
Abstract
Previous research has shown that genes play a substantial role in determining a person’s susceptibility to age-related hearing impairment. The existing studies on this subject have different results, which may be caused by difficulties in determining the phenotype or the limited number of participants involved. Here, we have gathered the largest sample to date (discovery n = 9,675; replication n = 10,963; validation n = 356,141), and examined phenotypes that represented low/mid and high frequency hearing loss on the pure tone audiogram. We identified 7 loci that were either replicated and/or validated, of which 5 loci are novel in hearing. Especially the ILDR1 gene is a high profile candidate, as it contains our top SNP, is a known hearing loss gene, has been linked to age-related hearing impairment before, and in addition is preferentially expressed within hair cells of the inner ear. By verifying all previously published SNPs, we can present a paper that combines all new and existing findings to date, giving a complete overview of the genetic architecture of age-related hearing impairment. This is of importance as age-related hearing impairment is highly prevalent in our ageing society and represents a large socio-economic burden.
Similar content being viewed by others
Introduction
Hearing loss with age is highly prevalent and accounts for a large socio-economic burden1,2. Presbycusis or age-related hearing impairment (ARHI) may lead to loss of productivity at work3, social withdrawal4 and depression5. ARHI is associated with cognitive decline6 and dementia although the precise relationship between the two is debated7. ARHI typically affects hearing thresholds bilaterally which is most pronounced in the higher frequency range8, while the age of onset and rate of progression are variable. The cochlea plays a vital role in its pathophysiology and signs of cochlear degeneration are present in cases with ARHI9,10.
The etiology of ARHI is multifactorial and includes genetic factors, environmental factors, and their interaction11. Heritability estimates of ARHI vary depending on the precise phenotype studied but are substantial, ranging from 36–70%12,13,14,15. However, the genetic architecture of ARHI remains unclear and to date few genetic variants have been convincingly identified in humans. There are many known genetic variants associated with hearing loss of different types16, but few have been identified as underlying ARHI, which include TJP217, MYO618, and WFS119.
Owing to the difficulty of obtaining human cochlea tissue, much research into ARHI has been performed in mice where genetic manipulation and biochemical studies are relatively easy to perform20. Several loci – named Ahl - have been associated with ARHI, resulting in the identification of one causative gene: cadherin 2321. Mutations in the human homologue of Ahl are implicated in Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB1222. Evidence of their contribution to ARHI in humans is still lacking.
ARHI is not easily quantifiable. The gold standard measure of hearing, pure tone audiometry, provides an audiogram with hearings thresholds at multiple frequencies. Normal values for thresholds are dependent on age and sex and show a skewed distribution. In previous genome-wide association studies (GWAS) investigators have used a variety of traits, including different ways of deriving information from the pure tone audiogram: absolute thresholds23, pure tone averages23, principal components23,24,25,26, and Z-scores27. Health record ICD-9 diagnoses28 have also been used in absence of audiometry. This has led to associations reported between ARHI and variants in the following genes: GRM724,27, IQGAP224, SIK326, ISG20, TRIOBP, EYA4 and ILDR128. There is a lack of consistency between these studies though, which is probably explained by relatively modest sample sizes, given the likely small effect sizes of the variants. However, suboptimal definition of the phenotype, large genetic heterogeneity and the study of isolated populations may also play a role in the failure to date to replicate many of the reported findings.
Here we present a large, 1000Gv3 imputed GWAS meta-analysis of ARHI using pure tone audiometry from multiple cohorts in Northern Europe and the USA, providing a total discovery cohort of 9,675 individuals. We were interested in exploring correlates of high and low/mid frequency hearing loss across samples of different ethnic backgrounds. Because previous work suggested an influence of ethnicity on ARHI, we performed the analyses both stratified by ancestry and in all ancestries combined.
Results
Discovery
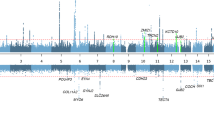
Ten unique loci with suggestive or significant genome-wide P-values (Table 1 and Fig. 1) were identified. The HIGH phenotype yielded 5 associations (4 suggestive, 1 genome-wide significant), the LOW/MID phenotype also yielded 5 associations (3 suggestive, 2 significant). For the HIGH phenotype, the genome-wide significant SNP (rs2332035, P = 7.83*10−10) was located on 3q13.33 in the intron region within the ILDR1 gene. For LOW/MID, the two genome-wide significant SNPs identified were rs6740893 (P = 3.22*10−08), located on 2p16.2 in the intron region within the SPTBN1 gene, and rs9298078 (P = 3.36*10−08), located on 8q12.3 in an intronic non-coding RNA region. Interestingly, the HIGH and LOW/MID phenotypes did not show any overlap in associated signals, with all suggestive and significant loci being different between the two (Fig. 1). A Manhattan plot of all 4 phenotypes is included in Supplementary Fig. S1, while locus zoom plots of all genome-wide significant loci are available in Supplementary Fig. S2.

Manhattan plots for high and low/mid frequency hearing loss. The significant (P < 5*10−8; red horizontal line) and suggestive (P < 1*10−6; blue horizontal line) associations are coloured green for HIGH (high frequency hearing loss) and yellow for LOW/MID (low and mid frequency hearing loss). This colour scheme illustrates that high and low/mid frequency hearing loss have different genetic backgrounds. We found three genome-wide significant SNPs: one at chromosome 2 (rs6740893; LOW/MID phenotype), one at chromosome 3 (rs2332035; HIGH phenotype), and one at chromosomes 8 (rs9298078; LOW/MID phenotype).
Replication
Replication was performed for European ancestry only as well as all genetic ancestries combined (Table 1). Of the 5 SNPs found in the HIGH phenotype, 3 replicated at nominal significance levels (P < 0.05) in European ancestry only. One locus, rs6500458, was significantly replicated when combining replication samples from all ancestries (P = 0.002). For the LOW/MID phenotype 1 out of 5 SNPs replicated (rs10403118). One other SNP (rs56203268) was also significantly replicated, but the direction was inconsistent. Overall, we were able to replicate 4 SNPs lying on 4 separate chromosomes. Of note, the genome-wide significant SNP from the HIGH phenotype (rs2332035) was not formally replicated, but reached significance level in the meta-analysis of discovery and replication cohorts of European ancestry combined (P = 2.29*10−8; Supplementary Table S1).
Clinical validation
The use of UK Biobank questionnaire data (as well as to identify other phenotypes linked to ARHI), was validated by exploring the phenotype genetic correlations using LDHub (Supplementary Table S2). A highly significant genetic correlation was identified between the questionnaire-based hearing loss phenotype in the UK Biobank and phenotype HIGH (rG = 0.751, P = 5.75*10−7). Of note, the second and third strongest trends from the LD Hub analyses were also related to hearing loss (‘Hearing aid user’ and ‘Hearing difficulty/problems with background noise’).
Ten independent loci were examined in UK Biobank using a phenotype derived from responses to questions about self-reported hearing loss. Of the 10 independent loci identified at the discovery phase, 5 loci (3 for HIGH and 2 for LOW/MID) achieved significance after Bonferroni correction (P < 0.005; Table 1). When considering both the formal replication with pure tone audiometry and the validation using UK Biobank, a total of 7 independent loci were identified for ARHI: 4 for high, and 3 for low/mid frequency hearing loss.
Gene expression in the auditory system
A total of 20 genes were marked as candidate genes. In humans, 6 genes showed reproducible expression in the cochlea, 5 were marginally expressed, 3 were absent and data on 6 genes were not available (Table 2). In mice a majority of the genes (n = 15) were expressed within (inner or outer) hair cells. This number was somewhat lower in spiral ganglion cells: 10 out of 20 genes.
The following genes showed signs of differential expression in the inner ear: MAST2 (utricle and postnatal period), ILDR1 (hair cells), TRIL (supporting cells), SCUBE2 (postnatal period), DOCK9 (hair cells and postnatal period), ISG20 (supporting cells, utricle, postnatal period), SHC2 (hair cells) and ODF3L2 (hair cells, cochlea, postnatal period). The greatest differential expression was noted for ILDR1, which is 7.65 times more expressed in hair cells than in supporting cells (FDR = 1.33*10−4).
Genetic correlations
All 10 genetic variants identified as genome-wide suggestive or significant were either on different chromosomes or were far apart from each other (>10 Mb) and can therefore be considered independent loci. Only a very minor attenuation of signal was observed in the joint analysis (Supplementary Table S3).
A significant genetic correlation was identified between HIGH and LOW/MID phenotypes (rG = 0.69, P = 8*10−5) with SNP-based heritability (h2) more or less equal for the two: HIGH = 0.15 (P = 0.002); LOW/MID = 0.11 (P = 0.032). This implies that between 0.11–0.15 of the phenotypic variance can be explained by common SNPs with minor allele frequency > 0.05.
Candidate gene approach
Results of the 7 candidate gene variants previously linked to ARHI are given in Table 3. No evidence for association between the two hearing phenotypes and the GRM7 gene (rs11928865, rs779706 and rs779701) or the IQGAP2 gene (rs457717) was found. We were unable to replicate the SIK3 SNP (rs681524), which had been identified in the G-EAR consortium and TwinsUK. However, positive support was established for ILDR1 (rs2877561 was highly significant in HIGH and LOW/MID), ISG20 (rs4932196, highly significant in LOW/MID, and less so in HIGH) and TRIOBP (rs5756795, highly significant in LOW/MID) (Table 3). The direction of effect of these SNPs was the same in both studies. The SNP in EYA4 (rs9493627) just failed to reach significance (P = 0.006 in LOW/MID).
Discussion
This study is the largest GWAS meta-analysis of ARHI to date and identified 7 associated loci, of which 5 are novel (FXYD5, IPP, SPIRE2, SPTBN1 and TRIL) and 2 have been previously related to hearing loss (ILDR1, ISG20). Suggestive and significant associations showed no overlap between the low/mid and high frequency hearing loss phenotypes, indicating different pathophysiological mechanisms. In addition, we have confirmed some of the SNPs previously reported as related to ARHI (ILDR1, ISG20 and TRIOBP), while others were not replicated (EYA4, GRM7, IQGAP2 and SIK3). Our study has again shown that ARHI is highly polygenic as many genes - each with small effect - contribute to the estimated heritability of 36–70%12,13,14,15. In this study we identify a SNP-based heritability of 11–15%. This is in line with the observation that twin and family studies produce higher heritability estimates of common complex traits because GWAS estimates only the contribution made by common variants while other heritable variation (such as indels and CNVs) are largely ignored29.
At the outset, we chose to include only those cohorts having collected pure tone audiograms, as this is the gold standard measure of hearing ability and provides the best opportunity to define sub-phenotypes by which to interrogate putative pathologic pathways. Although ARHI initially impairs high frequency hearing, a correlate for low/mid frequency hearing loss was also included. Indeed, we found genetic variants associated with both HIGH and LOW/MID phenotypes and consistent with current understanding of cochlea pathology the associated variants were mutually exclusive.
The present study has a number of limitations. As our data are cross-sectional, we cannot exclude a contribution from other causes of hearing loss besides ARHI. However, congenital forms and other cochlea diseases will probably represent only a very small heterogeneous subset of the total number of participants. Second, replication was performed in a mixed ethnicity sample including European, African American and Hispanic heritage. We were unable to replicate most of our 10 discovery hits in non-Europeans, most likely due to a lack of power owing to the small sample size and maybe a different genetic architecture of ARHI. Indeed prevalence of ARHI differs by ethnicity, with African Americans having lower rates compared to non-Hispanic whites and Hispanics30,31. The prevalence of ARHI in Hispanics varies between different backgrounds32, but on average appears to be similar to non-Hispanic whites33.
The UK Biobank was used to validate our results based on responses to questions regarding hearing loss. Previous work on this dataset had examined the speech-in-noise phenotype and found it unreliable. A significant correlation was found between our HIGH phenotype and self-reported hearing loss in the UK Biobank, indicating that these responses may be a useful alternative to pure tone audiometry, in line with literature34. The Rotterdam study provides similar reassurance, where a high correlation between self-reported hearing loss and pure tone audiometry has been demonstrated (A.P.N., A.G., unpublished data). Despite different approaches to phenotyping, the UK Biobank questionnaire responses are useful in that they indicate the genetic variants responsible for pure tone audiometry changes are relevant to individuals’ clinical symptoms, something that is increasingly important in publically funded research.
Data on gene expression in the auditory system can provide supportive evidence, although such data should be interpreted with caution. The effects of aging may be manifest through other mechanisms, for example via the circulation, and may have an important influence on hearing function. Almost all associated genes identified showed signs of expression within the human or mouse cochlea, spiral ganglion cells or both. Of specific interest is the differential expression because most genes involved in hearing loss to date have been shown to be overexpressed within cochlear hair cells35.
The most highly associated SNP in our meta-analysis lies on chromosome 3 in close proximity to two genes: SLC15A2 and ILDR1. The SLC15A2 gene was initially considered a candidate gene for the nonsyndromic hearing loss locus, and was designated DFNB4236. However, sequencing of the SLC15A2 gene yielded no causal variants. Six years later, the Ig-like domain containing receptor 1 (ILDR1) gene was identified as the causal gene37. Several mutations within the ILDR1 gene have been associated with autosomal recessive nonsyndromic hearing loss to variable extent37,38,39,40,41. In addition, this gene has been linked previously to ARHI through a candidate gene approach28. The ILDR1 protein mediates the recruitment of tricellulin to tight tricellular junctions, which plays a crucial role in the epithelial barrier function. ILDR1 knock-out mice initially show normal development of inner and outer hair cells and the organ of Corti42. At 2 weeks of age however, outer hair cells begin to degenerate at the basal turn of the cochlea, corresponding to the higher frequencies. Later, this progresses to outer hair cells of the lower frequencies as well, and hearing function is severely diminished at 3 weeks of age. This process describes an accelerated version of the biology of ARHI in humans.
SPTBN1, encoding the spectrin beta, non-erythrocytic 1 protein, also known as βII spectrin, contains the top SNP of one of the low/mid frequency hearing loss loci. Spectrins are a major component of the cell membrane cytoskeleton and are located in hair and supporting cells of the cochlea43. As the cytoskeleton has a close relationship with outer hair cell electromotility, namely a shortening and elongation of the cell in response to sound stimuli, this may provide a mechanism through which SPTBN1 contributes to hearing loss.
Cystathionine-γ-lyase (CTH) is involved in the formation of hydrogen sulfide and this gas has been demonstrated to regulate cochlear blood flow and has the ability to protect against noise-induced hearing loss44. Odf3l2 has been listed as a candidate gene involved in mild hearing loss in a large-scale screen in mice45 and is differentially expressed in hair cells35. To our knowledge, no relationship has been established yet between hearing loss and any of the other annotated genes (DOCK9, EML6, FXYD5, GPBP1L1, IPP, MAST2, NASP, SCUBE2, SHC2, SPIRE2, TMEM69, TRIL, and VPS9D1). The precise mechanism through which they act on hearing function is yet unknown. Pathway analysis performed on our significantly associated variants was unrewarding.
Published genome-wide associations with ARHI are also of particular interest, as it confirmed associated variants in ILDR1, ISG20 and TRIOBP, but calls into question the association of GRM7, which for a long time was considered proven because of the number of studies supporting its association. Considering differences that may arise through different phenotyping, the first study to identify the association between GRM7 and ARHI also employed a Z-score approach to pure tone audiometry, but a slightly different method of calculation was used by normalizing according to the ISO standard27. It would seem unlikely that this difference in methodology should account for the contrasting results. Alternative explanations include a false positive finding based on smaller sample size or differences in the populations studied.
The IQGAP2 gene, which also failed to replicate in our dataset was reported associated in one of the earlier GWA studies24. There are two major study differences: principal components of the pure tone audiogram were used instead of the Z-score method we employed. Second, the Finnish study was performed in an isolated population of the Saami using a small sample, so results may not be pertinent to the outbred Northern European sample of our work. Similarly, a study in a Han Chinese male sample investigating the relationship between IQGAP2 and ARHI did not show any significant association46.
SIK3 was identified in a GWAS meta-analysis of the G-EAR consortium and TwinsUK which included samples from outbred and inbred populations26. All samples except one contributed to the signal in the same direction and three samples were nominally significant. While the imputation quality of some of the samples was not high, this signal was confirmed in samples where whole genome sequencing was available. This finding was made in principal component 2, representing the slope of the audiogram, which corresponds to our HML phenotype. As the HML phenotype and some other studies that employed principal component 223,24,25 did not produce the same results, the SIK3 might also be a false positive finding, though it is expressed in mouse cochlea.
ISG20 was replicated in our dataset, but the mechanism by which this gene affects hearing is unknown. It is barely expressed within the cochlea but is demonstrable in spiral ganglion cells, especially at later stages of development47. Interestingly, more interferon-related genes show signs of differential expression during late development and adulthood. Interferons are required for central nervous system homeostasis48, perhaps also in spiral ganglion cells.
TRIOBP (TRIO- and F-actin binding protein) is another gene that was replicated in our discovery cohort and is a well-established hearing loss gene, labelled as DFNB2849. The TRIOBP protein is localized in the rootlets of hair cell stereocilia and provides stability and rigidity. In knock-out mice, stereocilia develop normally but are easier to deflect and damage50. Initially thought to be responsible for profound, early onset hearing loss, mutations in TRIOBP have been recently shown to be also linked to late onset28 and more moderate hearing loss51, fitting the description of ARHI well.
To conclude, this study provides a large step forward in unravelling the genetic architecture of ARHI. Future studies have to confirm the associated loci and elucidate the pathophysiological pathways in which they may lead to hearing loss.
Methods
Six population-based studies from the CHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology) consortium52 that have collected pure tone audiometry were included. Participants from Age, Gene/Environment Susceptibility Study (AGES; n = 3,104), Cardiovascular Health Study (CHS; n = 327), Framingham Heart Study (FHS; n = 1,978), Health ABC (HABC; n = 1,174), and the Rotterdam study (RS cohorts II and III; n = 3,092) were combined to provide a discovery sample of n = 9,675. The replication samples had also obtained pure tone audiometry and included the Antwerp study (n = 1,161), the G-EAR consortium (n = 1,339), the Jackson Heart Study (JHS; n = 735), Hispanic Community Health Study/Study of Latinos (HCHS/SOL; n = 6,909) and TwinsUK (n = 819), leading up to a total available sample of n = 10,963. Findings were further validated in the UK Biobank sample (https://www.ukbiobank.ac.uk/) using responses to questions on self-reported hearing loss because the speech-in-noise measures were found to be unreliable. In every analysis, only males and females aged 45 years or older were included. All participants provided written informed consent. Ethics approval was obtained locally at each study site: the Icelandic National Bioethics Committee (AGES); the institutional review boards at the University of Pittsburgh, the Johns Hopkins School of Public Health, Wake Forest University Health Sciences and the University of California Davis (CHS); the Boston Medical Center Institutional Review Board (FHS); the institutional review boards at the University of Pittsburgh and the University of Tennessee, Memphis (HABC); the Medical Ethics Committee of the Erasmus Medical Center (RS); the Committee for Medical Ethics UZA-UAntwerp; the Institutional Review Board of IRCCS Burlo Garofolo (G-EAR); the institutional review boards at the San Diego State University, the University of Miami, the University of North Carolina, the University of Illinois and the Albert Einstein College of Medicine (HCHS/SOL); the Institutional Review Board of the University of Mississippi Medical Center (JHS); and the National Research Ethics service London-Westminster (TwinsUK). The Declaration of Helsinki was adhered to. A more detailed description of the discovery and replication cohorts is available in the supplementary information.
Phenotype description
Pure tone audiometry (air conduction thresholds at frequencies 0.5, 1, 2, 4 and 8 kHz) was collected on participants in all studies. We elected to examine hearing as a quantitative trait, rather than arbitrarily assign participants to case or control status. For each participant, the results of the better ear were used, defined as having the lowest threshold averaged over 0.5, 1, 2 and 4 kHz; when these were equal across ears we took the ear with the lowest threshold averaged over 4 and 8 kHz; if these were similar, the ear with the lowest threshold averaged over 0.5, 1 and 2 kHz was chosen; and if these too were similar, the results of the left ear were selected.
We found that ISO 7029 mean and standard deviation of hearing53 did not summarise our data well so decided that a standardised approach would allow each cohort to use its own data to generate means and standard deviations. Thus, each cohort provided its own reference panel and the effect of variations between audiometry centers was hereby minimised. Definition and calculation of the phenotypes was established before commencing the genetic analysis and not altered post hoc. Age-related hearing impairment mainly affects thresholds of the higher frequencies (Supplementary Fig. S3), showing a marked increase in hearing thresholds above 2 kHz (Supplementary Table S4, data from the Rotterdam Study). This has led to the decision to define a phenotype for high frequency hearing loss above 2 kHz and a second, separate one, that reflects hearing loss at the remaining low and mid frequencies. Age- and sex-adjusted weighted Z-scores of combinations of air conduction thresholds at frequencies 0.5 to 8 kHz were generated and mean thresholds of the following combinations were used to define:
-
HIGH: high frequency hearing loss: 4 and 8 kHz
-
LOW/MID: low and mid frequency hearing loss: 0.5, 1 and 2 kHz
Z-scores were generated by plotting mean thresholds against age using a linear regression model, with males and females considered separately. In contrast to the ISO 7029 standard, a quadratic function did not result in lower residuals so the simplest function, namely a linear function, was chosen. Data distributions were positively skewed, so a separate standard deviation was calculated for participants above and below the regression line. The residuals were each divided by the appropriate standard deviation, resulting in the phenotype. A positive value thus reflects a larger hearing loss than expected for age. An example R code to calculate the phenotype is provided in the supplementary information. For clarity purposes, the analysis on two additional phenotypes was excluded from the main text but made available in the supplementary information.
Genotyping by center
Details of genotyping platforms, quality control and imputation to 1000Gv3 are available in Supplementary Table S5. Linear regression analysis was performed by each cohort adjusting only for cohort specific covariates (e.g. principal components, center, relatedness), because age and gender were already incorporated within the phenotype definition. Cohort summary statistics underwent quality control using EasyQC based on the standard protocol54, using a 1% minor allele frequency threshold.
Discovery
Inverse variance-based meta-analysis of summary statistics was performed in METAL55. The most highly associated SNPs were considered for replication if they were genome-wide significant (P < 5*10−8) or suggestively associated (P < 1*10−6). Comparison of findings between the phenotypes was considered to be informative of the genetic relationship between specific characteristics of the audiogram (low/mid versus high frequency hearing loss).
Replication
The same phenotype definition was used in five independent studies in which we attempted to replicate the most highly associated SNPs: the Antwerp study, the G-EAR consortium, JHS, HCHS/SOL and TwinsUK (details on genotyping are listed in Supplementary Table S5). They provided a total of 10,963 participants. These cohorts were of European ancestry except for JHS (African American ancestry; n = 735) and HCHS/SOL (Hispanic ancestry; n = 6,909). A combined meta-analysis of discovery and replication was performed both ancestry-specific, as well as for all ancestries combined (Supplementary Table S1).
All suggestive and significant loci from the discovery meta-analysis (P < 1*10−6) were considered for replication. Inverse variance-based meta-analysis of summary statistics was performed in METAL.
Clinical validation
We sought to validate our findings further using UK Biobank. This national bioresource in the UK comprises ~500,000 participants aged 40–69 years registered with a general practitioner, and has been extensively described elsewhere56. Ethics approval was provided by the North West Multi-Center Research Ethics Committee (MREC). Participants were invited to local examination centers and underwent a battery of clinical tests and completed online questionnaires including questions pertaining to hearing ability. We elected at the outset to use responses to hearing ability questions to see if we could validate the genetic findings using a subjective measure that reflects clinical hearing disability. Participants had been asked the question ‘Do you have any difficulty with your hearing?’ Cases were defined as responding ‘Yes’ or ‘I am completely deaf’ while control status was assigned to those answering ‘No’. Participants with missing data and those aged below 45 years at the time of participation were excluded from the present study. GWAS was performed using Northern Europeans alone as defined by the response to the ethnicity question and principal component analysis.
PLINK2 logistic regression models were used to test for association adjusting for age, genetic sex (inferred from genotype data), UK Biobank PCs 1–10 and genotyping platform. Data were pre-processed by UK Biobank before release, and further QC was performed based on their recommended filters for excess relatedness (<10 putative 3rd degree relatives in the kinship table), putative sex chromosome aneuploidy, heterozygosity and missing rates57. The final sample, selecting Northern Europeans as determined genetically, comprised n = 98,816 cases and n = 257,325 controls. Since 10 independent SNPs were investigated results were considered significant if P < 0.005, with Bonferroni correction.
Gene expression in the auditory system
All suggestive and significant SNPs from the discovery cohort were included. Given that eQTL data were not available for the cochlea, candidate genes were selected in two ways. First, a candidate gene was assigned to each locus based on proximity to the top SNP. Second, in FUMA-GWAS58, a MAGMA gene-based test was used to link associated SNPs to genes and gene-phenotype association was examined using a burden test59. Genes with P < 2.8*10−6 were considered significantly associated with the phenotype of interest.
The cochlea is a distinct, highly differentiated organ with its own specific gene expression pattern. Human cochlea material is difficult to obtain so expression data are relatively lacking. We assessed a database, the only existing one as far as we are aware, derived from three human cochlear specimens60. Threshold for expression was set at FPKM > 1 (fragments per kilobase of exon per million reads mapped). Interpretation was performed in a qualitative manner, assigning results to one of four categories: not available (data on specific gene not available or did not pass quality control); absent (expression < 1 FPKM); marginal (expression > 1 FPKM in only one sample); or present (expression > 1 FPKM in more than one sample).
The Shared Inner Ear Laboratory Database (SHIELD) was also consulted, as it contains a wide variety of information on gene expression in the mouse auditory system that is unavailable in the human variant61. We looked at whether the candidate genes were expressed in adult cochlear inner and outer hair cells, at P25–P3062, and spiral ganglion cells of the cochlear nerve, at P1547. Expression was designated “present” when, respectively, levels exceeded 10.9 or >=50% of detection calls were positive. In addition, differential expression in the inner ear, defined as >2-fold change, was noted, looking at: cell type (hair cells vs supporting cells); tissue source (cochlea vs utricle); and developmental stage (embryonic vs postnatal period)35. False discovery rate was used to assess statistical significance.
Genetic correlations
Independence of all suggestively and significantly associated variants was examined. Genome-wide Complex Trait Analysis (GCTA) was used to identify secondary signals employing the cojo-slct function63. A combined dataset of the Rotterdam II and III cohorts was used as genetic background.
LD score regression was employed to investigate the genetic correlation between the phenotypes, as well as to examine the genetic correlation between our phenotypes and publically available data on LD Hub64. Only traits investigated in European ancestry samples were considered. If a trait was included multiple times, the largest dataset was considered. Traits for which no estimates could be obtained were excluded. This approach resulted in 518 traits being investigated, including 339 unpublished traits from the UK Biobank, of which three were hearing-related phenotypes. Significance was set at Bonferroni adjusted P < 9.65*10−5.
Candidate gene approach
We were interested to determine the strength of evidence in this dataset supporting SNPs previously identified as associated with ARHI. We used the discovery cohort to examine 9 genetic variants in 7 genes that have been previously associated with ARHI using agnostic study methods and replicated, or identified through GWAS meta-analysis: GRM727; IQGAP224; SIK326; EYA4, ILDR1, ISG20, and TRIOBP28. Significance level was Bonferroni adjusted for 9 SNPs and set at P = 0.0056.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Olusanya, B. O., Neumann, K. J. & Saunders, J. E. The global burden of disabling hearing impairment: a call to action. Bull World Health Organ 92, 367–373 (2014).
Wilson, B. S., Tucci, D. L., Merson, M. H. & O'Donoghue, G. M. Global hearing health care: new findings and perspectives. Lancet 390, 2503–2515 (2017).
Kooser, C. Hearing loss and employment in the United States. Work 46, 181–186 (2013).
Ciorba, A., Bianchini, C., Pelucchi, S. & Pastore, A. The impact of hearing loss on the quality of life of elderly adults. Clin Interv Aging 7, 159–163 (2012).
Li, C. M. et al. Hearing impairment associated with depression in US adults, National Health and Nutrition Examination Survey 2005–2010. JAMA Otolaryngol Head Neck Surg 140, 293–302 (2014).
Yuan, J., Sun, Y., Sang, S., Pham, J. H. & Kong, W. J. The risk of cognitive impairment associated with hearing function in older adults: a pooled analysis of data from eleven studies. Sci Rep 8, 2137 (2018).
Pichora-Fuller, M. K., Mick, P. & Reed, M. Hearing, Cognition, and Healthy Aging: Social and Public Health Implications of the Links between Age-Related Declines in Hearing and Cognition. Semin Hear 36, 122–139 (2015).
Zwaardemaker, H. Der Verlust an hohen Tönen mit zunehmendem Alter: ein neues Gesetz. Arch Ohrenheilk 32, 53 (1891).
Nelson, E. G. & Hinojosa, R. Presbycusis: a human temporal bone study of individuals with flat audiometric patterns of hearing loss using a new method to quantify stria vascularis volume. Laryngoscope 113, 1672–1686 (2003).
Nelson, E. G. & Hinojosa, R. Presbycusis: a human temporal bone study of individuals with downward sloping audiometric patterns of hearing loss and review of the literature. Laryngoscope 116, 1–12 (2006).
Van Eyken, E., Van Camp, G. & Van Laer, L. The complexity of age-related hearing impairment: contributing environmental and genetic factors. Audiol Neurootol 12, 345–358 (2007).
Kvestad, E., Czajkowski, N., Krog, N. H., Engdahl, B. & Tambs, K. Heritability of hearing loss. Epidemiology 23, 328–331 (2012).
Wolber, L. E., Steves, C. J., Spector, T. D. & Williams, F. M. Hearing ability with age in northern European women: a new web-based approach to genetic studies. PLoS One 7, e35500 (2012).
Gates, G. A., Couropmitree, N. N. & Myers, R. H. Genetic associations in age-related hearing thresholds. Archives of otolaryngology–head & neck surgery 125, 654–659 (1999).
Christensen, K., Frederiksen, H. & Hoffman, H. J. Genetic and environmental influences on self-reported reduced hearing in the old and oldest old. J Am Geriatr Soc 49, 1512–1517 (2001).
Van Camp, G. & Smith, R. J. H. Hereditary Hearing Loss Homepage, https://hereditaryhearingloss.org.
Walsh, T. et al. Genomic duplication and overexpression of TJP2/ZO-2 leads to altered expression of apoptosis genes in progressive nonsyndromic hearing loss DFNA51. Am J Hum Genet 87, 101–109 (2010).
Oonk, A. M. et al. Progressive hereditary hearing impairment caused by a MYO6 mutation resembles presbyacusis. Hear Res 299, 88–98 (2013).
Kytovuori, L., Hannula, S., Maki-Torkko, E., Sorri, M. & Majamaa, K. A nonsynonymous mutation in the WFS1 gene in a Finnish family with age-related hearing impairment. Hear Res 355, 97–101 (2017).
Bowl, M. R. & Dawson, S. J. The mouse as a model for age-related hearing loss - a mini-review. Gerontology 61, 149–157 (2015).
Di Palma, F. et al. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nature genetics 27, 103–107 (2001).
Bork, J. M. et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet 68, 26–37 (2001).
Girotto, G. et al. Hearing function and thresholds: a genome-wide association study in European isolated populations identifies new loci and pathways. J Med Genet 48, 369–374 (2011).
Van Laer, L. et al. A genome-wide association study for age-related hearing impairment in the Saami. Eur J Hum Genet 18, 685–693 (2010).
Fransen, E. et al. Genome-wide association analysis demonstrates the highly polygenic character of age-related hearing impairment. Eur J Hum Genet 23, 110–115 (2015).
Wolber, L. E. et al. Salt-inducible kinase 3, SIK3, is a new gene associated with hearing. Hum Mol Genet 23, 6407–6418 (2014).
Friedman, R. A. et al. GRM7 variants confer susceptibility to age-related hearing impairment. Hum Mol Genet 18, 785–796 (2009).
Hoffmann, T. J. et al. A Large Genome-Wide Association Study of Age-Related Hearing Impairment Using Electronic Health Records. PLoS Genet 12, e1006371 (2016).
Vinkhuyzen, A. A., Wray, N. R., Yang, J., Goddard, M. E. & Visscher, P. M. Estimation and partition of heritability in human populations using whole-genome analysis methods. Annu Rev Genet 47, 75–95 (2013).
Agrawal, Y., Platz, E. A. & Niparko, J. K. Prevalence of hearing loss and differences by demographic characteristics among US adults: data from the National Health and Nutrition Examination Survey, 1999–2004. Arch Intern Med 168, 1522–1530 (2008).
Lin, F. R. et al. Association of skin color, race/ethnicity, and hearing loss among adults in the USA. J Assoc Res Otolaryngol 13, 109–117 (2012).
Cruickshanks, K. J. et al. Hearing Impairment Prevalence and Associated Risk Factors in the Hispanic Community Health Study/Study of Latinos. JAMA Otolaryngol Head Neck Surg 141, 641–648 (2015).
Dillon, C. F., Gu, Q., Hoffman, H. J. & Ko, C. W. Vision, hearing, balance, and sensory impairment in Americans aged 70 years and over: United States, 1999–2006. NCHS Data Brief, 1–8 (2010).
Valete-Rosalino, C. M. & Rozenfeld, S. Auditory screening in the elderly: comparison between self-report and audiometry. Braz J Otorhinolaryngol 71, 193–200 (2005).
Scheffer, D. I., Shen, J., Corey, D. P. & Chen, Z. Y. Gene Expression by Mouse Inner Ear Hair Cells during Development. J Neurosci 35, 6366–6380 (2015).
Aslam, M. et al. A novel autosomal recessive nonsyndromic hearing impairment locus (DFNB42) maps to chromosome 3q13.31-q22.3. American journal of medical genetics 133A, 18–22 (2005).
Borck, G. et al. Loss-of-function mutations of ILDR1 cause autosomal-recessive hearing impairment DFNB42. Am J Hum Genet 88, 127–137 (2011).
Ramzan, K. et al. ILDR1: Novel mutation and a rare cause of congenital deafness in the Saudi Arabian population. Eur J Med Genet 57, 253–258 (2014).
Tlili, A., Fahd Al Mutery, A., Mahfood, M., Kamal Eddine Ahmad Mohamed, W. & Bajou, K. Identification of a novel frameshift mutation in the ILDR1 gene in a UAE family, mutations review and phenotype genotype correlation. PLoS One 12, e0185281 (2017).
Talebi, F., Mardasi, F. G., Asl, J. M. & Sayahi, M. Next-generation sequencing identifies three novel missense variants in ILDR1 and MYO6 genes in an Iranian family with hearing loss with review of the literature. Int J Pediatr Otorhinolaryngol 103, 103–108 (2017).
Kim, N. K. et al. Downsloping high-frequency hearing loss due to inner ear tricellular tight junction disruption by a novel ILDR1 mutation in the Ig-like domain. PLoS One 10, e0116931 (2015).
Sang, Q. et al. ILDR1 deficiency causes degeneration of cochlear outer hair cells and disrupts the structure of the organ of Corti: a mouse model for human DFNB42. Biol Open 4, 411–418 (2015).
Legendre, K., Safieddine, S., Kussel-Andermann, P., Petit, C. & El-Amraoui, A. alphaII-betaV spectrin bridges the plasma membrane and cortical lattice in the lateral wall of the auditory outer hair cells. J Cell Sci 121, 3347–3356 (2008).
Li, X. et al. Protective role of hydrogen sulfide against noise-induced cochlear damage: a chronic intracochlear infusion model. PLoS One 6, e26728 (2011).
Bowl, M. R. et al. A large scale hearing loss screen reveals an extensive unexplored genetic landscape for auditory dysfunction. Nat Commun 8, 886 (2017).
Luo, H. et al. The European GWAS-identified risk SNP rs457717 within IQGAP2 is not associated with age-related hearing impairment in Han male Chinese population. Eur Arch Otorhinolaryngol 273, 1677–1687 (2016).
Lu, C. C., Appler, J. M., Houseman, E. A. & Goodrich, L. V. Developmental profiling of spiral ganglion neurons reveals insights into auditory circuit assembly. J Neurosci 31, 10903–10918 (2011).
Blank, T. & Prinz, M. Type I interferon pathway in CNS homeostasis and neurological disorders. Glia 65, 1397–1406 (2017).
Shahin, H. et al. Mutations in a novel isoform of TRIOBP that encodes a filamentous-actin binding protein are responsible for DFNB28 recessive nonsyndromic hearing loss. Am J Hum Genet 78, 144–152 (2006).
Kitajiri, S. et al. Actin-bundling protein TRIOBP forms resilient rootlets of hair cell stereocilia essential for hearing. Cell 141, 786–798 (2010).
Wesdorp, M. et al. Broadening the phenotype of DFNB28: Mutations in TRIOBP are associated with moderate, stable hereditary hearing impairment. Hear Res 347, 56–62 (2017).
Psaty, B. M. et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet 2, 73–80 (2009).
International Organization for Standardization. Acoustics–Statistical distribution of hearing thresholds related to age and gender. ISO 7029:2000 (2000).
Winkler, T. W. et al. Quality control and conduct of genome-wide association meta-analyses. Nat Protoc 9, 1192–1212 (2014).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779 (2015).
Bycroft, C. et al. Genome-wide genetic data on ~500,000 UK Biobank participants. bioRxiv (2017).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8, 1826 (2017).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 11, e1004219 (2015).
Schrauwen, I. et al. A comprehensive catalogue of the coding and non-coding transcripts of the human inner ear. Hear Res 333, 266–274 (2016).
Shen, J., Scheffer, D. I., Kwan, K. Y. & Corey, D. P. SHIELD: an integrative gene expression database for inner ear research. Database (Oxford) 2015, bav071 (2015).
Liu, H. et al. Characterization of transcriptomes of cochlear inner and outer hair cells. J Neurosci 34, 11085–11095 (2014).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88, 76–82 (2011).
Zheng, J. et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017).
Hoffmann, T. J. et al. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nature genetics 49, 54–64 (2017).
Acknowledgements
The acknowledgements and funding sources are listed per cohort in the supplementary information. The authors wish to thank all individuals for their participation. The funders had no role in study design, analysis or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptual design and phenotype definition was done by A.P.N., N.L.H.-C., D.S.E., S.R.P., C.-M.L., W.T.L. Jr., S.S., G.J.T., V.G., H.J.H. and A.G. Phenotype calculation, data management and GWAS in each of the discovery cohorts was performed by A.P.N., L.B., J.J., M.W.C., N.L.H.-C., D.S.E., S.R.P., T.R.P., G.E., M.A.I., C.-M.L., W.T.L. Jr., M.N., S.S., N.S., G.J.T., A.G.U., V.G., H.J.H. and A.G. Replication cohort data was maintained, analysed and provided by C.B., M.B., M.C., Y.G., N.P., C.S., M.R.S., K.V., D.V., H.W., E.F., C.S., G.v.C., A.C., K.J.C., P.G., G.G., R.C.K., J.M.S., J.G.W. and F.M.K.W. GWAS meta-analysis was performed by L.B. and J.J. UK Biobank validation was done by H.W. and F.M.K.W. Data on gene expression in the human cochlea was provided by E.F. and G.v.C. Interpretation of results was done by A.P.N., L.B., N.R.Z.N., J.J., N.P., A.G.U., H.J.H., F.M.K.W. and A.G. The manuscript was written by A.P.N., L.B., N.R.Z.N. and F.M.K.W. All other authors reviewed and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nagtegaal, A.P., Broer, L., Zilhao, N.R. et al. Genome-wide association meta-analysis identifies five novel loci for age-related hearing impairment. Sci Rep 9, 15192 (2019). https://doi.org/10.1038/s41598-019-51630-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51630-x
This article is cited by
-
Sex differences in the polygenic architecture of hearing problems in adults
Genome Medicine (2023)
-
Rare-variant association analysis reveals known and new age-related hearing loss genes
European Journal of Human Genetics (2023)
-
Imputation of SNPs associated with presbycusis through linkage disequilibrium analysis in the ILDR1 gene
Journal of Genetics (2023)
-
A genome-wide association study of frailty identifies significant genetic correlation with neuropsychiatric, cardiovascular, and inflammation pathways
GeroScience (2023)
-
The hearing-impaired patient: what the future holds
Human Genetics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
