
Abstract
Translation elongation factor EF1A delivers aminoacyl-tRNA to the ribosome in a GTP-bound form, and is released from the ribosome in a GDP-bound form. This association/dissociation cycle proceeds efficiently via a marked conformational change in EF1A. EF1A function is dependent on the ribosomal “stalk” protein of the ribosomal large subunit, although the precise mechanism of action of the stalk on EF1A remains unclear. Here, we clarify the binding mode of archaeal stalk aP1 to GTP-bound aEF1A associated with aPelota. Intriguingly, the C-terminal domain (CTD) of aP1 binds to aEF1A•GTP with a similar affinity to aEF1A•GDP. We have also determined the crystal structure of the aP1-CTD•aEF1A•GTP•aPelota complex at 3.0 Å resolution. The structure shows that aP1-CTD binds to a space between domains 1 and 3 of aEF1A. Biochemical analyses show that this binding is crucial for protein synthesis. Comparison of the structures of aP1-CTD•aEF1A•GTP and aP1-CTD•aEF1A•GDP demonstrates that the binding mode of aP1 changes markedly upon a conformational switch between the GTP- and GDP-bound forms of aEF1A. Taking into account biochemical data, we infer that aP1 employs its structural flexibility to bind to aEF1A before and after GTP hydrolysis for efficient protein synthesis.
Similar content being viewed by others

Introduction
Protein synthesis on the ribosome is regulated by the action of several translational GTPase factors on the ribosome with their action being coupled to GTP hydrolysis1,2,3. The archaeal GTPase factor aEF1A (eEF1A in eukaryotes, EF-Tu in bacteria) delivers aminoacyl-tRNA to the ribosome in its GTP-bound form4. After codon recognition by the aminoacyl-tRNA and GTP hydrolysis, aEF1A is then released from the ribosome in its GDP-bound form. aEF1A also delivers aRF1 and aPelota in translation termination and mRNA surveillance pathways, respectively5. Therefore, the progress of the ribosome•aEF1A association/dissociation cycles mediated by GTP hydrolysis is related to efficient and accurate translation3.
The ribosomal protein called the “stalk” which is in the ribosomal large subunit plays a crucial role in efficient action of translational GTPases3,6,7,8. In archaea, the ribosomal stalk protein, aP1, and its anchor protein aP0 form a heptameric complex aP0•(aP1)2•(aP1)2•(aP1)29,10. Our biochemical studies have indicated that the flexible C-terminal end region of aP1 (aP1-CTD) directly binds to both the GTP- and GDP-bound forms of aEF1A11,12, and suggest that it plays important roles in activity, including in recruitment of aEF1A•GTP to the ribosomal factor binding center and in activation of GTP hydrolysis12,13. In a previous study, we solved the crystal structure of the complex between aP1 and the GDP-bound form of aEF1A12. The structure showed that aP1-CTD forms an α-helix in the complex and interacts with a hydrophobic space between domains 1 and 3 of aEF1A•GDP. Comparison of the structures of the GDP- and GTP-bound conformations of aEF1A demonstrates a difference in the location of domain 1 relative to domains 2 and 3. Such a change is also observed for bacterial EF-Tu (these GDP- and GTP-bound conformations of EF1A/EF-Tu are sometimes referred to as the open and closed conformations, respectively)14,15,16,17,18. In the GTP-bound form of aEF1A (closed conformation), the space between domains 1 and 3, to which aP1-CTD binds in aP1•aEF1A•GDP (open conformation), is completely disrupted. Therefore, the stalk is expected to bind to aEF1A•GTP in a completely different mode to that when it is bound to aEF1A•GDP. However, no data is available on the structure of the aP1•aEF1A•GTP complex.
Elucidation of the molecular mechanism of binding between the stalk and aEF1A•GTP is important, because it would provide evidence for the action of the ribosomal stalk in recruitment to the ribosome of aEF1A•GTP associated with ligands (including aminoacyl-tRNA, aRF1, and aPelota), and also in the coupled GTP hydrolysis. Using archaeal samples, we here describe X-ray crystal structural analysis of the complex of aP1•aEF1A•GTP. We use a 17-mer peptide as an aP1-CTD sample and aPelota as a ligand of aEF1A to stabilize the GTP-bound state, and have determined the complex structure at 3.0 Å resolution. The structure shows that aP1-CTD binds to a hydrophobic cleft between domains 1 and 3 formed uniquely in the GTP-bound form of aEF1A. Moreover, the interactions between aP1-CTD and aEF1A•GTP seen in the crystal structure are evaluated by biochemical analysis using aEF1A mutants. The results suggest that, despite the large conformational change of aEF1A before and after GTP hydrolysis, interaction between aP1-CTD and aEF1A is maintained by switching of binding modes.
Results
Binding of aP1 to GTP-bound form of aEF1A
The ribosomal stalk protein aP1 promotes translation by recruiting aEF1A•GTP associated with its ligand (aminoacyl-tRNA, aRF1, or aPelota) to the ribosome. Therefore, we have analyzed the binding between aP1 and aEF1A•GTP in the presence of one of its ligands, aPelota. We initially attempted to prepare Pyrococcus horikoshii aPelota (PhoPelota), because we were able to show binding of PhoP1 to the GDP-bound form of elongation factor aEF1A using P. horikoshii PhoEF1A and elucidated the crystal structure of the complex12. Unfortunately, E. coli expression of PhoPelota was poor. However, we found that Pyrococcus furiosus aPelota (PfuPelota) was expressed at a sufficiently high enough level for the analysis. Furthermore, there is high homology in amino acid sequences between PhoPelota and PfuPelota (Fig. S1). Therefore, we used PfuPelota with PhoP1 and PhoEF1A for further biochemical binding assays.
Our previous study showed that PhoP1 binds to the PhoEF1A•GTP•aminoacyl-tRNA complex via its C-terminal region12. Therefore, we tested whether the C-terminal region of PhoP1 binds to the PhoEF1A•GTP•PfuPelota complex, using a pull-down assay with maltose-binding protein fused to the C-terminal 14 amino acid sequence of PhoP1 (MBP-PhoP1C14). As shown in Fig. 1A, MBP-PhoP1C14 co-precipitated not only with PhoEF1A•GTP (lane 10), but also with both PhoEF1A•GTP and PfuPelota (lane 12). However, PfuPelota was not precipitated in the sample lacking PhoEF1A•GTP (lane 11). The results indicate that the C-terminal region of PhoP1 binds to PhoEF1A within the PhoEF1A•GTP•PfuPelota complex. The binding between the C-terminal region and the PhoEF1A•GTP•PfuPelota complex was also detected by fluorescent polarization using a C-terminal 14-mer peptide of PhoP1, which was labeled with fluorescein isothiocyanate (FITC) at its N-terminus (Fig. 1B). Effective binding was not detected with a 11-mer peptide lacking the C-terminal 3 amino acids of the 14-mer peptide (PhoP1C14∆3). The Kd value for the binding of the C-terminal peptide to the PhoEF1A•GTP•PfuPelota complex was estimated as 23.6 μM, a value which is comparable to the value of 10.4 μM determined for binding of the same C-terminal peptide to the PhoEF1A•GDP complex (Fig. 1C). The results indicate that the C-terminal region of PhoP1 binds both the PhoEF1A•GTP•PfuPelota and PhoEF1A•GDP complexes with a similar affinity.

Biochemical analyses of the interaction between aEF1A•GTP and aP1. (A) In vitro pull-down assay using amylose resin. PhoEF1A, MBP-PhoP1C14, and PfuPelota indicated at the top of the gel were mixed without (lanes 1–6) or with amylose resin (lanes 7–12). A portion of input samples (lanes 1–6) and protein samples bound to amylose resin (lanes 7–12) were subjected to SDS-PAGE. The proteins were detected by CBB staining. For reference, the uncropped gel image is included as Fig. S9A. (B) Fluorescence polarization binding assay. The FITC-labeled peptide PhoP1C14 (WT) or PhoP1C14∆3 lacking the C-terminal 3 residues from PhoP1C14 (∆C3) was mixed with increasing concentrations of the PhoEF1A •GTP•PfuPelota complex, and mP values were determined for individual protein concentrations. Error bars represent SD values (n = 3). (C) The same Fluorescence polarization assay as (B), except that the binding of PhoP1C14 to the PhoEF1A•GTP•PfuPelota complex (red line) was compared with that of the PhoEF1A•GDP complex (blue line). The mP values were determined as in (B). The Kd values are presented under the curves.
Structural basis for interaction of aP1 stalk protein with aEF1A•GTP•aPelota complex
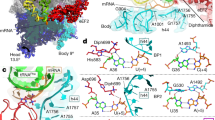
To elucidate the molecular details of the interaction between the C-terminal region of aP1 and the GTP-bound form of aEF1A, we attempted crystallization of the P1•EF1A•GTP•Pelota tetrameric complex using samples from several species including P. horikoshii, P. furiosus, and Aeropyrum pernix. The results of these efforts were a crystal of the A. pernix tetrameric complex ApeP1C17•ApeEF1A•GTP•ApePelota that diffracted to 3.0 Å resolution. The data collection and structure refinement statistics are summarized in Tables S1 and S2. The quality of the electron density maps surrounding aP1 and GTP in the complex is shown in Fig. S2. The overall structure of the tetrameric complex is shown in Fig. 2A. As reported by Kobayashi et al.16, the orientation of domains 1, 2, and 3 of ApeEF1A•GTP in the presence of ApePelota is that of the closed state (Fig. 2B), as observed in the tRNA-bound form of bacterial EF-Tu•GTP14. A part of the ApeP1C17 peptide (S103 to F111) was observed in a hydrophobic space between domains 1 and 3 in an extended unstructured form (Fig. 2A). This P1 binding site was ~25 Å away from the GTP/GDP-binding site in domain 1.

Crystal structure of the aP1-CTD•aEF1A•GTP•aPelota complex. (A) Overall structure of the GTP-bound complex ApeP1C17•ApeEF1A•GTP•ApePelota. ApeEF1A•ApePelota, ApeP1C17, and GTP are represented by surface, ribbon, and stick models, respectively. Colors in ApeEF1A are as follows, domain 1 (pink), domain 2 (orange) and domain 3 (yellow); ApePelota is blue; aP1 is green. The N- and C-termini of ApeP1C17 are indicated as ‘N’ and ‘C’, respectively. (B) Structural comparison of ApeEF1A•GTP in the complex described here with PhoEF1A•GDP [PDB ID: 3WY9]12 superposing domains 2 and 3. The colors of domains of aEF1A are as in (A), except for domain 1 of PhoEF1A•GDP (grey). (C) Structure of the binding interface between ApeP1C17 and ApeEF1A in the ApeP1C17•ApeEF1A•GTP•ApePelota complex. ApeEF1A and ApeP1C17 are represented by a ribbon model, and the amino acid residues of ApeEF1A and ApeP1C17 that participate in the interaction by stick models. The color coding is the same as in (A). (D) Schematic diagram of the interactions between ApeP1C17 and ApeEF1A. The van der Waals contacts and hydrogen bonds are represented by black and red lines, respectively. Amino acid residues and numbering are for A. pernix samples, and those in parentheses are for P. horikoshii samples.
Comparison of the structures of ApeP1C17-bound and ApeP1C17-free ApeEF1A•GTP•ApePelota revealed that, although the orientations of several amino acid side chains in the aP1 binding site of aEF1A differ markedly (details of the interaction between aEF1A and aP1 are described below), the overall structures of the two forms are highly similar (rms deviation of 0.49 Å for equivalent Cα atoms) (Fig. S3A,B). The conformations of regions including the P-loop and switch I and II elements, which are related to GTP hydrolysis, are also similar in the two structures (Fig. S3C). These results indicate that aP1 binding has no substantial effect on the overall structure of ApeEF1A including the GTP hydrolysis site in the domain 1.
Detailed interaction between aP1 and the GTP-bound form of aEF1A
The structure of the ApeP1C17•ApeEF1A•GTP•ApePelota complex revealed that the three hydrophobic amino acids, M108, F109, and F111 of ApeP1 make contact with several amino acid residues of ApeEF1A in the complex (Fig. 2C,D). Thus, M108 binds to R140 in domain 1 of ApeEF1A via van der Waals interaction; F109 binds to R329, F331, I421, and I423 in domain 3 of ApeEF1A via van der Waals interactions; F111 binds to R140 via hydrogen bonding, and to F401, R409, and I423 in domain 3 of ApeEF1A via van der Waals interactions. It is particularly noteworthy that the F109 residue of ApeP1, which is highly conserved among eukaryotes/archaea (Fig. S4), is surrounded by hydrophobic amino acids (F331, I421, and I423 in the case of ApeEF1A) (Fig. 2C), as is the case with the binding to aEF1A•GDP, aEF2•GMPPCP, aIF5B•GDP, and aABCE1•ADP12,19,20,21. It is also notable that ApeP1 has an extra amino acid, F111, at its C-terminus, which is not present in other archaeal aP1 molecules (Fig. S4) and which does not seem to be essential for factor binding as described below.
Functional effect of mutations at the aEF1A•stalk binding sites
The crystal structural analysis indicates contact of M108, F109 and F111 of ApeP1 with ApeEF1A. The results are consistent with binding data shown in Fig. 1B, whereby deletion of the three C-terminal amino acids (L106, F107, and G108) of PhoP1 disrupted PhoEF1A binding. However, as described above, ApeP1 has an extra phenylalanine residue F111 at the C-terminus which is unique to ApeP1 (Fig. S4). We confirmed that deletion of F111 of ApeP1 resulted in no marked effect in binding to the ApeEF1A•GTP complex (Fig. S5). It is therefore likely that M108 and F109 of ApeP1 have the primary role in binding to ApeEF1A. Notably, the highly conserved F109 of ApeP1 binds to a hydrophobic pocket that is formed by the hydrophobic amino acids F331, I421, and I423 of ApeEF1A. To confirm the importance of this feature, we constructed a single mutant, F331A (the closest residue to F109 in ApeEF1A), and triple mutant F331A/I421A/I423 A (3A-mutant) of ApeEF1A, and tested their binding to ApeP1 (MBP-ApeP1C15) by pull-down assay (Fig. 3). Both the mutants associated with GTP and ApePelota showed a markedly reduced ability to bind to MBP-ApeP1C15 (lanes 9 and 10), compared with the wild type ApeEF1A (lane 8).

Effect of mutations at F331, I421, and I423 of ApeEF1A, which were identified as the binding site of the conserved F109 of ApeP1. In vitro pull-down assay using amylose resin. ApeEF1A, MBP-ApeP1C15, and ApePelota indicated at the top of the gel were mixed without (lanes 1–5) or with amylose resin (lanes 6–10). A portion of input samples (lanes 1–5) and protein samples bound to amylose resin (lanes 6–10) were subjected to SDS-PAGE. The proteins were detected by CBB staining. For reference, the uncropped gel image is included as Fig. S9B.
The present structural study showed that 7 amino acid residues (R140, R329, F331, F401, R409, I421, and I423) of ApeEF1A interact with the C-terminal region of ApeP1 (Fig. 2). Amino acid residues at these positions are conserved in archaea at least in terms of their properties (Fig. S6). Indeed, it is remarkable that R409 is highly conserved from archaea to eukaryotes. We next investigated the effect of mutations of these amino acid residues of EF1A on translation elongation. By using P. horikoshii elongation factors aEF1A/aEF2 with P. furiosus 70 S ribosomes, we have previously established a high temperature polyphenylalanine synthesis system13. Here we introduced six mutations in P. horikoshii EF1A, namely at R132, Q326, I328, V398, R406, and M420, which correspond to R140, R329, F331, F401, R409, and I423 in ApeEF1A, respectively. In the present crystal structure, all these amino acid residues interacts with ApeP1 via their side chains (A418 of PhoEF1A, which corresponds to I421 of ApeEF1A, was excluded from the mutagenesis study because it is an alanine residue). We then tested these mutants for poly(U)-dependent polyphenylalanine synthesis activity (Fig. 4). The R132A and I328S point mutations and I328S/M420S double mutation (which are presumed to interact with F107 of PhoP1, a highly conserved phenylalanine residue) caused considerable reduction in activity, while the M420S mutation resulted in only slightly reduced activity. In addition, the effects of these mutations of PhoEF1A were consistent with data on binding between PhoEF1A•GTP•PfuPelota and PhoP1C14 peptide, assayed by fluorescent polarization (Table S3). Although the effects of the point mutations on the binding assay were small, the results were reproducible. These biochemical analyses support the crystal structural data presented here.

Effects of the amino acid substitutions in aEF1A on aEF1A/aEF2-dependent polyphenylalanine synthesis. P. furiosus 70S ribosomes were incubated with P. horikoshii aEF2 and P. horikoshii wild-type aEF1A (bar 2) or each of the aEF1A mutants (bars 3–9) in the presence of poly(U) and E. coli [14C]Phe-tRNA. The amount of polyphenylalanine synthesized in the 10 min reaction was assayed as described in Materials and Methods. Error bars represent SD values (n = 3).
Discussion
The ribosomal stalk protein plays an important role in the recruitment of the GTP-bound form of elongation factor EF-Tu in bacteria, eEF1A in eukaryotes, and aEF1A in archaea, and thus in the delivery of the associated aminoacyl-tRNA to the A site of the ribosome. It had been expected that the stalk binding mode for GTP-bound aEF1A would differ from that for the GDP-bound form which has been reported previously12, because the relative location of domain 1 of aEF1A would be expected to differ in the closed GTP-bound and open GDP-bound conformations. In the present study using archaeal samples, we show that aP1-CTD binds to aEF1A•GTP with similar affinity to aEF1A•GDP. Furthermore, we have determined the crystal structure of the complex of the C-terminal aP1 peptide with aEF1A•GTP•aPelota, which has revealed that the binding mode of aP1 changes markedly upon the conformational switch between the closed and open conformations of aEF1A.
In the aP1-CTD•aEF1A•GTP•aPelota complex, an extended form of the aP1 peptide bound to the interface of domains 1 and 3 of aEF1A in the GTP-bound closed conformation (Fig. 2). This observation is supported by binding experiments (Fig. 3 and Table S3). All mutations of the putative aP1-binding site of aEF1A, which reduced the binding of aP1 to aEF1A•GTP, also decreased the ribosome-dependent polyphenylalanine synthesis (Fig. 4). Therefore, it is likely that the binding of aP1 to aEF1A•GTP as described in this study is functionally important and presumably contributes to recruitment of the ternary complex of aEF1A•GTP•aminoacyl-tRNA to the ribosome. It is interesting and worthwhile to consider the significance of why aP1 binds to the interface of domains 1 and 3 in aEF1A•GTP. A recent study using fluorescence resonance energy transfer (FRET) showed that EF-Tu•GTP is not locked in the closed conformation in solution, but adopts a dynamic conformation in which domain 1 can swing, even in the ternary complex with aminoacyl-tRNA18. Furthermore, this study also showed that the ribosomal factor binding center selectively accepts the EF-Tu•GTP•aminoacyl-tRNA complex in the closed conformation18. Based on these findings, we infer that aP1 facilitates the formation of the closed conformation of aEF1A•GTP by bridging domains 1 and 3, probably with the aid of aminoacyl-tRNA/aPelota/aRF1, and promotes the selective binding of the closed-conformation aEF1A•GTP•aminoacyl-tRNA/aPelota/aRF1 complex to the factor binding center of the ribosome.
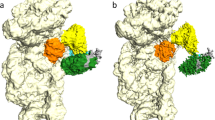
To reinforce the idea that the binding of the C-terminal region of aP1 facilitates factor recruitment to the factor binding center of the ribosome, we constructed a docking model, using the structures of the bacterial 70 S ribosome•EF-Tu•GDPCP•aminoacyl-tRNA complex22 and the archaeal heptameric stalk protein complex10 (Fig. 5). The model shows that the aP1-binding site within aEF1A is facing outward, and that the C-terminal region of aP1 can reach the ribosome-associated aEF1A•GTP via the flexible C-terminal half of aP1 without steric hindrance. Thus, this docking supports our view that aP1 directly binds to aEF1A via the C-terminal region of aP1, and contributes to the recruitment of aEF1A to the factor binding center of the ribosome.

Model of aP1•aEF1A•GTP•aPelota complex docking with the 70S ribosome. Colors are as follows: 50S (white), 30S (light yellow), stalk complex (green), aPelota (blue), aEF1A domain 1 (pink), aEF1A domain 2 (orange) and aEF1A domain 3 (yellow). In this docking model, we superimposed the aP1•aEF1A•GTP•aPelota complex described in this paper onto the bacterial 70 S•EF-Tu•GDPCP•aminoacyl-tRNA complex [PDB ID: 4V5G]22 (Voorhees et al. 2010). Subsequently the aP0•aP1 stalk complex core from Pyrococcus horikoshii [PDB ID: 3A1Y]10 was superimposed onto the model. The aP0•aP1 stalk complex core and aP1C17 peptide bound to aEF1A were connected by the flexible hinge region of aP1. Other hinge regions of aP0 and aP1 were modeled arbitrarily.
Comparison of the current results describing aP1 binding to the GTP-bound form of aEF1A with previous results that describe binding to the GDP-bound form demonstrates that as expected the binding modes differ greatly in the two cases (Figs 6 and S7). In the results described here, the extended C-terminal peptide of aP1 binds to the interface of domains 1 and 3 of the closed GTP-bound conformation of aEF1A and this involves 3 residues of aP1 and 7 residues of aEF1A (Fig. S7). In the aP1•aEF1A•GDP, the C-terminal 27 residues of aP1 form an α-helix, and binds to a different site of the interface between domains 1 and 3 in the opened GDP-bound form of aEF1A, via 8 residues of aP1 and 9 residues of aEF1A (Fig. S7). Of these residues, only the conserved M108 and F109 of ApeP1 (L106 and F107 in PhoP1, respectively) are involved in binding of aP1 to both aEF1A•GDP and to aEF1A•GTP (Fig. S7). These findings suggest that the flexible C-terminal region of aP1 can bind to the two conformations of aEF1A in the process of GTP hydrolysis (aEF1A•GTP and aEF1A•GDP), and that it adopts an induced conformation dependent on the structures of aEF1A (Figs 6 and S8). This presumption is applicable to stalk binding to the other translational GTPases aIF5B20, aEF219, and the translational ATPase aABCE121. These crystal structural analyses have revealed that the conformation of the C-terminal region of aP1 in these complexes differs (Fig. S8). The variable structural properties of the C-terminal region which depends on associated proteins seems to be related to the functional role of the stalk proteins in efficient recruitment of various translational GTP/ATPases to the ribosome in translation.

Comparison of the structures of the GTP- and GDP-bound forms of aP1•aEF1A complexes. Comparison of crystal structures of aP1•aEF1A•GTP (A,C) and aP1•aEF1A•GDP (B,D) [PDB ID: 3WY9]12. Schematic representations of the interaction in the GTP-bound and GDP-bound forms of aP1•aEF1A complexes are shown in (C,D), respectively. Panel (A,B) are shown from the same orientation and direction with respect to domains 2 and 3.
The question arises as to why aP1 maintains the binding to switched GTP/GDP-bound conformations of aEF1A, and we propose a hypothesis as follows. As described above, the binding of aP1 to the GTP-bound form of aEF1A would promote the selective binding of the closed-conformation aEF1A•GTP•aminoacyl-tRNA/aPelota/aRF1 complex to the factor binding center of the ribosome. Upon the binding, GTP hydrolysis occurs and the conformational change of aEF1A from the closed conformation to the open conformation takes place on the ribosome. In this process, aP1 would temporarily release aEF1A, and subsequently aP1 could bind again to aEF1A to stabilize, in turn, the open GDP-bound conformation by bridging domains 1 and 3 in a different position from that in aEF1A•GTP (Figs 6 and S7). Because aEF1A•GDP is released form the ribosome in the open conformation, this stabilization of the open conformation of aEF1A by aP1 would promote the release of aEF1A•GDP from the ribosome. Furthermore, we infer that the switching of the binding of the stalk mentioned above also takes place in eukaryotes (eEF1A•GTP/GDP) and eubacteria (EF-Tu•GTP/GDP). Further studies are needed to validate these hypotheses.
Methods
Plasmid construction
The coding sequences for elongation factor aEF1A of the hyperthermophilic archaeon Aeropyrum pernix (ApeEF1A) were amplified from the genome by PCR, and inserted between the NdeI and XhoI sites in the E. coli expression vector, pET-15b containing a six His-tag. The coding sequences for aPelota were amplified from Pyrococcus furiosus and A. pernix genomes, and inserted between the NdeI and BamHI sites in the pET-15b vector. The plasmid for expression of P. horikoshii aEF1A (PhoEF1A) and aEF2 (PhoEF2) were as described previously12,23. The plasmid for expression of N-terminal maltose binding protein (MBP) fused with the C-terminal 14 amino acid residues of P. horikoshii aP1 (MBP-PhoP1C14) was constructed by removing the coding sequence for residues 61–94 from pMAL-c4x-aP1[61–108]21 by PCR. Although pMAL-c4x-aP1[61–108] was prepared using the P. furiosus genome, the amino acid sequence for the C-terminal 14 amino acid residues of P. furiosus aP1 is identical to that of P. horikoshii aP1 (Fig. S4). The plasmid for expression of N-terminal MBP fused with the C-terminal 15 amino acid residues of A. pernix aP1 (MBP-ApeP1C15) was constructed by insertion of the PCR-amplified sequence corresponding to the C-terminal 15 amino acid residues of A. pernix aP1 between the EcoRI and PstI sites in the pMAL-c4x vector. Site-directed mutagenesis of PhoEF1A or ApeEF1A was performed by PCR using the plasmid carrying each EF1A gene, as described previously12. The PCR primers are listed in Table S4.
Protein expression and purification
ApeEF1A, PhoEF1A, their mutants, PfuPelota, ApePelota, and the fusion proteins MBP-PhoP1C14 and MBP-ApeP1C15 were all expressed in E. coli BL21 (DE3) codon plus RIL or BL21 (DE3) by adding 0.5 mM IPTG. After incubation at 37 °C for 3 h (for PhoEF1A, its mutants, and MBP-PhoP1C14) or 20 °C for 18 h (for ApeEF1A, its mutants, ApePelota, PfuPelota, and MBP-ApeP1C15), cells were harvested by centrifugation, resuspended in Lysis buffer (20 mM HEPES-KOH pH7.6, 1 M NH4Cl, 5% (v/v) glycerol, 7 mM 2-ME) and disrupted by sonication. After sedimenting cell debris and other insoluble materials, PhoEF1A, ApeEF1A and their mutants were purified by heat treatment (80 °C for 20 min) of cell extracts, Ni-affinity chromatography and size-exclusion chromatography. The His-tag of aEF1A samples and its mutants was cleaved with thrombin before anion-exchange chromatography. To prepare the GTP-bound form of aEF1A, tightly bound GDP was removed from purified aEF1A by bacterial alkaline phosphatase treatment, as described previously12. MBP-PfuP1C14 and MBP-ApeP1C15 were purified by amylose-affinity chromatography and size-extraction chromatography. ApePelota and PfuPelota were purified by heat treatment (70 °C for 20 min) of cell extracts, Ni-affinity chromatography and anion-exchange chromatography.
All peptides used were synthesized by Hokkaido System Science Co., Ltd (Sapporo, Japan). The C-terminal 17-mer peptide of A. pernix aP1 (ApeP1C17) [sequence: KKEEEVDLSGLSGMFGF] was used for crystallization (See Fig. S4). The C-terminal 14-mer peptide of P. horikoshii aP1 (PhoP1C14) [sequence: SEEEALAGLSALFG] and the 11mer, the same peptide as the 14-mer, but lacking the C-terminal 3 residues (PhoP1C14∆3) [sequence: SEEEALAGLSA], were labeled with fluorescein isothiocyanate (FITC) at the N-termini and used for binding experiments.
Crystallization and structure determination
Crystals of the ApeP1C17•ApeEF1A•GTP•ApePelota complex were prepared by the sitting drop vapor diffusion method at 20 °C by mixing 1 µl of protein solution (1.55 mg/ml ApeP1C17 peptide, 6.5 mg/ml ApeEF1A, 5.4 mg/ml ApePelota, 2.5 mM GTP, 25 mM Tris-HCl, pH 8.0, 500 mM NaCl, 5 mM MgCl2, 1 mM DTT) and an equal volume of reservoir solution (100 mM Tris-HCl, pH 8.0, 200 mM Li2SO4, 16% (w/v) PEG3350). Diffraction data were collected with a wavelength of 1.000 Å at 95 K on the BL-5A of the Photon Factory (Tsukuba, Japan) using an ADSC Quantum 315r CCD detector. Diffraction data were processed with the program HKL2000 (HKL Research). The initial model of the ApeP1C17•ApeEF1A•GTP•ApePelota complex was obtained by the molecular replacement method using MOLREP24, using the structures of ApeEF1A•GTP•ApePelota [PDB ID: 3WXM]16 as a search model. The model was then manually refined using COOT and REFMAC525,26. The resultant data collection and refinement statistics are summarized in Supplementary Tables S1 and S2. Structural figures were prepared using CueMol2 (http://www.cuemol.org/en/) and PyMOL (https://pymol.org/2/).
Pull-down assay
EF1A (1,000 pmol) and Pelota (1,000 pmol) were mixed with equal amounts of MBP-P1 in 100 µl of PD buffer (25 mM Tris-HCl pH 8.0, 200 mM NaCl, 10 mM MgCl2, 1 mM GTP). Subsequently, 20 µl of amylose resin (New England Biolabs) was added to the mixture. After incubation with rotation at 4 °C for 90 min, the beads were washed with PD buffer and then bound proteins were co-eluted with 50 µl of PD buffer containing 50 mM maltose. The eluted proteins were analyzed by SDS-PAGE, followed by CBB staining21.
Fluorescent polarization binding assay
The FITC-labeled peptide (50 nM) was mixed with 0, 2.5, 5, 10, 20, 40, 80, 160 µM of the PhoEF1A•GTP•PfuPelota, or ApeEF1A•GTP•ApePelota complex in 100 µl of FP buffer (20 mM HEPES-KOH pH 7.6, 10 mM MgCl2, 150 mM KCl, 0.1 mg/ml BSA). All measurements were performed at 25 °C using a Pan Vera Beacon 2000 fluorescence polarization instrument (Invitrogen), as described in the manufacturer’s protocol. Curve fitting was performed using nonlinear regression using saturation binding–one site specific binding in GraphPad Prism 6, GraphPad Software (San Diego, California USA).
Polyphenylalanine synthesis
P. furiosus ribosomes were prepared, as described previously27, except the buffer for preparation and storage contained 0.5 mM spermine. Poly(U)-dependent polyphenylalanine synthesis was assayed using the isolated ribosomes, PhoEF1A/PhoEF2, and [14C]Phe-tRNA, as described previously13. Functional effects of mutations of PhoEF1A were tested on the polymerization activity.
Data Availability
The structural coordinates of the refined model and the structure factors have been deposited in the RCSB Protein Data Bank (www.rcsb.org/) and assigned the identifier 6JI2. All other data supporting the findings of this study are available within the paper and Supplementary Information files.
References
Rodnina, M. V. & Wintermeyer, W. Recent mechanistic insights into eukaryotic ribosomes. Curr. Opin. Cell Biol. 21, 435–443 (2009).
Schmeing, T. M. & Ramakrishnan, V. What recent ribosome structures have revealed about the mechanism of translation. Nature 461, 1234–1242 (2009).
Liljas, A. & Ehrenberg, M. The catalysts––translation factors. Structural Aspects of Protein Synthesis (2nd edition). 149–228 (World Scientific, Singapore, 2013).
Stark, H. et al. Ribosome interactions of aminoacyl-tRNA and elongation factor Tu in the codon-recognition complex. Nat. Struct. Biol. 9, 849–854 (2002).
Saito, K. et al. Omnipotent role of archaeal elongation factor 1 alpha (EF1α) in translational elongation and termination, and quality control of protein synthesis. Proc Natl Acad Sci USA 107, 19242–19247 (2010).
Liljas, A. & Sanyal, S. The enigmatic ribosomal stalk. Q. Rev. Biophys. 51, e12 (2018).
Mohr, D., Wintermeyer, W. & Rodnina, M. V. GTPase activation of elongation factors Tu and G on the ribosome. Biochemistry 41, 12520–12528 (2002).
Diaconu, M. et al. Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell 121, 991–1004 (2005).
Maki, Y. et al. Three binding sites for stalk protein dimers are generally present in ribosomes from archaeal organism. J. Biol. Chem. 282, 32827–32833 (2007).
Naganuma, T. et al. Structural basis for translation factor recruitment to the eukaryotic/archaeal ribosomes. J. Biol. Chem. 285, 4747–4756 (2010).
Nomura, N. et al. Archaeal ribosomal stalk protein interacts with translation factors in a nucleotide-independent manner via its conserved C terminus. Proc. Natl. Acad. Sci. USA 109, 3748–3753 (2012).
Ito, K. et al. Molecular insights into the interaction of the ribosomal stalk protein with elongation factor 1α. Nucleic Acids Res. 42, 14042–14052 (2014).
Imai, H. et al. Functional role of the C-terminal tail of the archaeal ribosomal stalk in recruitment of two elongation factors to the sarcin/ricin loop of 23S rRNA. Genes Cells 20, 613–624 (2015).
Nissen, P. et al. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science 270, 1464–1472 (1995).
Polekhina, G. et al. Helix unwinding in the effector region of elongation factor EF-Tu–GDP. Structure 4, 1141–1151 (1996).
Kobayashi, K. et al. Strucural basis for mRNA surveillance by archaeal Pelota and GTP-bound EF1α complex. Proc. Natl. Acad. Sci. USA 107, 17575–17579 (2010).
Kobayashi, K., Saito, K., Ishitani, R., Ito, K. & Nureki, O. Structural basis for translation termination by archaeal RF1 and GTP-bound EF1α complex. Nucleic Acids Res. 40, 9319–9328 (2012).
Johansen, J. S. et al. E. coli elongation factor Tu bound to a GTP analogue displays an open conformation equivalent to the GDP-bound form. Nucleic Acids Res. 46, 8641–8650 (2018).
Tanzawa, T. et al. The C-terminal helix of ribosomal P stalk recognizes a hydrophobic groove of elongation factor 2 in a novel fashion. Nucleic Acids Res. 46, 3232–3244 (2018).
Murakami, R. et al. The interaction between the ribosomal stalk proteins and translation initiation factor 5B promotes translation initiation. Mol. Cell Biol. 38, e00067–18 (2018).
Imai, H. et al. The ribosomal stalk protein is crucial for the action of the conserved ATPase ABCE1. Nucleic Acids Res. 46, 7820–7830 (2018).
Voorhees, R. M., Schmeing, T. M., Kelley, A. C. & Ramakrishnan, V. The mechanism for activation of GTP hydrolysis on the ribosome. Science 330, 835–838 (2010).
Nomura, T. et al. In vitro reconstitution of the GTPase-associated centre of the archaebacterial ribosome: the functional features observed in a hybrid form with Escherichia coli 50S subunits. Biochem. J. 396, 565–571 (2006).
Vagin, A. & Teplyakov, A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 (1997).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011).
Benelli, D. & Londei, P. In vitro studies of archaeal translational initiation. Methods Enzymol. 430, 79–109 (2007).
Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research (B) (JP16H04741 to T.U.); a Grant-in-Aid for JSPS Fellows (JP16J01001 to H.I.); a Grant-in-Aid for Scientific Research (C) (JP15K06964 to K.I.) from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Contributions
The authors T.U. and K.I. conceived and designed the experiments. K.M., H.I. and M.K. performed the experiments, and K.M. and H.I. made equal contributions. S.I. and Y.I. provided cultured P. furiosus cells and gave useful advice on archaeal protein samples. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maruyama, K., Imai, H., Kawamura, M. et al. Switch of the interactions between the ribosomal stalk and EF1A in the GTP- and GDP-bound conformations. Sci Rep 9, 14761 (2019). https://doi.org/10.1038/s41598-019-51266-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51266-x
This article is cited by
-
TaEF1A is involved in low phosphorus stress responses and affects root development
Plant Growth Regulation (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
