
Abstract
In this study we investigated the impact of dietary protein and carotene levels on microbial functions and composition during the last month of purebred fattening Duroc pigs. Fecal microbiota was characterized using 16S ribosomal RNA sequencing at two points of live, 165 (T1) and 195 (T2) days. From 70 to 165 days of age, 32 pigs were divided into two groups fed either a standard-protein (SP) or a low-protein (LP) diet. In the last month (165–195 days), all pigs received a LP diet, either carotene-enriched (CE) or not (NC). Significant differences were observed between T1 and T2 at Amplicon Sequences Variants (ASVs), phylum and genus levels. In T1 group, Prevotella, Faecalibacterium and Treponema were the genera most influenced by dietary protein, together with predicted functions related with the degradation of protein. In contrast, the CE diet did not impact the microbiome diversity, although 160 ASVs were differentially abundant between CE and NC groups at T2. Weak stability of enterotype clusters across time-points was observed as consequence of medium-term dietary interventions. Our results suggest that during the last month of fattening, dietary protein have a stronger effect than carotenes on the modulation of the compositional and functional structure of the pig microbiota.
Similar content being viewed by others
Introduction
The microbiota is a heterogeneous ecosystem composed of thousands of microbial species that influence on host physiological functions such as food intake, production of vitamins, metabolism, immune system activation, and resistance to infection1. Previous studies in pigs described the composition of microbiota at growing-finishing and fattening phases2,3,4,5,6. Modulations of the gut microbial community can have beneficial effects on pig’s health through direct stimulation of the immune system and also through microbiota-generated nutrients such as aminoacids, vitamins or short-chain fatty acids7.
As in humans, pig microbiota diversity starts being established at birth2 and reaches some stability at adulthood2,5. It has been postulated that microbiota could be modulated to improve the health or/and the productivity of pigs7,8. According to bacterial relative abundance, the pig gut microbial composition is structured in two well-defined enterotypes2,9,10 that remain more stable in post-weaning pigs than in younger animals2. This classification can change with the influence of many factors including the age of the animal, hygienic and health conditions and the diet7,8. For instance, in model animals, the level of protein and Vitamin A in feed can affect the gut bacterial ecosystem, which in turn can disturb the physiology of the host11,12,13,14. The amount of protein in the diet has a direct effect on gut microbiota with a positive correlation with the microbial diversity15,16. This correlation depends directly on the quantity and quality of the protein15. Protein-enriched diets results in longer transit time and a higher microbiota concentration in the large intestine as this section plays a relevant role in protein degradation into peptides and amino acids via extracellular bacterial proteases and peptidases17. In addition, low‐protein diets can be used to reduce the cost and environmental burdens of livestock production without affecting performance18. For instance, low protein diets can impact pig microbiota by decreasing microbial metabolites such as ammonia19. On the other hand, restriction on dietary vitamin A has been proposed as a dietary intervention to improve intramuscular fat content, a trait related to meat quality, in beef20 and pork. Provitamins A such as β-carotenoids are transformed into the active vitamin A mainly in the gut epithelial cells, a process that is enhanced by the action of colon microbiota over fat solubilisation11. Modified crops such as high-carotenoid corn can impact directly in the levels of vitamins A in the feed21. In a previous work we have shown the combinatory effect of dietary protein and vitamin A on intramuscular fat content and composition22. In the present study we question whether pig microbiota diversity has been affected by these dietary interventions and can relate to some of the phenotypic changes. Therefore, in this study we used 16S rRNA gene sequencing to evaluate the impact of different levels of protein and high-carotene diets on the fecal microbiota of 32 Duroc pig in terms of taxonomic and functional composition across two age-points during the fattening period.
Results and Discussion
In this study 16S rRNA gene sequences from 64 fecal samples of Duroc pig collected in two time-points (T1, 165d of live and T2, 195d of live) were used to determine the influence of dietary protein (T1: SP, standard-protein vs LP, low-protein diets) and dietary carotenes (T2: CE, carotene-enriched vs NC, not carotene diet) on the microbiota composition. After quality control, a total of 3,286,123 contiguous sequences were retained to detect 3,141 Amplicon Sequences Variants (ASVs). In agreement with previous findings in Duroc pigs4, as well as in other pig breeds2,9,10,23,24,25, the most abundant phyla in both time-points were Firmicutes (55.16%, in T1 and 46.48%, in T2) and Bacteroidetes (33.58%, in T1 and 36.47%, in T2). In both time-points, the less abundant phyla were Proteobacterius (5–6%), Spirochaetes (2.3–6%) and Verrucomicrobia (0.9–1.3%). At genus level, the predominant genera were Prevotella (18.68%, in T1 and 14.96%, in T2) followed by the uncategorized group (5.23%, in T1 and 5.82%, in T2) and Oscillospira (4.56%, in T1 and 4.54%, in T2). The abundance of Treponema genus was higher (p = 1.16E05) in T2 (5.48%) than in T1 (2.40%).
Dietary intervention affects fecal microbiota composition rather than diversity
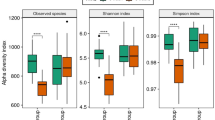
Before estimating diversity indexes (alpha, beta and richness), samples were rarefied at 6,000 reads of depth to allow an equal depth in all samples. Observed similarities at taxonomic levels between time points were also reflected at diversity index level. There were no significant differences (p > 0.05) between the T1 and T2 groups, neither within each treatment group in T1 (SP vs LP) or T2 (CE vs NC). It is well known that diet, environment, lifestyle and resource availability can influence the composition and function of the gut microbiota26,27,28,29,30,31. However, similarly to our results, other groups have reported no changes in the ileal, cecal and colonic microbiota diversity between pigs fed with different inclusion level of protein32,33, which may be partially explained by the smaller difference (3%) of protein inclusion between diets in our and some of those studies32,33. In line with these results, reduction level of protein inclusion in piglets can result in lower intestinal ammonia concentration without significance changes of the bacterial community34,35. Indeed, in pigs, changes in dietary protein inclusion levels can affect microbiota composition rather than diversity, even during drastic dietary challenges such as those taking place at weaning36.
Despite the absence of relevant changes in the diversity indexes, compositional differences between groups were observed. According to the Non-metric multidimensional scaling (NMDS) and Multivariate analysis of variance (PERMANOVA) analyses, time groups (T1 vs T2) exhibited the most important effects over the microbiota composition. The NMDS analysis showed two clear clusters at ASVs, phylum and genus levels (Fig. 1) suggesting a dietary effect on the pig microbiota composition due to the shift from SP to LP between T1 to T2. Group factor (T1 vs T2) explained around 42% of total variability of the relative abundance data. The significance of this finding was confirmed with the PERMANOVA analysis (p < 0.0001), suggesting a strong impact of time-points at all analyzed levels (ASVs, phylum and genus). In addition to the time group effect, other confounding factors such as diet composition or other environmental stressors must coexist to modulate the microbiota composition. For instance, several studies agree that diet is one of the most relevant factors modulating microbiota composition26,37,38. In our study, different diets were employed between T1 and T2. Pigs at T1 diet were fed at two protein intakes, whereas at T2 all of them were fed at a low-protein level, although with two different levels of carotenes, a fact that might modulate the composition of microbial gut ecosystem. This dietary modification may also affect environmental conditions and therefore alter microbial populations11,12,13,14. PERMANOVA analysis was used to assess the effect of the dietary protein at T1 (SP vs LP) and the carotene level at T2 (CE vs NC). Significant effects were observed at genus (p < 0.05) and suggestive at ASVs level (p = 0.07) in T1, whereas no significant effect associated with the carotene level was observed at T2 (p > 0.05). This will confirm that changes in dietary protein play a more relevant effect than carotene level on gut microbiota composition during the last month of fattening. The impact of dietary protein on microbiota composition has been reported in pigs18,32,33 and in other mammalian species15,29,30,39,40. For example, in mice, lower dietary protein levels reduce the faecal bacterial concentration30. Moreover, microbial metabolism of protein-based diet can result in the negative effect of the epithelial cell in the gastrointestinal tract41 as product of the formation of deleterious metabolites as phenols, indoles, amines, sulfides, ammonia, and monocarboxylic acids, particularly as a result of fermentation in the large intestine, which are cytotoxic to the cells36,42.

Non-Metric Multidimensional Scaling (NMDS) plots of bacterial communities at (a) Amplicon Sequences Variants, (b) phylum and (c) genus levels by samples taken at 165 days (T1) and 195 days (T2).
Time and diet-specific modulation of fecal microbiota composition
We also performed a presence-absence (PA) test to identify specific features in T1 and T2 at ASVs, phyla and genus levels (Supplementary Table S1). According to PA, a total of 7 ASVs were only found in T1 and 25 in T2. At phylum and genus levels the PA test did not show any time-point-specific taxa. In addition, a differential abundance (DA) analysis was done to test significant differences in the abundance of features between time-point-groups of animals. The DA between groups (T1 vs T2) identified 6 phyla, 30 genera and 777 ASVs (Supplementary Tables S2, S3 and S4). Amongst the DA phyla, Deferribacteres, Euryarchaeota and Spirochaetes were the most abundant at T1 while Synergistetes, Tenericutes and Spirochaetes were at T2. At genera level, the most abundant at T1 were Bulleidia, Ruminococcus and Lachnospira and at T2, Treponema, CF231 and RFN20 were identified in a higher proportion.
To identify features linked to the different dietary protein (T1) and pro-Vitamin A (T2) levels, we focused the DA analysis within each time-point. At T1 (SP vs LP), 160 ASVs were detected as DA at false discovery rate of 5%. Within genus, Dorea (higher in SP) and Peptococcus (higher in LP diet) were DA, whereas no significant differences were found at the phylum level. On the other hand, within T2 group (CE vs NC) a total of 162 ASVs were identified, but only the genus Epulopiscium was DA, showing higher abundance in CE. According to DA, a similar number of differentially abundant ASVs was identified at each time-point (160 ASVs, in T1 and 162 ASVs, in T2). However, the overlapping between the ASVs reported as DA across time-points (T1 and T2) was low. For instance, only 12% (20) of ASVs identified as DA in T1 were also reported in T2. Moreover, no overlapping was observed at phylum or genus level. The low overlapping suggests diet-specific gut microbiota modulation as a consequence of the different dietary protein (T1) and carotene (T2) levels.
Changes in fecal microbiota can discriminate pigs by diet et each time point
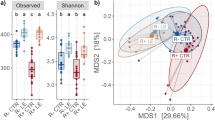
To complement the DA as well as to identify discriminant ASVs which contribute to classify samples according to the protein (SP vs LP) or carotene (CE vs NC) intake, a partial least squares discriminant analysis (PLS-DA) was also implemented. Samples were clearly discriminated by diet using the first component (Fig. 2). The relative abundance of 139 and 151 non-overlapping ASVs were sufficient to classify samples in T1 (Fig. 2a) and T2 (Fig. 2b), respectively.

Sparse Partial‐least‐squares Discriminant Analysis plots of T1 group (a) and T2 group (b) after Amplicon Sequences Variants selection (139 and 151, respectively). The feed factor standard protein (SP) vs low protein (LP) in T1 and carotene-enriched (CE) vs control diet (NC) in T2 factors were used to distinguish groups. At time-point T1, purple color represents the low-protein diet and pink color the standard-protein diet. At time-point T2, purple color represents the carotene-enriched diet and pink color the control diet.
We then applied a more conservative approach and only retained the ASVs that were simultaneously detected by PLS-DA and DA. In T1, 62 ASVs (44%) (Fig. 3a) were commonly identifies in both analyses. The taxonomic classification of the most abundant features in the SP diet included genera such as Dorea, Oscillospira, Faecalibacterium, Campylobacter and Treponema. Conversely, the LP diet was associated with a higher abundance of Anaerovibrio, Bulleidia, CF231, Clostridium, Gemmiger, Phascolarctobacterium, Prevotella, Roseburia and Succinivibrio. Noticeably, Faecalibacterium, Treponema, Clostridium and Prevotella have been previously reported as sensitive genera to dietary protein in pigs43. Thus, in agreement with the observed differences, in pigs, a high-protein diet reduced the abundance of Faecalibacterium prausnitzii and Clostridium coccoides in cecum and colon43, while a low-protein diet increased the proportion of Prevotella in cecum33.

Heat map showing sample distribution using the most commonly identified Amplicon Sequence Variants (ASVs) in the differentially abundant and the sparse partial least squares regression discriminant analyses. At time-point T1, purple color represents the low-protein diet and green color the standard- protein diet. At time-point T2, purple color represents the carotene-enriched diet and green color the control diet.
On the other hand, at T2, 62 ASVs (38%) were commonly identified by both sPLS-DA and DA procedures (Fig. 3b). The taxonomic classification of these ASVs revealed members of Prevotella, Treponema and YRC22 amongst the most DA. In humans, Prevotella species dominate in the gut microbiome of long-term exposure to low-protein high-fibers diets44, Moreover, increased Prevotella abundance has been reported in autistic children treated with very high levels of vitamin A45. As previously noted, Prevotella was among the most discriminant group of bacterial also in T1, were 37% of the AVSs were taxonomic classified as Prevotella. In T2, the most represented genus was also Prevotella (33%) followed by Treponema (8.6%). The ASVs classified as Prevotella were most abundant in the LP diet in T1, in line with the pattern described in humans44. In contrast, in T2 a non-clear taxonomic pattern between diets (CE or NC) was observed. The lack of evidence for a differential distribution of Prevotella genus between CE and NC groups may be explained by the fact that protein intake was the same for these two diets in addition to, as suggested by the PERMANOVA analysis, or by the low impact of dietary carotene on microbiota composition.
Functional metagenomic prediction of dietary modulation of the fecal microbiota
In order to complement the taxonomic analysis (ASVs, phylum and genus) and to identify functions that may be modulated by the dietary interventions, a metagenomic prediction was done using PICRUSt against the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways and COGs (Clusters of Orthologous Groups). The metagenomic prediction based on KOs (KEGG Orthology) revealed that globally the gut microbiota was enriched mainly with functions related to membrane transport, metabolism of carbohydrate and amino acid, replication and repair process, translation and energy metabolism (Supplementary Table S5).
For diversity indexes, the Tukey HSD test46,47 only led to significant differences for the beta-diversity index (p = 0.002) of predicted KOs between T1 and T2. Similarly to taxonomic abundance, the strongest effect for diversity was associated with time-points (T1 vs T2). We also applied a DA test, identifying a total of 1,277 KOs and 1,470 COGs, which happened to be differentially abundant between T1 and T2. Within each time-point, differences between diets were only observed at T1, with 31 KOs and 47 COGs showing differentially abundance. Again, this result suggests that protein rather carotene intake has a higher impact on microbiota. To gain a deeper insight into their functions, we annotated the KO in it corresponding functional classification module. The 31 KOs belonged to functions mainly related with branched-chain amino acid transport system and NADH: quinone oxidoreductase. Interestingly, the module M00237 (branched-chain amino acid transport system), which was the most overrepresented in SP are directly related with products of protein degradation48. This result could be partly explained by the fact that the increased protein intake in SP could trigger the microbial activities related to protein degradation that take place in the colon, where proteolytic bacteria are present17.
The global patterns between T1 and T2 for COGs were similar to those found for KOs. The main differences between time groups include functions related with translation, ribosomal structure and biogenesis, cell wall/membrane/envelope biogenesis and transport and metabolism of amino acid (Supplementary Fig. S1). In T1, the main functions associated to the diets were related to the transport and metabolism of amino acids, coenzymes, carbohydrates and lipids (Supplementary Fig. S2). The analysis of the most significant DA COGs at T1 revealed that members of the family fucose permease (COG0738) were more abundant in LP than in SP. Members of this COG family are involved in the fucose uptake, a monosaccharide related to host–microbe interactions49. To be noted, recent studies suggest that nitrogen availability is likely to be shaped by host–microbe interactions29,30. In addition, long-term reduction of nitrogen have been associated with a higher carbohydrates intake29 that may explained the observed higher COG0738 abundance as well as the higher relative abundance of genera associated with carbohydrate metabolism such as Prevotella, Succinivibrio and Roseburia in the group of pigs fed with LP compared to SP diet.
Medium-term dietary interventions may alter enterotypes stability
In order to evaluate the presence of enterotypes as well as the dietary effect on the enterotype stability, a clustering analysis was done within each time-point. Previous reports have described that there are two main enterotypes in pigs2,9,10, unlike humans, where there are at least three dominant enterotypes50. In agreement with this, the analysis of our data showed that the optimal number of cluster both at T1 and T2 was two (Fig. 4). The classification in enterotype type A (EPA) and enterotype type B (EPB) was not linked to the diet neither at T1 nor at T2. To characterize the cluster taxonomic composition, a DA was done at genera level. Within T1, EPA and EPB samples showed significantly DA of 19 genera, 17 of which were more abundant in EPA than in EPB. The most DA genera in T1 were Turicibacter, Butyrivibrio and Paraeggerthella, whereas the Catenibacterium genus was identified as predominant in EPB (Tables 1 and 2). Interestingly, Lu et al. (2018) reported the genus Turicibacter as the dominating genus of one of the pig enterotypes in 15-week-old pigs. Similar patterns, in terms of the number of DA genera were observed at T2, with 14 DA genera, 13 of which were more abundant in T2-EPA including Faecalibacterium, Dorea and Succinivibrio. The Alistipes genus was the only one that was more abundant in the T2-EPB.

Evolution/transition of pigs from time-point T1 (165 days of age) to time-point T2 (195 days of age). The red color represents EPA: Enterotype like cluster A; the blue color represents EPB: Enterotype like cluster B. CE: carotene-enriched, NC: not carotene, control diet.
In our study we used two time points (T1 vs T2), therefore, we have the opportunity to test the sample stability in each enterotype across T1 and T2. Within T1, 14 samples clustered in the EPA and 13 in EPB. This classification did not match the protein diet groups. However, at T2 the sample assignation was not as balanced as observed in T1 (Fig. 4). In addition, transitions between the cluster assignations were observed indicating low enterotype stability across time. For instance, only ≈62% of samples remained in the corresponding enterotype in T2 compared to T1 (Fig. 4). In human, the effect of dietary interventions on enterotype resilience has been investigated51. In short-term dietary interventions, the microbial community recovers its original status in a few days, including the enterotypes, which remain stable, particularly as compared to long-term perturbations51. In healthy pigs, after weaning and under regular management conditions (i.e., diet and absence of antibiotic treatment), the diversity of microbiota increases6 which, once the enterotype is established, does not change substantially over time2,28. However, in a recent research10, working with samples taken at weaning (18 d), 15 weeks after weaning (≈118 d) and 196 d, reported a similar percentage of enterotype shift between samples as we have seen here between 165 d and 195 d. Therefore, the low enterotype stability observed in our study may be explained by the perturbation due to the dietary changes occurred from T1 to T2 as well as the effect of the inclusion of different levels of proteins, the interaction between diet or the age of animals studied. In fact, in line with the enterotype taxonomic composition at T2, the most abundant genera in the SP diet were Dorea, Oscillospira and Faecalibacterium. Faecalibacterium is a commensal bacterium, commonly reported in the gastrointestinal tracts of animals and humans as one of the main butyrate producers bacteria in the intestine52,53, that has been suggested as a marker of gut health and host wellbeing in humans54,55. As mentioned before, in pigs, high-protein diets reduce the proportion of Faecalibacterium43 and low-protein diets increase the proportion of Prevotella33. At T2, all pigs were subjected to a low-protein diet, which may explain the observed higher proportion of both Faecalibacterium and Prevotella. Moreover, co-occurrence (i.e., positive correlations) between both genera9 have also been reported in pigs. Supporting previous findings, a moderate positive correlation between Prevotella and Faecalibacterium was observed, both at T1 (R = 0.44) and T2 (R = 0.26).
To the best of our knowledge, the present study is the first to provide a comprehensive overview of the effect of dietary intervention of protein and pro-Vitamin A carotenoids on the structural and functional composition of pig gut microbiota. Although these factors do not have a relevant impact on community diversity, our results show that dietary protein variations have a stronger effect than carotenes on the compositional and functional structure of the pig gut microbiota. Prevotella, Faecalibacterium, Treponema and Clostridium were the genera most influenced by dietary protein, which contributed to the functional classification related with the degradation of protein. Our findings also suggest that enterotype stability can be affected even after medium-term dietary interventions. Additional longitudinal studies are needed to better characterize enterotype stability across fattening, for instance overcoming one of the limitations of our experiment, which was the different duration of protein and carotene interventions. In summary, our results contribute to the knowledge of microbial modulation in pigs and can be useful to study the inclusion of optimal dietary protein to reduce the cost and the environmental impact of pig production without affecting growth performance.
Method
Ethics statement
All experimental procedures were approved by the Ethics Committee for Animal Experimentation of the University of Lleida (agreement CEEA 02-04/14) and were performed in accordance with authorization 7704 issued by the Catalan Ministry of Agriculture, Livestock, Fisheries and Food, Spain.
Animals and experiment design
Feces samples were collected from 32 barrows of a pure Duroc line produced by 23 dams and 7 sires. A detailed description of the management and feeding conditions of these pigs during the experiment have been previously reported22. Briefly, all pigs were born within the range of 4 days (d) and castrated within the first week of age. At 70 d of age, they were moved to an experimental research center (Centre d’Estudis Porcins, Torrelameu, Lleida, Spain). There, they were allocated in 8 pens of 4 pigs and raised under identical conditions except the diet (Supplementary Fig. S3). During the growing-finishing phase, from 70 to 165 d of age, a different cereal-based protein diet was given to four alternate pens. Thus, half of the pigs were fed with a standard-protein (SP) diet (17%, 16%, and 15% of crude protein, from 70 d to 110 d, from 110 d to 140 d, and from 140 d to 165 d, respectively) and half with a low-protein (LP) diet (15%, 14%, 13% of crude protein from 70 d to 110 d, from 110 d to 140 d and from 140 d to 165 d, respectively). The 2% differences in protein content are not expected to impact pig performance but can help reduce nitrogen emissions56. At the start of the finishing phase, pigs were redistributed across pens so that in each pen there were two pigs fed with the SP diet and two pigs fed with the LP diet. During the finishing phase, from 165 to 195 d, a different cereal-based carotene diet (12% crude protein, without supplemented vitamin A) was given to four alternate pens. The two diets supplied in this phase were identical except for the corn line used in the feed formulation. Thus, half of the pigs were fed with a carotene-enriched (CE) diet (20% of M37W-Ph3 carotenoid-fortified corn)21 and half with a control (NC) diet (20% of a near-isogenic M37W corn, which only contains traces of carotenoids). The NC and CE diets were estimated to provide 1,300 IU and at least 2,100 IU of vitamin A/kg. At all phases, sires were equally represented by diet. Fecal samples were collected at 165 d (T1) and 195 d (T2), i.e. at the end of the growing-fattening and finishing phases, respectively, from all the pigs in the experiment. The diet composition in each group (SP vs LP) were formulated following the recommendation of Spanish Foundation for the Development of Animal Nutrition (FEDNA, 2013)56. A detailed description of the ingredients and nutrient content of the diets used during the experiment can be found in Supplementary Tables S6 and S7.
DNA extraction, PCR amplification, and sequencing
Total bacterial DNA from feces was isolated using the DNeasy PowerSoil Kit (QIAGEN, Hilden, Germany), according to the manufacturer instructions with the following modifications: Frozen wet fecal samples (0.1 g) were disrupted with 3 glass beads in a bead homogenizer (BeadMill 4, ThermoFisher) in presence of buffer C1, and incubated at 95 °C for 5 minutes. These steps were repeated twice. Sample tubes were spun at 10,000 g for 30 s and the supernatant transferred to a new tube to follow with the manufacturer instructions. In the final step, the DNA was eluted from the column in a volume of 100 µl. The DNA concentration and quality were estimated by spectrometry (Nanodrop-100) and fluorometry with the dsDNA HS Assay kit (Qubit 4, Invitrogen, Carlsbad, CA). The microbiota composition was assessed by amplifying the V3 region of the 16S rRNA gene using primers adapted from57 with NEXTERA dual index specific tails for the forward: 5′TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG and Reverse: 5′GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG primers. Purified amplicons were paired-end sequenced (2 × 250 nt) on an Illumina MiSeq (Illumina, San Diego, CA, USA) at FISABIO (Valencia, Spain).
Bioinformatics and statistical analysis of 16S rRNA gene sequences data
The fastq files were processed using QIIME2 software58. Cleaned sequencing reads were obtained after remove the primers, barcode sequences, and the low quality reads from the raw data. Paired-end reads were merged, and quality control implementation allowed the retention of sequences with a length of 250 bp and with a mean sequence quality score >30. Sequences with mismatched forward and/or reverse primers were omitted. The cleaned 16S rRNA gene sequences were process into Amplicon Sequences Variants (ASVs) at 99% of identity. All reads were classified to the lowest possible taxonomic rank using QIIME258 and the GreenGenes Database v13.5 as reference. Samples with less than 3000 reads (n = 3) were excluded and not considered in posteriors analysis.
The diversity index of bacterial community was assessed with the Vegan package59. Alpha and richness diversity indexes were evaluated with the Chao160 and Shannon61 index. Between-sample (beta) diversity was calculated using the unweighted and weighted UniFrac62 distances and Bray-Curtis63 dissimilarities. Principal-coordinate analysis (PCoA) was used to visualize these distances using Emperor. The zero-inflated Gaussian mixture model implemented in the fitZig function of the MetagenomeSeq R package64 was used to identify the ASVs that differed significantly between (T1 and T2) and as function of the diets within each time-points group (SP vs LP, in T1 and CE vs NC, in T2). The Adonis function implemented in Vegan package59 was employed to perform a permutational multivariate analysis of variance (9,999 permutations) to the Bray-Curtis63 dissimilarities. In addition, to identify a subset of ASVs that discriminate samples according to diets T1 (SP vs LP) and T2 (CE vs NC), Sparse Partial‐least‐squares Discriminant Analysis (sPLS-DA) was implemented using the Mixomics package65.
Finally, enterotype-like cluster detection was done using the genera abundance in each sample as described in Arumugam50. In brief, samples clusters were detected using the probability distribution distance metric related to Jensen-Shannon divergence and the Partitioning Around Medoids Arumugam50. The optimal number of cluster was determined following the Calinski-Harabasz (CH) Index66 (R Script available at: http://enterotype.embl.de/enterotypes.html).
Functional prediction
We explored the functional capacity of the microbiota by inferring metagenomics functionality from the 16S rRNA gene sequencing data using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) software (version 1.1.2)67. The corresponding know OTUs (Greengenes v13.5 database) from the ASVs were used to predict the functionality of our sequenced 16S rRNA against the precalculated files corresponding to KEGG (Kyoto Encyclopedia of Genes and Genomes, version 87.1)68 pathways and COGs (Clusters of Orthologous Groups).
Data Availability
All data supporting the findings of this study are available within the article and Supplementary Information, or are available from the corresponding author upon reasonable request.
References
Flint, H. J., Scott, K. P., Louis, P. & Duncan, S. H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 9, 577–589 (2012).
Mach, N. et al. Early-life establishment of the swine gut microbiome and impact on host phenotypes. Environ. Microbiol. Rep. 7, 554–569 (2015).
Dou, S. et al. Characterisation of early-life fecal microbiota in susceptible and healthy pigs to post-weaning diarrhoea. PLoS One 12, e0169851 (2017).
Pajarillo, E. A. B. et al. Characterization of the fecal microbial communities of Duroc pigs using 16S rRNA gene pyrosequencing. Asian-Australasian J. Anim. Sci. 28, 584–91 (2015).
Yang, L., Bian, G., Su, Y. & Zhu, W. Comparison of faecal microbial community of Lantang, Bama, Erhualian, Meishan, Xiaomeishan, Duroc, Landrace, and Yorkshire sows. Asian-Australas. J. Anim. Sci. 27, 898–906 (2014).
Xiao, L. et al. A reference gene catalogue of the pig gut microbiome. Nat. Microbiol. 1, 16161 (2016).
Richards, J. D., Gong, J. & de Lange, C. F. M. The gastrointestinal microbiota and its role in monogastric nutrition and health with an emphasis on pigs: Current understanding, possible modulations, and new technologies for ecological studies. Can. J. Anim. Sci. 85, 421–435 (2005).
Yang, H. et al. Unraveling the fecal microbiota and metagenomic functional capacity associated with feed efficiency in pigs. Front. Microbiol. 8, 1555 (2017).
Ramayo-Caldas, Y. et al. Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J. 10, 2973–2977 (2016).
Lu, D. et al. Host contributes to longitudinal diversity of fecal microbiota in swine selected for lean growth. Microbiome 6, 4 (2018).
Biesalski, H. K. Nutrition meets the microbiome: micronutrients and the microbiota. Ann. N.Y. Acad. Sci. 1372, 53–64 (2016).
McCullough, F. S., Northrop-Clewes, C. A. & Thurnham, D. I. The effect of vitamin a on epithelial integrity. Proc. Nutr. Soc. 58, 289–93 (1999).
Amit-Romach, E., Uni, Z., Cheled, S., Berkovich, Z. & Reifen, R. Bacterial population and innate immunity-related genes in rat gastrointestinal tract are altered by vitamin A-deficient diet. J. Nutr. Biochem. 20, 70–77 (2009).
Cha, H. R. et al. Downregulation of Th17 Cells in the small intestine by disruption of gut flora in the absence of retinoic acid. J. Immunol. 184, 6799–6806 (2010).
Singh, R. K. et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 15, 73 (2017).
Fan, P. et al. Metabolites of dietary protein and peptides by intestinal microbes and their impacts on gut. Curr. Protein Pept. Sci. 16, 646–54 (2015).
Macfarlane, G. T., Cummings, J. H. & Allison, C. Protein Degradation by Human Intestinal Bacteria. Microbiology 132, 1647–1656 (1986).
Qiu, K. et al. Dietary protein level affects nutrient digestibility and ileal microbiota structure in growing pigs. Anim. Sci. J. 89, 537–546 (2018).
Nyachoti, C. M., Omogbenigun, F. O., Rademacher, M. & Blank, G. Performance responses and indicators of gastrointestinal health in early-weaned pigs fed low-protein amino acid-supplemented diets. J. Anim. Sci. 84, 125–34 (2006).
Park, S. J. et al. Genetic, management, and nutritional factors affecting intramuscular fat deposition in beef cattle - A review. Asian-Australas. J. Anim. Sci. 31, 1043–1061 (2018).
Zhu, C. et al. Combinatorial genetic transformation generates a library of metabolic phenotypes for the carotenoid pathway in maize. Proc. Natl. Acad. Sci. USA 105, 18232–7 (2008).
Henriquez-Rodriguez, E. et al. Carotenoid intake and SCD genotype exert complementary effects over fat content and fatty acid composition in Duroc pigs. J. Anim. Sci. 95, 2547–2557 (2017).
Xiao, Y. et al. Comparative biogeography of the gut microbiome between Jinhua and Landrace pigs. Sci. Rep. 8, 5985 (2018).
Isaacson, R. & Kim, H. B. The intestinal microbiome of the pig. Anim. Heal. Res. Rev. 13, 100–109 (2012).
Kim, H. B. et al. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet. Microbiol. 153, 124–133 (2011).
Zhang, N., Ju, Z. & Zuo, T. Time for food: The impact of diet on gut microbiota and human health. Nutrition 51–52, 80–85 (2018).
Ghosh, S. et al. Short chain acyl-CoA dehydrogenase deficiency and short-term high-fat diet perturb mitochondrial energy metabolism and transcriptional control of lipid-handling in liver. Nutr. Metab. (Lond). 13, 17 (2016).
Han, G. G. et al. Tracing of the fecal microbiota of commercial pigs at five growth stages from birth to shipment. Sci. Rep. 8, 6012 (2018).
Holmes, A. J. et al. Diet-microbiome interactions in health are controlled by intestinal nitrogen source constraints. Cell Metab. 25, 140–151 (2017).
Reese, A. T. et al. Microbial nitrogen limitation in the mammalian large intestine. Nat. Microbiol. 3, 1441–1450 (2018).
Fragiadakis, G. K. et al. Links between environment, diet, and the hunter-gatherer microbiome. Gut Microbes 10, 216–227 (2019).
Fan, P., Liu, P., Song, P., Chen, X. & Ma, X. Moderate dietary protein restriction alters the composition of gut microbiota and improves ileal barrier function in adult pig model. Sci. Rep. 7, 43412 (2017).
Zhou, L., Fang, L., Sun, Y., Su, Y. & Zhu, W. Effects of the dietary protein level on the microbial composition and metabolomic profile in the hindgut of the pig. Anaerobe 38, 61–69 (2016).
Heo, J. M. et al. Gastrointestinal health and function in weaned pigs: a review of feeding strategies to control post-weaning diarrhoea without using in feed antimicrobial compounds. J. Anim. Physiol. Anim. Nutr. (Berl). 97, 207–237 (2013).
Bikker, P. et al. Dietary protein and fermentable carbohydrates contents influence growth performance and intestinal characteristics in newly weaned pigs. Livest. Sci. 108, 194–197 (2007).
Rist, V. T. S., Weiss, E., Eklund, M. & Mosenthin, R. Impact of dietary protein on microbiota composition and activity in the gastrointestinal tract of piglets in relation to gut health: a review. Animal 7, 1067–1078 (2013).
Haenen, D. et al. A diet high in resistant starch modulates microbiota composition, SCFA concentrations, and gene expression in pig intestine. J. Nutr. 143, 274–283 (2013).
Frese, S. A., Parker, K., Calvert, C. C. & Mills, D. A. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome 3, 28 (2015).
Zhu, Y. et al. Beef, chicken, and soy proteins in diets induce different gut microbiota and metabolites in rats. Front. Microbiol. 8, 1395 (2017).
Kar, S. K. et al. Dietary protein sources differentially affect microbiota, mTOR activity and transcription of mTOR signaling pathways in the small intestine. PLoS One 12, e0188282 (2017).
Kim, E., Kim, D. B. & Park, J. Y. Changes of mouse gut microbiota diversity and composition by modulating dietary protein and carbohydrate contents: A pilot study. Prev. Nutr. food Sci. 21, 57–61 (2016).
Kim, E., Coelho, D. & Blachier, F. Review of the association between meat consumption and risk of colorectal cancer. Nutr. Res. 33, 983–994 (2013).
Liu, X. et al. High-protein diet modifies colonic microbiota and luminal environment but not colonocyte metabolism in the rat model: the increased luminal bulk connection. Am. J. Physiol. Liver Physiol. 307, G459–G470 (2014).
Wu, G. D. et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108 (2011).
Liu, J. et al. Effect of vitamin A supplementation on gut microbiota in children with autism spectrum disorders - a pilot study. BMC Microbiol. 17, 204 (2017).
Yandell, B. Practical data analysis for designed experiments. (Chapman & Hall, 1997).
Miller, R. Simultaneous statistical inference, https://doi.org/10.1007/978-1-4613-8122-8 (Springer-Verlag, 1985).
Davila, A. M. et al. Intestinal luminal nitrogen metabolism: role of the gut microbiota and consequences for the host. Pharmacol. Res. 68, 95–107 (2013).
Becker, D. J. & Lowe, J. B. Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R–53R (2003).
Arumugam, M. et al. Enterotypes of the human gut microbiome. Nature 473, 174–180 (2011).
Costea, P. I. et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 3, 8–16 (2018).
Barcenilla, A. et al. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl. Environ. Microbiol. 66, 1654–61 (2000).
Stewart, C. S., Hold, G. L., Duncan, S. H., Flint, H. J. & Harmsen, H. J. M. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 52, 2141–2146 (2002).
Lopez-Siles, M., Duncan, S. H., Garcia-Gil, L. J. & Martinez-Medina, M. Faecalibacterium prausnitzii: from microbiology to diagnostics and prognostics. ISME J. 11, 841–852 (2017).
Miquel, S. et al. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 16, 255–261 (2013).
FEDNA|Spanish foundation for the development of animal nutrition. Fundacionfedna.org at, http://www.fundacionfedna.org (2019).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1 (2013).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology 37, 852–857 (2019).
Oksanen, J. et al. Vegan: community ecology package. R package version 2, 4–6 (2018).
Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 11, 265–270 (1984).
Shannon, C. E. A mathematical theory of communication. Bell Syst. Tech. J. 27, 379–423 (1948).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–35 (2005).
Beals, E. W. Bray-Curtis ordination: an effective strategy for analysis of multivariate ecological data. Adv. Ecol. Res. 14, 1–55 (1984).
Paulson, J. N., Stine, O. C., Bravo, H. C. & Pop, M. Differential abundance analysis for microbial marker gene surveys. Nat. Methods 10, 1200–1202 (2013).
Rohart, F., Gautier, B., Singh, A. & Lê, C. K. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLOS Computational Biology 13, e1005752 (2017).
Calinski, T. & Harabasz, J. A dendrite method for cluster analysis. Commun. Stat. - Theory Methods 3, 1–27 (1974).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Acknowledgements
This study supported by the Spanish Ministry of Economy and Competitiveness and the European Union Regional Development Funds (AGL2015-65846-R). Y. Ramayo-Caldas was funded by the European Union H2020 Research and Innovation programme under Marie Skłodowska-Curie grant (P-Sphere) agreement No. 6655919. ES is a recipient of a PhD fellowship from the University of Lleida. RG is a recipient of a postdoctoral fellowship from the UdL-Impuls program. We acknowledge the staff at Selección Batallé for their cooperation.
Author information
Authors and Affiliations
Contributions
J.E. and R.N.P. conceived and designed the experiment; A.R. performed the experiment and extracted fecal samples. E.S. and R.N.P. analyzed the data in laboratory. R.G. and Y.R.C. realized the bioinformatics analysis of the data. R.G., R.N.P., J.E. and Y.R.C., wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41598_2019_51136_MOESM2_ESM.xlsx
Supplementary Table S1. Amplicon Sequences Variants (ASVs) identified in the presence-absence test between time-points for ASVs
41598_2019_51136_MOESM3_ESM.xlsx
Supplementary Table S2. Amplicon Sequences Variants (ASVs) identified in the differentially abundant test between time-points for ASVs.
41598_2019_51136_MOESM4_ESM.xlsx
Supplementary Table S3. Amplicon Sequences Variants (ASVs) identified in the differentially abundant test between time-points for phyla
41598_2019_51136_MOESM5_ESM.xlsx
Supplementary Table S4. Amplicon Sequences Variants (ASVs) identified in the differentially abundant test between time-points for genera
41598_2019_51136_MOESM6_ESM.xlsx
Supplementary Table S5. Functional modules of Kyoto Encyclopedia of Genes and Genomes Orthology (KOs) identified as differentially abundant
41598_2019_51136_MOESM7_ESM.pdf
Supplementary Tables S6 and S7. Detailed description of the ingredients and nutrient content of the diets used during the experiment.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
González-Prendes, R., Pena, R.N., Solé, E. et al. Modulatory Effect of Protein and Carotene Dietary Levels on Pig gut Microbiota. Sci Rep 9, 14582 (2019). https://doi.org/10.1038/s41598-019-51136-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51136-6
This article is cited by
-
Impacts of weaning weights and mycotoxin challenges on jejunal mucosa-associated microbiota, intestinal and systemic health, and growth performance of nursery pigs
Journal of Animal Science and Biotechnology (2022)
-
Metagenomic Analysis of the Faecal Microbiome of Rats with 1, 2-Dimethylhydrazine Induced Colon Cancer and Prophylactic Whole-Cell Carotenoid Intervention
Indian Journal of Microbiology (2021)
-
A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
