidipine hydrochloride on oxidative stress-injured neural stem cells. Brain Res. 1551, 1–12 (2014).
Yamagata, K., Ichinose, S. & Tagami, M. Amlodipine and carvedilol prevent cytotoxicity in cortical neurons isolated from stroke-prone spontaneously hypertensive rats. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 27, 271–282 (2004).
Mogi, M. et al. Amlodipine Treatment Reduces Stroke Size in Apolipoprotein E–Deficient Mice. Am. J. Hypertens. 19, 1144–1149 (2006).
Siesjö, B. K. & Bengtsson, F. Calcium Fluxes, Calcium Antagonists, and Calcium-Related Pathology in Brain Ischemia, Hypoglycemia, and Spreading Depression: A Unifying Hypothesis. J. Cereb. Blood Flow Metab. 9, 127–140 (1989).
Lukic-Panin, V. et al. Prevention of neuronal damage by calcium channel blockers with antioxidative effects after transient focal ischemia in rats. Brain Res. 1176, 143–150 (2007).
Kawai, H. et al. Protection against ischemic stroke damage by synergistic treatment with amlodipine plus atorvastatin in Zucker metabolic rat. Brain Res. 1382, 308–314 (2011).
Thored, P. et al. Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells Dayt. Ohio 24, 739–747 (2006).
Pivovarova, N. B. & Andrews, S. B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 277, 3622–3636 (2010).
Norenberg, M. D. & Rao, K. V. R. The mitochondrial permeability transition in neurologic disease. Neurochem. Int. 50, 983–997 (2007).
Trotta, A. P. & Chipuk, J. E. Mitochondrial dynamics as regulators of cancer biology. Cell. Mol. Life Sci. CMLS 74, 1999–2017 (2017).
Hartley, D. M., Kurth, M. C., Bjerkness, L., Weiss, J. H. & Choi, D. W. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J. Neurosci. Off. J. Soc. Neurosci. 13, 1993–2000 (1993).
Eimerl, S. & Schramm, M. The quantity of calcium that appears to induce neuronal death. J. Neurochem. 62, 1223–1226 (1994).
Friel, D. D. Mitochondria as regulators of stimulus-evoked calcium signals in neurons. Cell Calcium 28, 307–316 (2000).
Giacomello, M., Drago, I., Pizzo, P. & Pozzan, T. Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ. 14, 1267–1274 (2007).
Starkov, A. A., Chinopoulos, C. & Fiskum, G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 36, 257–264 (2004).
Friberg, H. & Wieloch, T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie 84, 241–250 (2002).
Baumgartner, H. K. et al. Calcium Elevation in Mitochondria Is the Main Ca2+ Requirement for Mitochondrial Permeability Transition Pore (mPTP) Opening. J. Biol. Chem. 284, 20796–20803 (2009).
Ishimitsu, T. et al. Amlodipine, a long-acting calcium channel blocker, attenuates morning blood pressure rise in hypertensive patients. Clin. Exp. Pharmacol. Physiol. 26, 500–504 (1999).
Tanaka, T. et al. Blockade of Calcium Influx through L-Type Calcium Channels Attenuates Mitochondrial Injury and Apoptosis in Hypoxic Renal Tubular Cells. J. Am. Soc. Nephrol. 15, 2320–2333 (2004).
Kale, O. E., Awodele, O., Ogundare, T. F. & Ekor, M. Amlodipine, an L-type calcium channel blocker, protects against chlorpromazine-induced neurobehavioural deficits in mice. Fundam. Clin. Pharmacol. 31, 329–339 (2017).
Wang, C., Wei, Z., Jiang, G. & Liu, H. Neuroprotective mechanisms of miR-124 activating PI3K/Akt signaling pathway in ischemic stroke. Exp. Ther. Med. 13, 3315–3318 (2017).
Li, Z., Gang, Z. Z., Shuang, L. X., Ann, H.-S. & Michael, C. The PI3K/Akt Pathway Mediates the Neuroprotective Effect of Atorvastatin in Extending Thrombolytic Therapy After Embolic Stroke in the Rat. Arterioscler. Thromb. Vasc. Biol. 27, 2470–2475 (2007).
Lee, Y. J. et al. Cilnidipine mediates a neuroprotective effect by scavenging free radicals and activating the phosphatidylinositol 3-kinase pathway. J. Neurochem. 111, 90–100 (2009).
Cantley, L. C. The phosphoinositide 3-kinase pathway. Science 296, 1655–1657 (2002).
Pap, M. & Cooper, G. M. Role of translation initiation factor 2B in control of cell survival by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta signaling pathway. Mol. Cell. Biol. 22, 578–586 (2002).
Bian, Y. et al. Amlodipine treatment prevents angiotensin II-induced human umbilical vein endothelial cell apoptosis. Arch. Med. Res. 42, 22–27 (2011).
Vervliet, T., Parys, J. B. & Bultynck, G. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene 35, 5079–5092 (2016).
Scarpulla, R. C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278 (2011).
Shao, D. et al. PGC-1 beta-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERR alpha. Mitochondrion 10, 516–527 (2010).
Jahani-Asl, A. & Slack, R. S. The phosphorylation state of Drp1 determines cell fate. EMBO Rep. 8, 912–913 (2007).
Chen, H., Chomyn, A. & Chan, D. C. Disruption of Fusion Results in Mitochondrial Heterogeneity and Dysfunction. J. Biol. Chem. 280, 26185–26192 (2005).
Currle, D. S., Hu, J. S., Kolski-Andreaco, A. & Monuki, E. S. Culture of mouse neural stem cell precursors. J. Vis. Exp. JoVE 152, https://doi.org/10.3791/152 (2007).
Choi, Y. K. et al. Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat. Med. 22, 1335–1341 (2016).
Chojnacki, A. & Weiss, S. Production of neurons, astrocytes and oligodendrocytes from mammalian CNS stem cells. Nat. Protoc. 3, 935–940 (2008).
Studer, L., Tabar, V. & McKay, R. D. Transplantation of expanded mesencephalic precursors leads to recovery in parkinsonian rats. Nat. Neurosci. 1, 290–295 (1998).
Park, H.-H., Lee, K.-Y., Kim, S. H., Lee, Y. J. & Koh, S.-H. l-DOPA-induced neurotoxicity is reduced by the activation of the PI3K signaling pathway. Toxicology 265, 80–86 (2009).
Koh, S.-H., Noh, M. Y., Cho, G. W., Kim, K. S. & Kim, S. H. Erythropoietin increases the motility of human bone marrow-multipotent stromal cells (hBM-MSCs) and enhances the production of neurotrophic factors from hBM-MSCs. Stem Cells Dev. 18, 411–421 (2009).
Choi, H., Choi, N.-Y., Lee, K.-Y., Lee, Y. J. & Koh, S.-H. Candesartan Restores the Amyloid Beta-Inhibited Proliferation of Neural Stem Cells by Activating the Phosphatidylinositol 3-Kinase Pathway. Dement. Neurocognitive Disord. 16, 64–71 (2017).
Acknowledgements
This research was supported by the Basic Science Research Program of the National Research Foundation of Korea, which is funded by the Ministry of Science, ICT, and Future Planning (2018R1D1A1A09082825, 2018R1A2A2A15023219, and 2018R1D1A1B07047722), by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI18C1254), and by the Medical Research Center (2017R1A5A2015395).
Author information
Authors and Affiliations
Contributions
Conception and design of the study: S.H.K., H.H.P. and M.H.H. Conducting a research and performing the experiments: H.H.P. Acquisition and analysis of data: S.H.K., H.H.P. and M.H.H. Contributed reagents/materials/analysis tools: S.H.K. and H.H.P. Wrote the manuscript: H.H.P. and M.H.H. Study supervision: H.J.C., K.Y.L., Y.J.L., J.M.K., J.H.C. and J.I.R. Re-examined and revising this paper: H.J.C., K.Y.L., Y.J.L., J.M.K., J.H.C. and J.I.R. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, HH., Han, MH., Choi, H. et al. Mitochondria damaged by Oxygen Glucose Deprivation can be Restored through Activation of the PI3K/Akt Pathway and Inhibition of Calcium Influx by Amlodipine Camsylate. Sci Rep 9, 15717 (2019). https://doi.org/10.1038/s41598-019-52083-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52083-y
This article is cited by
-
Inhibition of the NLRP3 Inflammasome Activation/Assembly through the Activation of the PI3K Pathway by Naloxone Protects Neural Stem Cells from Ischemic Condition
Molecular Neurobiology (2023)
-
Posttreatment with Ospemifene Attenuates Hypoxia- and Ischemia-Induced Apoptosis in Primary Neuronal Cells via Selective Modulation of Estrogen Receptors
Neurotoxicity Research (2023)
-
PDIA3 inhibits mitochondrial respiratory function in brain endothelial cells and C. elegans through STAT3 signaling and decreases survival after OGD
Cell Communication and Signaling (2021)
-
Telmisartan Inhibits the NLRP3 Inflammasome by Activating the PI3K Pathway in Neural Stem Cells Injured by Oxygen-Glucose Deprivation
Molecular Neurobiology (2021)
-
Are antibacterial effects of non-antibiotic drugs random or purposeful because of a common evolutionary origin of bacterial and mammalian targets?
Infection (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
